
ABSTRACT
Introduction
Obstructive sleep apnea (OSA) is a common and serious breathing disorder. Several pathophysiological factors predispose individuals to OSA. These factors are quantifiable, and modifiable pharmacologically.
Areas covered
Four key pharmacotherapeutic targets are identified and mapped to the major determinants of OSA pathophysiology. PubMed and Clinicaltrials.gov were searched through April 2023.
Expert opinion
Target #1: Pharyngeal Motor Effectors. Increasing pharyngeal muscle activity and responsivity with noradrenergic-antimuscarinic combination is central to recent breakthrough OSA pharmacotherapy. Assumptions, knowledge gaps, future directions, and other targets are identified. #2: Upper Airway Sensory Afferents. There is translational potential of sensitizing and amplifying reflex pharyngeal dilator muscle responses to negative airway pressure via intranasal delivery of new potassium channel blockers. Rationales, advantages, findings, and potential strategies to enhance effectiveness are identified. #3: Chemosensory Afferents and Ventilatory Control. Strategies to manipulate ventilatory control system sensitivity by carbonic anhydrase inhibitors are supported in theory and initial studies. Intranasal delivery of agents to stimulate central respiratory activity are also introduced. #4: Sleep-Wake Mechanisms. Arousability is the fourth therapeutic target rationalized. Evolving automated tools to measure key pathophysiological factors predisposing to OSA will accelerate pharmacotherapy. Although not currently ready for general clinical settings, the identified targets are of future promise.
1. Introduction
Sleep problems significantly diminish health, alertness, safety, and quality of life. The root cause of the most common and serious sleep disorder is impaired breathing. In North America, obstructive sleep apnea (OSA) occurs in approximately 5% of women and 15% of men, with obesity worsening this prevalence [Citation1–3]. Globally, it is estimated that over 935 million people have mild to severe sleep apnea, and 425 million have moderate to severe sleep apnea [Citation4]. This public health burden of sleep-disordered breathing increases the risk for a wide range of adverse health outcomes including diabetes, obesity, cardiovascular disease, and cognitive impairment [Citation5,Citation6].
Continuous positive airway pressure (CPAP) is effective in OSA treatment through keeping the upper airspace open as a conduit for airflow [Citation7], but is limited by patient compliance. For example, approximately 50% of OSA patients do not tolerate or use CPAP or use it less than 4 h per night [Citation8–10]. Other therapies include targeting anatomical predisposition to OSA via upper airway surgery, intra-oral devices, avoiding the supine sleeping position, weight loss, as well as surgical implantation of devices to stimulate motor nerve activity to the tongue musculature [Citation11–16]. Given the recent significant breakthroughs in pharmacological manipulation of pharyngeal collapsibility and reductions in OSA severity, this narrative review will focus mainly on ‘druggable’ therapeutic targets for OSA and the physiological principles and approaches, challenges, and emerging strategies for their use. Such pharmacotherapies may be especially useful in OSA patients not accepting or tolerating CPAP.
2. Key targets in physiological principle
It is an important, and now widely recognized, key principle in OSA pathophysiology and treatment that one (or more) factors can be critical to OSA within and between patients [Citation17–22]. In the context of within a patient, for example, some of the major predisposing factors can vary in their contributions across sleep states and body positions and as such vary over time across the night [Citation22]. Overall, one (or more) of these pathophysiological factors are amenable to targeted manipulation if they are recognized as significant to pathophysiology in that individual, i.e. a ‘personalized medicine’ approach to treatment.
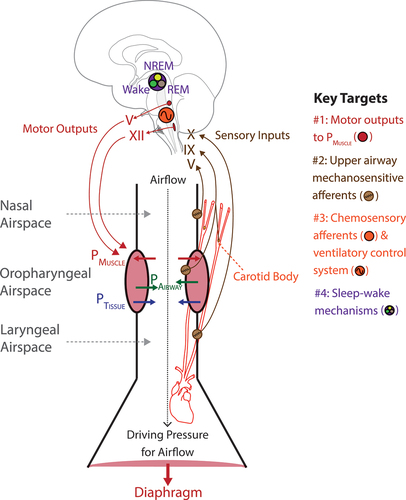
Ultimately, key targets and strategies for therapeutic interventions for OSA are based on the structural organization of the respiratory control system relevant to OSA pathophysiology. identifies such key therapeutic targets for OSA mapped to this structural organization.
Figure 1. Schematic diagram identifying the key targets for OSA pharmacotherapy mapped to the structural organization of the respiratory control system relevant to OSA pathophysiology. The major targets are #1, pharyngeal motor effectors; #2, upper airway sensory afferents mediating reflex pharyngeal dilator muscle responses to sub-atmospheric airway collapsing pressures; #3, chemosensory afferents and the ventilatory control system; and #4, sleep-wake mechanisms. Airway narrowing forces include sub-atmospheric pharyngeal airway pressure generated during inspiration (PAirway) and the positive pressure of the tissues surrounding the airspace (PTissue). These airway-collapsing and closing forces are opposed by the pressure generated by pharyngeal muscle activation (PMUSCLE) which can take the form of tonic and phasic activity which stiffen and enlarge the airspace. The cranial nerves mediating sensory inputs and motor outputs are indicated. See text for further details. Other abbreviations: REM, rapid eye movement sleep, NREM, non- rapid eye movement sleep.
2.1. Pharyngeal motor effectors
The upper airspace is an obligatory conduit for airflow into the lungs, but it also has the property of a collapsible tube (). This property is essential to its non-breathing functions that require dynamic changes in airway size to move air, liquids, and solids during behaviors such as vocalization, suckling, chewing, and swallowing. To enable such motor activities, the upper airspace is surrounded by a complex anatomical arrangement of skeletal muscles and soft tissues. This structural arrangement is unlike the trachea and bronchi, for example, which are more rigid and non-collapsible as they are supported by cartilage. This property of a collapsible pharynx can compromise the essential respiratory function as a conduit for airflow. The pharynx is particularly vulnerable to closure during sleep when pharyngeal muscle tone is diminished.
The mechanical properties of the upper airspace have been well characterized [Citation23–25] and only the key points relevant to this review focussing on therapeutic targets are identified here. Ultimately, the pharynx is the collapsible region of the upper airway situated between two non-collapsible regions above and below. Contraction of the diaphragm and other primary respiratory muscles creates the driving sub-atmospheric airway pressures necessary for airflow into the lungs (). The resulting pressure gradient along the conducting airways from nose to lung includes sub-atmospheric airway pressures in the pharynx that constitute an airway narrowing force (PAirway, ). The positive pressure applied on the collapsible airway by the surrounding tissue also constitutes a closing force (PTissue, ). PTissue results from the tissue and bony structures that surround the airspace, with the tissues constrained within those bony structures and compartment (i.e. the mandible and spinal vertebrae).
Importantly, the airway collapsing and closing forces are opposed by the pressure generated by pharyngeal muscle activation (PMUSCLE, ). Pharyngeal muscle tone is expressed as tonic activity (i.e. the prevailing continuous or background tone) and phasic activity (i.e. respiratory, and non-respiratory motor drives during breathing or other behaviors). This tonic and phasic expression of PMUSCLE acts to stiffen and enlarge the airspace. Key pharyngeal muscles include those of the tongue and soft palate innervated by the hypoglossal and trigeminal motor nuclei in the medulla and pons via cranial nerves XII and V respectively.
In the final analysis, the airway pressure required to close the pharyngeal airspace is identified by the critical closing pressure (PCrit) [Citation23–25]. As the pharyngeal airway is modeled effectively as a collapsible tube, maximum flow (VMAX) in this airway segment is determined by upstream nasal pressure (PN) and resistance (RN) [Citation25–27]. Airflow ceases in the collapsible segment of the upper airway at a value of critical pressure, i.e. the PCRIT. In the pharyngeal airspace, VMAX is determined by the following equation: VMAX = (PN - PCRIT)/RN. Based on this relationship, increases in PN lead to increases in VMAX, and this effect is the essential basis for effective nasal CPAP therapy. Overall, PCrit is strongly influenced by structural anatomy, with smaller upper airway size constituting a structural load that promotes upper airway collapsibility. In contrast, neuromuscular activity opposes the structural load and promotes upper airway stability [Citation22,Citation28].
Overall, this summary identifies the physiological rationale for PMUSCLE being a key modifiable determinant of upper airway collapsibility and a key therapeutic target for OSA via strategies to directly target hypoglossal and trigeminal motor outputs (Key Target #1. ).
2.2. Upper airway sensory afferents
The nasal, oropharyngeal, and laryngeal airspaces experience sub-atmospheric airway pressures generated during inspiration (). Sub-atmospheric airway pressure leads to reflex activation of the pharyngeal musculature [Citation29,Citation30] to promote upper airway stability via an increase in PMUSCLE ().
Sensory afferents in the nasal, oropharyngeal, and laryngeal mucosae and sub-mucosae mediate this reflex response via the trigeminal (V), glossopharyngeal (IX) and vagus (X) nerves () with nasal and laryngeal afferents providing the largest contribution [Citation29]. During OSA, closure of the oropharyngeal airspace leads to progressively larger inspiratory suction pressures generated against the occluded airway, and consequent exposure of the airspace below the site of obstruction (i.e. the laryngeal airspace) to those large sub-atmospheric pressures. Larger sub-atmospheric airway pressures elicit larger reflex activation of the pharyngeal muscles promoting potential airway reopening and stability [Citation31].
There are three central features of this protective reflex pharyngeal dilator muscle response to sub-atmospheric airway pressure that identify targeting the sensory afferents mediating this reflex as a rational therapeutic strategy for OSA. Firstly, the magnitude of the pharyngeal dilator muscle response to sub-atmospheric airway pressure exhibits considerable variability between individuals but lesser variability within subjects: i.e. individuals have a characteristic ‘strength’ of response [Citation30]. The consequence of this variation between people is that individuals with characteristically smaller responses will be less able to protect their airways from suction collapse compared to individuals with larger responses, i.e. will be more prone to airway narrowing and closure and less able to mount a compensatory neuromuscular response [Citation31]. Differences in neuromuscular compensatory effectiveness is a major determinant of the variability in OSA severity between patients [Citation22]: individuals with a robust neuromuscular response are better able to maintain or restore an open upper airway, even with a high structural (i.e. anatomical) load due to a small airspace, compared to individuals with low neuromuscular compensatory responses.
Secondly, pharyngeal dilator muscle responses to sub-atmospheric airway pressures are reduced in sleep [Citation32–34], further predisposing to airway collapsibility in sleep compared to wakefulness.
Thirdly, individuals with OSA exhibit upper airway tissue and sensory afferent nerve damage [Citation35,Citation36], edema [Citation37], inflammation [Citation38] and reduced upper airway mucosal sensation [Citation39–41]. Such decrements in upper airway sensory responses are likely mediated by the mechanical events associated with nights or years of snoring and repetitive nighttime obstructions such as the pressure swings, tissue distortions, vibrations, and shear forces.
Overall, any decrements in this protective reflex pharyngeal dilator muscle response to sub-atmospheric airway pressures over time may move an individual across the spectrum of upper airway narrowing from normal breathing with effective ventilation and airflow, to flow limitation, to obstructive hypopneas characterized by reduced inspiratory airflow, to OSA with upper airway closure. This summary analysis identifies the physiological rationale for the targeted augmentation of this protective reflex pharyngeal dilator muscle response to oppose suction collapse of the pharyngeal airspace. This can be achieved via direct targeting of the upper airway mechanosensitive afferents that mediate this reflex (Key Target #2. ), e.g. through an inhaled nasal and/or oropharyngeal spray before sleep.
2.3. Chemosensory afferents and modulation of respiratory network activity
Another rational strategy for OSA pharmacotherapy is via modulation of respiratory network activity more generally. This can be achieved by targeting components of the ventilatory control system, including its sensitivity to chemosensory stimulation and modulating overall ‘loop gain’ and ‘CO2 reserve’ [Citation17–19,Citation22,Citation42]. These terms and this strategy are explained below.
Loop gain reflects the overall sensitivity of a feedback control system. A high gain system responds quickly and vigorously to a perturbation, whereas a low gain system responds slowly and weakly. A significant proportion (approximately one-third) of OSA patients exhibited high loop gain when quantified [Citation17]. This finding emphasizes that although PCrit is an important determinant of OSA pathophysiology, other non-anatomical and non-upper airway muscle abnormalities such as loop gain are present to a significant degree in a significant number of OSA patients [Citation17]. Loop gain in the context of the respiratory control system is a modifiable trait and is determined by its two components: ‘plant gain’ and ‘controller gain.’
In control system terminology as related to breathing, the ‘plant’ is the volume of gases stored in the body, including the functional residual capacity of the lungs. The ‘gain’ component is how much a given change in ventilation causes a change in blood gases such as arterial CO2. The ‘CO2 reserve’ is the magnitude of increased ventilation that is sufficient to lower arterial CO2 to a threshold beyond which breathing ceases (i.e. the ‘apnea threshold’). A higher plant gain therefore results in smaller increases in ventilation being necessary to lower arterial CO2 beyond the apnea threshold, and unstable breathing results. Such an effect can occur from wakefulness to sleep, for example, when functional residual capacity decreases.
Likewise, in control system terminology as related to breathing, the ‘controller’ comprises all the chemosensory elements that comprise the ventilatory responses to hypercapnia and hypoxia. The gain component is how much a given change, for example, in arterial CO2 causes a change in ventilation. A higher controller gain increases the predisposition to unstable breathing and repetitive apneas via the effect of also lowering the CO2 reserve and thus making it more likely that the apnea threshold will be crossed by a respiratory disturbance that causes a hyperventilation and subsequent hypocapnia.
As a result, therefore, enhanced loop gain ultimately predisposes to respiratory instability. In the context of this review, loop gain can be modulated pharmacologically, e.g. via potassium (K+) channel modulation of peripheral chemoreceptor activity [Citation43,Citation44], by carbonic anhydrase inhibitors that produce a metabolic acidosis [Citation45–47], or by administration of inhaled O2 [Citation48]. Overall, this summary identifies the physiological rationale for the targeted manipulation of loop gain via pharmacological modulation of chemosensory control of breathing and overall loop gain (Key Target #3. ).
2.4. Reducing sleep disturbance
Arousals from sleep are both a consequence and a predisposing cause of respiratory instability and sleep apnea. Arousal-induced hyperventilation can predispose to subsequent apnea via the prevailing hypocapnia at sleep onset, i.e. depletion of the CO2 reserve and subsequent crossing of the apnea threshold. Arousals from sleep can be increased by certain central nervous stimulants or reduced by sedating agents, and thus are also a pharmacological target in individuals in whom this is a strong predisposing factor in their periodic breathing and OSA [Citation21,Citation49,Citation50]. How sedating agents may also permit increased PMuscle during an obstructive apnea via delaying the arousal response to increasing respiratory stimulation is summarized below from first principles [Citation21,Citation49,Citation50].
A certain degree of PMuscle is necessary for adequate airflow and lung ventilation during sleep within an individual, but in some people PMuscle is below this level resulting in obstructive hypopneas and OSA. During the hypoventilation and OSA there is progressively accumulating chemoreceptor-mediated respiratory stimulation leading to increased PMuscle. In an individual with a low threshold for arousal from sleep, the accumulating chemoreceptor stimulation may cause arousal before the required level of PMuscle is reached for adequate airflow. In this situation, the arousal from sleep itself leads to hyperventilation and thus further predisposition to unstable breathing and recurrent OSA [Citation21,Citation49,Citation50]. In such an individual with a low arousal threshold and OSA, therefore, delaying arousal from sleep with sedative hypnotics may permit the chemical respiratory drive to accumulate further, to the point that PMuscle exceeds the level required for adequate airflow and stable breathing. The one caveat here is that the sedating agent should not adversely affect the PMuscle response to respiratory stimulation per se, to not compromise the net PMuscle achieved during sleep.
Overall, this summary identifies the physiological rationale for the targeted manipulation of sedating agents to preferentially reduce sleep disturbance in OSA, via pharmacological modulation of sleep-wake mechanisms (Key Target #4. ).
2.5. Pharyngeal anatomy
While anatomical factors importantly contribute to PCrit and upper airway collapsibility, the pharmacological modification of anatomical factors is not a major focus of this review given the concentration on physiological factors and respiratory control mechanisms. Nevertheless, the effects on OSA severity of medications targeting upper airway anatomy have been investigated. Medications include nasal decongestants to reduce upper airway resistance, diuretics to reduce upper airway edema due to fluid retention, and weight loss medications [Citation42,Citation48]. The results of 12 such studies have recently been reviewed and summarized, with significant reductions in OSA severity as measured by the apnea-hypopnea index (AHI) in 10 of these studies covering each of nasal decongestants, diuretics, and weight loss medications [Citation42].
Weight loss medications are of particular interest given that obesity is an important and reversible risk factor for OSA [Citation25,Citation51]. In this context, tirzepatide, a glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 (GLP-1) receptor agonist administered subcutaneously once a week for 72-weeks led to large and sustained reductions in body weight in obese subjects [Citation52] (SURMOUNT-1, clinicaltrials.gov, NCT04184622). A phase 3 randomized, double-blind, placebo-controlled trial in obese patients with OSA is ongoing to identify the effect of tirzepatide over 52 weeks on indices of OSA severity, hypoxic burden, and other outcomes (SURMOUNT-OSA, clinicaltrials.gov, NCT05412004). A prior randomized, double-blind study with liraglutide, a GLP-1 analog with a 97% homology to human GLP-1, identified significantly larger reductions in AHI compared to placebo (mean reductions of 12.2 vs 6.1 events per hour respectively after 32 weeks) when administered as an adjunct to diet and exercise in OSA patients that were unwilling or unable to use CPAP [Citation53]. In addition, there were significant reductions in body weight, and improvements in glycated hemoglobin (HbA1c) and systolic blood pressure [Citation53]. Liraglutide is approved for the treatment of type 2 diabetes at once-daily doses up to 1.8 mg and the safety profile at the dose given to the OSA patients (3 mg) was similar to that observed with doses ≤1.8 mg, and the 3 mg dose was ‘generally well tolerated.’ Overall, these results suggest that weight loss medications can improve OSA and related parameters.
3. Challenges
There are a set of inescapable challenges, properties, and an overarching requirement that a therapeutic target must overcome and satisfy to be effective for OSA treatment. Ultimately, an intervention needs to be administered effectively to selectively target one (or more) of the key pathophysiological mechanisms that are primarily responsible for any individual’s OSA (i.e. Targets #1 to #4 as identified in and Section 2). This point becomes more significant because the principal target(s) can be expected to be different between OSA patients depending on which pathophysiological traits are the strongest contributors to OSA in that individual. The resulting caveat is that the interventions targeting the key pathophysiological mechanisms in OSA can vary between patients as well as within a patient in different sleep states and body positions [Citation22], including as a result of primary changes in ventilation and respiratory drive that themselves influence pathophysiology and can be a target for therapy [Citation54,Citation55].
Upon first inspection the above analysis poses a significant challenge and may be the reason that development of druggable targets and treatments for OSA has been a slow boil. Given the rational physiological targets identified in , interventions closest to the primary effectors of OSA pathophysiology and upper airway stability (e.g. motor efferents activating PMuscle and upper airway mechanosensitive afferents mediating reflex activation of PMuscle, Key Targets #1 and #2, ) may theoretically offer more potential strategic priority or effectiveness given their direct connectedness to local motor control and reflex mechanisms that cause and prevent upper airway obstruction in the first place. In contrast, targets further removed from such local pharyngeal motor control mechanisms (e.g. chemosensory afferents and the ventilatory control system, and sleep-wake mechanisms, Key Targets #3 and #4, ) may theoretically offer less direct and lesser initial strategic priority given their more widespread connectedness leading to potentially more off-target effects. Such off-target effects may include, for example, unwanted effects on autonomic activity or behaviors.
In summary, several pathophysiological factors can predispose an individual to OSA and these ‘endotypes’ are modifiable pharmacologically [Citation17–20,Citation56,Citation57]. Despite this reality, the end-result in all cases of OSA is that the upper airway closes only in sleep and not in wakefulness. As such, OSA is strongly impacted by the impact of sleep mechanisms on pharyngeal muscle tone and compensatory responses on a background of structural (anatomical) load that determines their initial predisposition to airway closure (i.e. small or crowded airways are more vulnerable to collapse in sleep than larger ones). Consequently, one desired result for OSA pharmacotherapy is to stimulate pharyngeal muscle activity during sleep to normal waking levels [Citation20,Citation58].
For this reason, identification of rational targets and strategies to directly manipulate PMuscle, and upper airway mechanosensitive afferents to activate PMuscle are of high priority and the primary focus of this review (). Targeted manipulation of loop gain, via pharmacological modulation of chemosensory afferents and the ventilatory control system, is of topical interest and supported by recent positive clinical studies, and as such is also a focus. Targeted manipulation of sedating agents to preferentially reduce sleep disturbance in OSA via pharmacological modulation of sleep-wake mechanisms has been covered in more detail elsewhere and the interested reader is alerted to these articles [Citation42,Citation48,Citation50,Citation59].
One final and major challenge to the field is how to identify and quantify the contributions of each of the key predisposing pathophysiological factors to OSA within and between individuals: (i) upper airway size and collapsibility; (ii) pharyngeal muscle responsiveness, (iii) ventilatory control sensitivity (‘loop gain’), and (iv) arousal threshold. Only after such identification and quantification would it be known which of the major contributing factor(s) to target and modify within a patient for individualized treatment. One particular challenge will be the development of a convenient method to accurately quantify these traits from standard sleep laboratory measurements that can be simply interpreted and broadly applied, rather than performed and interpreted by a few specialists in only a few laboratories. Steps have been made in this regard [Citation60,Citation61] and their further development and uptake will be a major contribution to diagnosis and treatment going forward.
Nevertheless, it is of topical and lively debate whether the major pathophysiological factors (traits) predisposing an individual to OSA can be reliably determined and quantified from data derived from routine polysomnograms, or whether a dedicated laboratory requiring sophisticated and established interventions and protocols is required [Citation62,Citation63]. In addition to the concerns raised regarding the assumptions behind the trait measurements from polysomnograms and their validation [Citation62], it is also acknowledged that single numerical quantification of each trait obtained from a single night does not necessarily reflect changes and variations in each trait that can occur within a subject overnight (e.g. trait measures can be different across sleep states and body positions) and potentially over days, weeks or years (e.g. with weight gain, alcohol use, medication use, clinical course, changes in sleep patterns and likely many other factors) [Citation62–65]. With recommended amounts of overnight data analyzed, within-night repeatability for trait measurements of upper airway collapsibility, loop gain, and arousal threshold were reported as strong (intraclass correlation coefficients ranging from 0.79 to 0.83) indicating reasonable stability of the measurements, with pharyngeal muscle responsiveness lower at 0.69 [Citation64]. Consistency of measurements over an average interval of 6.5 years was reported as modest, with intraclass correlation coefficients ranging from 0.30 to 0.61, but when accounting for body position and sleep state this improved to 0.36 to 0.63 [Citation64]. How much such consistency or variability of these pathophysiological factors that individually and collectively predispose an individual to OSA impacts the ‘personalized medicine’ approach to treatment via the targeted manipulation of those factors remains to be determined.
4. Therapeutic targets and strategies
4.1. Pharyngeal motor effectors -key target #1,
4.1.1. Noradrenergic-antimuscarinic drug combinations
2019 was a breakthrough year for OSA pharmacotherapy. A landmark paper identified significant improvements in OSA severity in a sample of 20 obese OSA patients treated with a combination of atomoxetine (noradrenergic reuptake inhibitor) and oxybutynin (muscarinic receptor antagonist) before sleep [Citation66]. Significantly, these individual patients were unselected for the pathophysiological traits that predispose them to their OSA (i.e. their ‘endotypes’). This patient group was studied for one night only (i.e. a proof-of-concept study), although a subset of six patients were studied for 1 week with similar findings [Citation66].
With the combination of atomoxetine and oxybutynin the reductions in indices of OSA severity (AHI and oxygen desaturations) were notable both in their consistency and magnitude. Most notably, the reductions in AHI were larger than all previous pharmacological strategies and interventions for OSA [Citation42]. The combination of atomoxetine and oxybutynin lowered AHI for the group of subjects by an average of 63% (interquartile range, 34–86%) from an average of 28.5 (range, 51.6–10.9) to 7.5 (range, 18.6–2.4) events per hour [Citation66]. In the 15 of 20 patients with an AHI over 10 per hour on placebo, the AHI was lowered by an average of 74% (range, 88–62%), with a reduction of ≥50% in all these patients. The drug combinations were effective in significantly reducing OSA severity largely via increasing pharyngeal muscle responsiveness (by ~ 3-fold), the latter identified as increasing genioglossus muscle activity in response to esophageal pressure swings [Citation66].
The findings received significant attention [Citation67,Citation68] and stimulated additional work from other groups that supported the findings using different agents with similar pharmacological targets, e.g. the noradrenergic reuptake inhibitor reboxetine and the muscarinic receptor antagonist scopolamine [Citation69,Citation70]. A number of subsequent studies using different noradrenergic-antimuscarinic drug combinations showed varying efficacy depending on the agents used, their pharmacological profile (e.g. muscarinic receptor subtype affinities), population of OSA patients tested (e.g. only a subset of responders in a sample of Japanese patients), and patients varying in their degrees of upper airway collapsibility and pharyngeal muscle responsiveness during sleep [Citation71–76]. Overall, pharyngeal muscle responsiveness appears to be one of the key pathophysiological traits in human OSA and the strongest contributor to improvements in OSA severity with noradrenergic-antimuscarinic combination pharmacotherapy [Citation17,Citation18,Citation21,Citation42,Citation66,Citation69,Citation71,Citation77–80].
Together, these results identify that meaningful efficacy in reducing OSA severity via pharmacotherapy is possible in theory and initial practice, at least in the short term. The noradrenergic-antimuscarinic combinational strategy illuminates this possibility and provides major impetus and a promising forward direction. This possibility of OSA pharmacotherapy supports examination of key OSA physiological and pathophysiological mechanisms to identify: (i) improved receptor targeting and pharmacologic profile of successful noradrenergic-antimuscarinic combinations, (ii) additional rational targets and strategies, (iii) the patients most likely to benefit as identified by their major pathophysiological traits predisposing them to OSA. Long term effects of such combinational pharmacotherapy on OSA severity, optimal dosing, and the choice of agents are areas of active investigation in large multicenter trials (MARIPOSA, clinicaltrials.gov, NCT05071612).
Potential side effects are also a concern. For example, noradrenergic-antimuscarinic pharmacotherapy can increase cardiovascular activity, although minimal or no changes were observed in the sample of OSA patients in the original study with atomoxetine and oxybutynin: median of 2.6 beats per minute in heart rate and no significant change in blood pressure [Citation66]. Increases in heart rate (average 8–9 beats per minute) with no clinically relevant changes in blood pressure were observed in a longer (1 month) study [Citation81]. Potential other adverse effects such as potentiating arousal and impairments of sleep quality are also of interest and possible importance. Despite the improvements in OSA severity with atomoxetine and oxybutynin, there were no overall significant improvements in subjective sleep quality for the whole group. However, in those patients with an AHI of >10 with placebo, there was a significant increase in objective and subjective sleep quality. The potential for arousal-inducing effects of atomoxetine may have been countered by oxybutynin [Citation66,Citation68]. The authors also reported that no patient experienced severe side effects or adverse events [Citation66].
4.1.2. Noradrenergic-antimuscarinic combinations: rationale and future directions
The choice of a noradrenergic reuptake inhibitor combined with a muscarinic receptor antagonist for testing OSA pharmacotherapy was based on initial animal studies: (i) a muscarinic-receptor-mediated cholinergic pathway linked to G protein-gated inwardly rectifying K+ (GIRK) channels mediates strong genioglossus muscle inhibition in rapid eye movement (REM) sleep [Citation82,Citation83]; (ii) an endogenous noradrenergic drive was identified as the major component of the ‘wakefulness stimulus’ to the hypoglossal motor nucleus, with this excitation being withdrawn in sleep, especially REM sleep [Citation84–88]. These findings from animal studies were the first identification of the major mechanisms controlling output to the pharyngeal muscle in sleep that is central to human OSA.
The noradrenergic-antimuscarinic drug combinations were effective in greatly reducing OSA severity largely via increasing pharyngeal muscle responsiveness (Section 4.1). The general assumption, at least initially, from the clinical studies [Citation42,Citation66,Citation69,Citation71,Citation77,Citation80] and their accompanying editorials [Citation67,Citation68,Citation70] was that the drug combinations were likely effective in producing such beneficial effects on pharyngeal muscle activity and OSA severity because of actions at the hypoglossal motor nucleus and/or other cranial motor nuclei as identified from animal studies. This assumption needs to be tested to pinpoint the mechanism of action and potentially identify more effective agents, refinements, and/or therapeutic targets, e.g. clinical efficacy varies with muscarinic receptor subtype affinities and likely other factors such as predominant endotypes contributing to an individual’s OSA [Citation42,Citation66,Citation69,Citation71,Citation77,Citation78]. What was subsequently identified from animal studies is that oxybutynin, a component of the successful OSA pharmacotherapy, is an effective muscarinic receptor antagonist at the hypoglossal motor nucleus, and that there is a sex-difference in acetylcholine-induced suppression of hypoglossal motor activity, with lesser suppression in females versus males [Citation89]. Nevertheless, the cholinergic cells and circuits responsible for sleep state-dependent modulation of hypoglossal motor activity are currently unidentified. This knowledge is important to help explain why noradrenergic-antimuscarinic combinations have been successful for OSA pharmacotherapy and identify strategies for improvement.
Another major finding from the initial clinical studies was that only combined noradrenergic-antimuscarinic drug administration was effective in reducing OSA severity, with individual administration of each agent alone being ineffective [Citation66]. A recent study, however, identified that the noradrenergic reuptake inhibitor reboxetine alone (single night study) was able to reduce AHI severity although effects were modest at 5.4 events per hour (95% confidence interval, −10.4 to −0.3 events per hour), with no additional effect of oxybutynin [Citation90].
It is known from animal studies that noradrenergic activity is highest in wakefulness, withdrawn in non-REM sleep and minimal in REM sleep [Citation85,Citation86,Citation88,Citation91], with a relative cholinergic inhibition featuring most strongly in REM sleep compared to non-REM sleep [Citation82]. As such, the noradrenergic mechanism may be mainly (but not exclusively) contributing to normal pharyngeal motor suppression in non-REM sleep whereas both noradrenergic and cholinergic mechanisms may be contributing in REM sleep. Consequently, it is to be expected that there is more than one mechanism contributing to net pharyngeal muscle activity in sleep with varying contributions of adrenergic disfacilitation and cholinergic inhibition operating in each sleep state, with both agents generally required to exert enough excitatory-stimulatory and inhibitory-blocking effects on pharyngeal muscle activity and responsiveness to improve AHI severity.
As the number of studies using noradrenergic-antimuscarinic drug combinations has grown there are some indications that an additional (or even major) effect of oxybutynin may be in reducing the arousal-effects of atomoxetine [Citation72,Citation77,Citation92] to promote stable sleep and breathing, and thus contribute to reduced AHI severity. It is even possible that instead of recruitment of a strong cholinergic mechanism that becomes dominantly active in REM sleep to inhibit hypoglossal motor activity, there is the potential for a tonic cholinergic inhibition that is present across all states but which appears strongest in REM sleep when noradrenergic (and other facilitating) inputs are withdrawn [Citation82,Citation93]. Identifying how and why the antimuscarinic component of noradrenergic-antimuscarinic drug combinations contributes to improvements in OSA severity is important to refining optimal targets and treatments and reducing side effects.
4.1.3. Strategy to identify other targets to activate pharyngeal motor effectors
In principle, pharmacological targets that are relatively highly expressed at cranial motoneuron pools (Key Target #1, ) compared to their expression in the rest of the brain offer a strategic opportunity to preferentially promote upper airway stability through their targeting. Such knowledge provides a potential roadmap to focus drug discovery and targeting efforts. Analysis of the Allen Mouse Brain Atlas online database of ~25,000 in situ hybridization experiments [Citation94] was performed to isolate genes that differentiate the hypoglossal motor nucleus and its primary afferent sources from the rest of the brain [Citation20]. There were 1,492 genes with enhanced expression (at least 2-fold) at the hypoglossal motor nucleus and its primary inputs, with expression enhanced up to 33-fold. A total of 99 genes were classified as probable modulators of neuronal activity, of which 18 were classified as G-protein coupled receptors and 37 as ion channels. Ultimately, this unbiased screen identified targets of high interest to modulate hypoglossal and/or pre-motor activity for OSA pharmacotherapy, and provided a database of them with FDA-approved drugs [Citation20].
4.1.4. Thyrotropin-releasing hormone analogs
From the Allen Mouse Brain Atlas analysis, it was identified that thyrotropin-releasing hormone (TRH) receptor ribonucleic acid shows relatively high differential expression at the hypoglossal motor nucleus (6.3‐fold, rank #4 of G-protein coupled receptors) compared to the rest of the brain [Citation20]. For comparison, the noradrenergic and muscarinic targets (α1 and M2 receptors respectively) that are strong controllers of hypoglossal activity in sleep and successful targets in combination OSA pharmacotherapy (Sections 4.1.1 and 4.1.2) were also identified from that screen, being expressed 3.7 and 3.6‐fold higher respectively (rank #11 and #12 of G-protein coupled receptors) at the hypoglossal motor nucleus [Citation20]. Anatomically there are strong projections of TRH-positive neurons to the hypoglossal motor nucleus with excitatory effects on motor activity [Citation95–97].
TRH itself is generally a poor drug candidate for potential therapeutic effects due to its short half-life (<10 min) because of rapid degradation, low intestinal and brain permeability, and endocrine side-effects [Citation98–100]. Taltirelin is a stable brain-penetrating TRH analog that is selective for non-endocrine actions [Citation98,Citation100]. Taltirelin is the first TRH analog approved for human use, in Japan, in the treatment of neurodegenerative disease [Citation98].
Recent animal studies have identified the effects of taltirelin on pharyngeal motor activity, sleep, and breathing [Citation101,Citation102]. The findings identify that taltirelin targeted directly to the hypoglossal motor nucleus elicits sustained and maintained tongue motor activation, unlike TRH itself which is transient and declines in minutes [Citation102]. The sustained activation of the tongue musculature with taltirelin at the hypoglossal motor nucleus persists in sleep [Citation102]. Based on those initial findings with targeted drug delivery to the hypoglossal motor nucleus, a pre-clinical study in rats was subsequently performed with systemic administration of taltirelin to determine effects on sleep and breathing [Citation101]. The protocol consisted of a randomized, within-subject, repeated measures design performed over six intervention study days (each separated by at least 72 h) in chronically instrumented male and female rats with (i) intraperitoneal administration of vehicle controls, (ii) the TRH analog taltirelin at two doses, (iii) all with and without co-administered trazodone. Trazodone was included due to clinical interest in the context of sleep apnea pharmacotherapy as it can suppress arousal without compromising pharyngeal muscle activity [Citation103–110]. The findings showed that systemically administered taltirelin increased tonic and within-breath phasic tongue muscle activity at least for 5 h, with little or no changes in diaphragm amplitude or respiratory rate with taltirelin [Citation101]. Taltirelin also promoted central nervous system arousal as evidenced by suppression of non-REM and REM sleep, increased wakefulness, increased postural muscle tone (as identified from the trapezius muscle) in non-REM sleep, and decreased total electroencephalogram power and delta (0.5–4 Hz) power [Citation101]. These arousal effects were not prevented by trazodone, at least at the dose administered.
Overall, those pre-clinical findings identify taltirelin as a stable upper airway-preferring respiratory stimulant with arousal properties. This property suggests potential utility to promote pharyngeal muscle activity and upper airway stability in some clinical situations that may include post-operative recovery and upper airway management, and/or to counter opioid or sedative-induced pharyngeal, respiratory, and arousal-state depression in the managed clinical setting [Citation43,Citation111–115]. The pre-clinical findings suggest, however, that taltirelin may not be useful by itself for OSA pharmacotherapy without the simultaneous effective targeting of arousability [Citation101]. Arousal threshold itself constitutes a predisposing factor for OSA pathophysiology in some individuals and is a pharmacologically modifiable trait [Citation42,Citation50].
4.1.5. Targeted modulation of select K+ channels in the central nervous system
Some ion channels also exhibit high differential expression at the cranial motor neuron pools such as the hypoglossal motor nucleus compared to the rest of the brain. A particularly notable example is the inwardly rectifying K+ (Kir) channel 2.4 with 28.0‐fold higher expression (rank #2 of ion channels after the N-methyl-D-aspartate (NMDA) receptor) from the Allen Mouse Brain Atlas analysis [Citation20].
Kir2.4 is the most restricted of all Kir subunits in the brain with Kir transcripts expressed almost exclusively in the cranial motoneurons relevant to the maintenance of upper airway stability [Citation116]. This near-exclusive expression of Kir2.4 to cranial motor pools together with the efficacy of Kir channel blockade in restoring waking levels of pharyngeal motor activity throughout sleep suggest that Kir2.4 is a target of interest for OSA [Citation117–119]. The fact that Kir2.4 channels exhibit markedly lower sensitivity to barium compared to other Kir channels [Citation116] indicates that there is something structurally and/or biochemically different about them. However, there are currently no available agents to specifically modulate Kir2.4 channel function [Citation120].
To model the effect of manipulating such a restricted target at cranial motor nuclei, ‘designer receptors’ have been targeted to the hypoglossal motor nucleus and selectively modulated with systemic administration of ‘designer drugs’ [Citation121–124]. These studies involved targeted expression (via adeno-associated viral vectors) of muscarinic receptors that have been engineered to no longer bind endogenous ligands but which can be activated (or inhibited) by synthetic ligands that are otherwise biologically inert: ‘chemogenetics’ [Citation125]. The results showed that selective activation of such a target whose expression is restricted to the hypoglossal motor pool in mice leads to (i) robust, selective, and sustained (up to 8–10 h) reactivation of tongue muscle tone throughout non-REM and REM sleep with no effects on diaphragm or postural muscle activities, or sleep-wake states [Citation122]; (ii) enlargement of the pharyngeal airspace as documented by magnetic resonance imaging [Citation121]; and (iii) improved upper airway patency and breathing during sleep as evidenced by reduced inspiratory flow limitation in obesity-induced sleep-disordered breathing in mice [Citation123].
Downstream effects of activation of the engineered muscarinic receptors (hM3Dq) by the synthetic ligand (clozapine-N-oxide, CNO) include stimulation of membrane bound phospholipase C-β which then causes hydrolysis of phosphatidylinositol bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol. The IP3 can trigger calcium release from the endoplasmic reticulum to activate voltage-dependent calcium channels and contribute to neural depolarization [Citation126,Citation127]. Voltage-gated K+ channels also show increased expression at the hypoglossal motor nucleus (5.8‐fold, rank #11 of ion channels) compared to the rest of the brain [Citation20], and their direct manipulation at the hypoglossal motor nucleus in animal models strongly increases tongue motor activity throughout sleep [Citation117]. In humans, there are small positive effects of targeting voltage-gated K+ channels on genioglossus muscle activity that are limited to REM sleep at the doses tested [Citation128]. However, doses are limited by potential adverse effects such as seizures thus limiting clinical efforts for OSA [Citation128].
Of relevance to Kir2.4, Kir channels require the docking of PIP2 for their activity [Citation129,Citation130] such that CNO-induced hydrolysis of PIP2 increases neuronal excitability through inactivation of such constitutively active K+ channels [Citation126,Citation127]. Accordingly, the activation of the ‘designer receptors’ that are experimentally selectively expressed at the hypoglossal motor nucleus [Citation121–124] may have produced selective activation of the tongue musculature in part by downstream effects that influenced the Kir channels that are naturally selectively expressed at the hypoglossal motor nucleus, with Kir2.4 a strong (albeit difficult) candidate for pharmacological targeting for OSA [Citation117–119].
There is ongoing advancement in the use of viral vectors targeting specific neuronal populations to influence function and disease [Citation131,Citation132]. These advancements support the proposal that the next generation of therapeutics in sleep medicine will be influenced by novel technologies [Citation133]. The use and validation of such genetic approaches via intraoral viral vector delivery to target motor pools that preferentially promote upper airway activity and stability is also broadly applicable to other disorders of motor function with tongue involvement such as motor neuron disease, dysphagia, dysarthria as well as OSA [Citation59,Citation134]. Even if successful in future years as targets and strategies in this therapeutic space are advanced, it remains to be determined if such chemogenetic targeting of central cranial motoneuron pools innervating the muscles relevant to OSA pathophysiology offer more, equivalent, or lesser effects, on upper airway stability compared to, for example, direct stimulation of the motor nerves also supplying these muscles [Citation11,Citation12,Citation16,Citation135].
TWIK-related acid-sensitive K+ (TASK)-1/3 channels also show high differential expression at the cranial motor neuron pools such as the hypoglossal motor nucleus (12.7‐fold, rank #4 of ion channels) compared to the rest of the brain [Citation20]. Theoretically, antagonism of TASK channels directly at the hypoglossal motor nucleus would be expected to augment tongue motor activity, especially during sleep. This is because noradrenaline, glutamate (via group I metabotropic receptors), serotonin, TRH, and Substance P (the latter two co-released with serotonin) all inhibit K+ leak via TASK-1/3 channels leading to motor excitation [Citation96,Citation136]. These neuromodulators are released from awake-active cell groups that contribute to awake hypoglossal motor activity in vivo [Citation84–88,Citation137]. As such, TASK channels are positioned to mediate a common downstream mechanism impacted by the collective reductions in the wake-active neuromodulator inputs to the hypoglossal motor pool in sleep.
Despite this strong theoretical rationale, however, there is remarkably robust maintenance of tongue motor activity despite various strategies for TASK channel manipulation directly at the hypoglossal motor nucleus (Key Target #1. ) in vivo, at least with the agents currently available in the public domain [Citation117,Citation138]. At the present time, data do not support this central target and direction for potential OSA pharmacotherapy. Nevertheless, the identification of TASK-1 channel mutations with causal links to sleep apnea does provide interest and further impetus to identify the physiological roles of TASK channels in respiratory control mechanisms and even providing therapeutic targets down the road [Citation139,Citation140] as potent and selective blockers are being identified [Citation141]. In this context the effects of TASK channel blockade delivered via topical nasal spray to sensitize and amplify reflex pharyngeal dilator muscle response to sub-atmospheric airway pressures are of interest and a specific focus of the following section.
4.2. Augmentation of reflex pharyngeal dilator muscle response to sub-atmospheric airway pressures - key target #2,
4.2.1. Rationale and findings
In the context of augmenting reflex pharyngeal dilator muscle responses to sub-atmospheric airway pressures (Key Target #2, ) via TASK channel blockade, a nasal spray provides for simple and direct delivery to the desired sites of action in the upper airway, with numerous advantageous compared to systemic application. Given the widespread expression of TASK-1/3 channels throughout the brain as well as carotid body chemosensors, and the heart, lung, and pulmonary circulation [Citation142–147], there is higher potential for diverse and unwanted actions with systemic delivery of TASK channel blockers compared to more local actions with direct application targeting upper airway mucosal sensory afferents with oronasal application via a spray [Citation148,Citation149].
Studies in supine anesthetized pigs provided initial evidence for effective augmentation of upper airway muscle reflex activity via targeted pharmacological manipulation of the oronasal airway and the potential for translation to OSA pharmacotherapy [Citation149,Citation150]. This initial study in pigs applied the K+ channel blocker AVE0118 via the nostrils in a slow-release formulation with steady release for over 4 h. AVE0118 showed strong efficacy in sensitizing and amplifying reflex genioglossus muscle responses to large sub-atmospheric airway pressures (−50 to −150 mbar) and prevention of airway collapse [Citation149]. Efficacy was apparent for at least 4 h (the duration of the study) and effective responses would have presumably gone on longer if tested [Citation149]. AVE0118 is a K+ channel blocker with some selectivity for TASK-1 channels, thus leading to reduced K+ leak and increased cellular excitability [Citation142,Citation151]. With intranasal administration, the aim is to preferentially target the superficial upper airway mechanosensitive afferents that mediate reflex pharyngeal dilator muscle response to negative airway pressure (Key Target #2. ).
Subsequent studies in OSA patients were performed in a multicenter, double-blind, randomized controlled trial (SANDMAN) using the selective TASK-1/3 channel blocker BAY 2253651 applied intranasally [Citation152]. However, no significant changes in polysomnographic variables were observed with BAY 2253651 compared to placebo. The final sample size was small (n = 16) as the trial was terminated early due to ineffectiveness. Importantly, it is not known if the protocol led to the hypothesized and anticipated sensitization and amplification reflex pharyngeal dilator muscle response to negative airway pressure, i.e. if the therapeutic target was mechanistically engaged to a meaningful degree with BAY 2253651 as it was in the pre-clinical study with AVE0118 [Citation149]. As such, at the time it was still an open question if targeting upper airway mechanosensitive afferents could lead to clinically meaningful changes in OSA severity if the target was effectively engaged in a clinical population.
A ‘proof of mechanism’ study with a chemically altered follow-up compound (BAY 2586116, a more potent TASK-1 and TASK-3 K+ channel blocker) has since been reported both in anesthetized pigs and OSA patients [Citation148,Citation153]. Application of BAY 2586116 via nasal spray in the anesthetized pigs led to consistent and sustained improvements in pharyngeal collapsibility to negative upper airway pressures that were dose-dependent [Citation148]. Likewise, pharyngeal collapsibility was also improved with BAY 2586116 in sleeping OSA patients (n = 12). As documented by the authors: (i) greater than 80% of patients showed improved PCrit with 160 μg intranasal BAY 2586116, (ii) ~60% showed an improvement of > 2cmH2O, and (iii) in this group the average improvement in PCrit was ~ 4cmH2O, (iv) leading to lower needed effective CPAP pressures, (v) with effects lasting throughout the sleeping period, i.e. at least 7 h of study [Citation148]. Effects of intranasal BAY 2586116 on indices of OSA severity, hypoxic burden, sleep quality and daytime sleepiness are pending further study (clinicaltrials.gov, NCT05527457).
Overall, these results identify that targeting upper airway mechanosensitive afferents to augment reflex pharyngeal dilator muscle response to negative airway pressure is promising at the present time as a potential strategy for OSA pharmacotherapy.
4.2.2. Summary and future directions
Modulation of TASK channels via local targeted pharmacological manipulation of the oronasal airway appears both feasible and effective in principle (Key Target #2. ). Initial evidence identifies that this approach can effectively sensitize upper airway mechanosensitive afferents to augment reflex pharyngeal dilator muscle responses to airway collapsing pressures. Initial studies are encouraging [Citation148–150,Citation153].
Given that individuals with OSA exhibit evidence of upper airway tissue damage, edema, and reduced sensation that can be improved with CPAP treatment [Citation35,Citation37–41], further clinical efficacy may be gained after some reversal of the mucosal damaging effects of years of snoring and apneas by an initial short course of CPAP therapy. Such an approach may further augment the ability of the sensory afferents to respond to the oronasal-applied pharmacological interventions.
4.3. Carbonic anhydrase inhibition to lower loop gain - Key Target #3,
In a recent four week randomized, placebo-controlled, double-blind, dose-guiding study [Citation47], the carbonic anhydrase inhibitor sulthiame led to an average reduction of apnea-hypopnea index of 32% and 41% for 200 mg and 400 mg sulthiame respectively, and >20 apnea-hypopneas per hour for both. There were also significant improvements in overnight oxygen saturation and frequency of arousals from sleep, albeit without significant improvements in daytime symptoms such as sleepiness [Citation47]. The authors report that sulthiame was safe and well-tolerated, although at the 400 mg dose there was a less favorable tolerability profile compared the dose of 200 mg; the most frequently occurring adverse event was short lasting paresthesia [Citation47]. This study was noteworthy as the magnitude and duration (4 weeks) of effect was sustained by a single pharmacological agent. Larger multicenter studies are ongoing (clinicaltrials.gov, NCT 05236842). The authors speculate that a potential future target patient group for sulthiame treatment include: (i) those with residual OSA after treatment with either CPAP, oral appliances, or surgery, or (ii) potential treatment with sulthiame in combination with CPAP or oral appliances [Citation47,Citation154].
Three other studies with the carbonic anhydrase inhibitor acetazolamide likewise identified significant improvements in OSA severity: (i) 1000 mg (250 mg four times per day) for two weeks significantly reduced apnea-hypopnea index by an average of 48% and >20 apnea-hypopneas per hour [Citation155]. However, as previously identified for sulthiame [Citation47], despite the significant improvements in OSA severity with acetazolamide there were no significant improvements in objective sleep quality or patient self-reported symptoms (daytime sleepiness, morning headache, and sleep disturbance). Paresthesia was also reported as common [Citation155]. (ii) 250 mg/day for 1 week significantly reduced apnea-hypopnea index by an average of 28% [Citation156]. However, as with the aforementioned study with acetazolamide there were no changes in objective sleep quality, and paresthesia was also reported [Citation156]. (iii) 500 mg twice a day for 1 week also significantly apnea-hypopnea index by an average of 52% [Citation46]. In the group of 13 subjects, 12 reported paresthesia and taste disturbances, and four reported no difference in sleep quality with the remainder reporting that they slept better. Overall, these consistent side-effects of carbonic anhydrase inhibitors observed over several studies may limit uptake in OSA patients, but larger studies will be able to document if this is a major limiting factor.
These effects of acetazolamide [Citation46,Citation155,Citation156] and sulthiame [Citation47] on OSA severity as judged by percent change in apnea-hypopnea index are typically of larger magnitude than other interventions targeting loop gain including inhaled O2 to lower responsivity of the peripheral chemoreceptors [Citation42]. Carbonic anhydrase inhibitors are thought to exert their positive treatment effects on OSA by decreasing overall loop gain and not via effects on the other contributing factors to OSA pathophysiology such as pharyngeal anatomy, pharyngeal muscle responses to apneas, or arousal threshold [Citation46]. How this beneficial effect on loop gain is achieved, however, is not immediately obvious and deserves some explanation.
Acetazolamide induces bicarbonaturea (i.e. increase HCO3− in the urine) via renal effects, thereby producing a metabolic acidosis (i.e. increased H+ in blood) [Citation45]. This metabolic acidosis increases ventilation, likely via H+ stimulation of the peripheral chemoreceptors () as the blood-brain barrier is impermeable to polar solutes. Recent meta-analyses and physiological model simulations suggest that the positive treatment effects of carbonic anhydrase inhibitors on OSA severity are primarily due to decreases in plant gain over and above any potential changes in controller gain (). The carbonic anhydrase-induced stimulation of ventilation causes a leftward shift of the ventilatory response to CO2 along an individual’s metabolic hyperbola such that eupneic ventilation occurs at a steeper portion of the metabolic relationship [Citation45] (). The physiological impact of this effect is that larger changes in ventilation are required to alter arterial PCO2 sufficiently to cause apneas: i.e. the ‘CO2 reserve’ has been increased via decreases in plant gain that dominates over any potential changes in controller gain (Section 2.3, ). This dominant effect on plant gain lowers overall loop gain and is a stabilizing effect on breathing and OSA [Citation45].
Figure 2. Schematic representation of how carbonic anhydrase inhibitors can exert positive effects on breathing stability through a net decrease in ‘loop gain’ via a decreased ‘plant gain.’ At eupnea the equilibrium point for resting breathing (A) exists at the intersection of the ventilatory response to increased arterial PCO2 (red line, i.e. ‘controller gain’) and the change in arterial PCO2 that results from a change in ventilation (i.e. the metabolic hyperbola; blue line). Any transient increase in ventilation (ΔV1, increase indicated by the upward black arrow, e.g. caused by a transient arousal from sleep) will reduce arterial PCO2 (magnitude of decrease indicated by the leftward black arrow) and elicit an apnea if the resultant hypocapnia reaches the ‘apnea threshold’ as extrapolated from ventilatory response to CO2 (dashed red line intersecting with the downward black arrow). The ‘CO2 reserve’ is the magnitude of change in arterial PCO2 required to reach the apnea threshold. Breathing in sleep is more stable and resilient to transient perturbations in ventilation with a larger CO2 reserve. In response to carbonic anhydrase inhibition there is a leftward shift on ventilation to a new equilibrium point for resting breathing (B) on a steeper region of the metabolic hyperbola. At this position, a larger increase in ventilation (ΔV2) is needed to deplete the CO2 reserve and reach the apnea threshold: i.e. CO2 reserve2 (i.e. after carbonic anhydrase inhibition) is greater than CO2 reserve1. Figure adapted and modified from Schmickl et al. [Citation45], under CC-BY.
![Figure 2. Schematic representation of how carbonic anhydrase inhibitors can exert positive effects on breathing stability through a net decrease in ‘loop gain’ via a decreased ‘plant gain.’ At eupnea the equilibrium point for resting breathing (A) exists at the intersection of the ventilatory response to increased arterial PCO2 (red line, i.e. ‘controller gain’) and the change in arterial PCO2 that results from a change in ventilation (i.e. the metabolic hyperbola; blue line). Any transient increase in ventilation (ΔV1, increase indicated by the upward black arrow, e.g. caused by a transient arousal from sleep) will reduce arterial PCO2 (magnitude of decrease indicated by the leftward black arrow) and elicit an apnea if the resultant hypocapnia reaches the ‘apnea threshold’ as extrapolated from ventilatory response to CO2 (dashed red line intersecting with the downward black arrow). The ‘CO2 reserve’ is the magnitude of change in arterial PCO2 required to reach the apnea threshold. Breathing in sleep is more stable and resilient to transient perturbations in ventilation with a larger CO2 reserve. In response to carbonic anhydrase inhibition there is a leftward shift on ventilation to a new equilibrium point for resting breathing (B) on a steeper region of the metabolic hyperbola. At this position, a larger increase in ventilation (ΔV2) is needed to deplete the CO2 reserve and reach the apnea threshold: i.e. CO2 reserve2 (i.e. after carbonic anhydrase inhibition) is greater than CO2 reserve1. Figure adapted and modified from Schmickl et al. [Citation45], under CC-BY.](/cms/asset/922a065f-32d7-44a1-82aa-a002a90b14ff/iett_a_2240018_f0002_oc.jpg)
Although high loop gain is the key mechanism typically underlying central sleep apnea it is also a significant contributor to OSA [Citation17,Citation157–159]. The meta-analysis performed by Schmickl et al. [Citation45], indicated that the carbonic anhydrase inhibitor acetazolamide decreased plant gain by 32% and overall loop gain by 26%, suggesting minimal or no changes in controller gain. Further modeling simulations indicated that the amount by which acetazolamide caused a leftward shift of the ventilatory response to CO2 along the individual’s metabolic hyperbola was a major determinant of the resulting change in loop gain via the effect on plant gain [Citation45]. Such a dominant effect of effect of carbonic anhydrase inhibition on lowering plant gain via this mechanism seems sufficient to also overcome any tendency for lower lung volumes in sleep or with obesity to increase plant gain or increase upper airway narrowing and resistance [Citation158]. In this context, in a sample of OSA patients, acetazolamide decreased loop gain but did not affect pharyngeal anatomy or pharyngeal muscle responses to apneas, or arousal threshold [Citation46].
4.4. Targeting central respiratory activity (key target #3) relevant to sleep-disordered breathing via other novel strategies
Intranasal delivery of pharmacological agents as a means of targeting selected mechanisms was introduced in Section 4.2 in the context of TASK channel blockers to sensitize and amplify oronasal reflex pharyngeal dilator muscle responses to sub-atmospheric airway pressures. Novel studies using intranasal delivery of other agents to target central respiratory activity (Key Target #3, ) may also have potential future relevance to pharmacotherapy for sleep-disordered breathing and OSA.
Studies identify that mice with diet-induced obesity develop sleep-disordered breathing that is reduced by intranasal administration of leptin [Citation160]. Intranasal leptin increased ventilation in non-REM and REM sleep and decreased oxygen desaturations in REM sleep, whereas intraperitoneal leptin had no effect [Citation160]. The intranasal administration of leptin effectively bypasses the blood-brain barrier across which leptin transport may be impaired in obesity-associated leptin resistance, and thus cleverly identifies that this intranasal route of delivery may be a future target for OSA pharmacotherapy and drug delivery [Citation59,Citation160,Citation161]. These findings further support the proposal that targeting central leptin receptors may be an effective and rational pharmacological strategy in the context of OSA as well as obesity hypoventilation [Citation59,Citation162,Citation163]. Further translational potential and utility of intranasal leptin is indicated by reduced opioid-induced sleep-disordered breathing in pre-clinical studies in mice: intranasal leptin decreased the frequency of morphine-induced apneas, periods of hypoventilation and inspiratory-flow limited breaths without reducing analgesia [Citation164].
Pre-clinical studies in mice also identify that intranasal oxytocin increases respiratory-related tongue motor activity and preferentially activates hypoglossal motoneurons driving the protrusor muscles of the tongue, effects that would be predicted to enlarge and stabilize the pharyngeal airspace [Citation165]. Although intranasal oxytocin has potential therapeutic relevance to several disorders including sleep-disordered breathing, effects on OSA severity and breathing are modest in initial clinical and proof-of-concept studies in OSA patients to date [Citation166–168].
Overall, this summary identifies the physiological rationale and initial positive pre-clinical studies supporting intranasal administration of agents targeting the central ventilatory control system and respiratory network (Key Target #3. ) in improving sleep-disordered breathing and OSA.
5. Conclusion
OSA is a common and serious breathing disorder with an unmet need for additional or alternate treatment options due to limited compliance with standard CPAP therapy. This article identifies four key targets and strategies for OSA pharmacotherapy that are mapped to the structural organization of the respiratory control system relevant to OSA pathophysiology. The specific targets are: #1, pharyngeal motor effectors; #2, upper airway sensory afferents; #3, chemosensory afferents and the ventilatory control system; and #4, sleep-wake mechanisms.
The pathway to OSA pharmacotherapy will be accelerated by, and ultimately require, development and validation of automated tools to identify and measure the contributions of each of the key predisposing pathophysiological factors to OSA within and between individuals. Having such analytical tools that can be broadly applied to standard sleep laboratory measurements, and freely accessible and interpretable, will rapidly advance the field. This development will lead to rational selection or targets, either alone or in combination, for directed treatment within an individual.
6. Expert opinion
A fundamental principle related to OSA pathophysiology has propelled recent advances toward OSA pharmacotherapy. This principle states that although all OSA patients exhibit some degree of upper airway closure exclusively during sleep as a universal attribute and cardinal diagnostic feature, different people end up with this final common outcome for different reasons.
Four principal factors predispose an individual to OSA: upper airway size and collapsibility, pharyngeal muscle responsiveness, ventilatory control sensitivity (‘loop gain’), and arousability from sleep. These factors are identifiable and quantifiable in a sleep laboratory (‘endotyping’) and are modifiable pharmacologically. One or more of these factors are amenable to targeted pharmacotherapy if they are found to be significant to OSA pathophysiology with an individual.
The current situation, however, is that specialized equipment and protocols are needed to quantify which of the major pathophysiological factors (alone or in combination) predispose an individual to OSA. Relatively few specially trained individuals in few laboratories are equipped to do so. One key challenge and opportunity, therefore, is the development of a convenient method to accurately quantify these pathophysiological factors from standard sleep laboratory measurements that can then be simply interpreted and broadly applied. This democratization of endotyping with freely available open-source tools will be a significant development. Their uptake will be a major accelerator to endotype-based targeted treatment. If this is achieved, then the journey to rational, personalized, and targeted OSA pharmacotherapy will be realistically closer, achievable in principle, and applicable to many afflicted people.
Four key targets for OSA pharmacotherapy are identified and mapped to the structural organization of the respiratory control system relevant to OSA pathophysiology. The major targets comprise: #1, pharyngeal motor effectors; #2, upper airway sensory afferents mediating reflex pharyngeal dilator muscle responses to sub-atmospheric airway collapsing pressures; #3, chemosensory afferents and the ventilatory control system; and #4, sleep-wake mechanisms.
The order of identification, review, and discussion was deliberate. The order was based, in part, on targets most closely and directly connected to the local motor control mechanisms that cause and prevent upper airway obstruction in the first place (targets #1 and #2). Based on these guiding factors, these first two targets may theoretically offer more potential strategic priority or effectiveness if targeted appropriately and specifically because of their direct connectedness to the upper airway. Subsequent targets (#3 and #4) are more distant upstream and affect a broader set of attributes related to the control of breathing and sleep-wakes states but not OSA primarily or necessarily. The latter two targets may then predispose to more off-target influences beyond the upper airway or OSA even if targeted appropriately and specifically as they are more upstream and have multiple downstream targets.
6.1. Target #1: pharyngeal motor effectors
Increasing pharyngeal muscle activity and responsivity with noradrenergic-antimuscarinic drug combination is central to recent breakthroughs and progress in OSA pharmacotherapy. One of the key assumptions, at least initially, was that the drug combinations were likely effective in producing beneficial effects on pharyngeal muscle activity and OSA severity because of actions at the relevant pharyngeal motor nuclei as identified from initial animal studies. This assumption needs to be tested in animal models to close the loop from the basic science findings to potential clinical effects and pinpoint the mechanism of action of these drugs on upper airway motor control. This knowledge will potentially open the pipeline to identify more effective agents, refinements, and/or therapeutic targets. Other key unknowns include the source and operation of the cholinergic cells and circuits responsible for sleep state-dependent suppression of pharyngeal motor activity. This knowledge may lead to identification of other potential strategies to interfere with this inhibitory mechanism without relying on antimuscarinic agents that can have uncomfortable side effects that limit tolerability and efficacy. Other knowledge gaps include long-term effects of noradrenergic-antimuscarinic combinational pharmacotherapy on OSA severity in large multicenter studies. Basic science studies continue to identify other potential targets and some of these may proceed to proof-of-concept studies. Advances in the use of novel technologies and their application to pharyngeal motor activity during sleep are increasing and reviewed herein. Such advancements support the notion that the next generation of therapeutics will be influenced by such novel technologies currently in pre-clinical studies.
6.2. Target #2: upper airway sensory afferents
There is high translational potential of sensitizing and amplifying reflex pharyngeal dilator muscle responses to negative airway pressure via intranasal delivery of newly developed and highly selective TASK channel blockers. There has been a long history of studies identifying the contribution of pharyngeal dilator muscle responses to negative airway pressure in the maintenance of upper airway patency since the discovery and characterization of this reflex in humans in 1991. It is only recently, however, that translational studies in animals and humans have targeted this reflex for OSA pharmacotherapy. Initial results are encouraging and support strong strategic interest for further development and testing. Decrements in this pharyngeal negative pressure reflex have been linked to the pathogenesis and progressive development of OSA. In this context, additional clinical efficacy or chances of sustained positive outcomes may be gained after some reversal of the mucosal damaging effects of years of snoring and OSA by an initial short course of CPAP therapy. Such an approach may restore and/or augment the ability of the sensory afferents to respond to the oronasal-applied pharmacological interventions. Oronasal application of the selected agents directly to their desired site of action at the upper airway mucosa may also lead to lesser unwanted side-effects compared to systemic delivery.
6.3. Target #3: chemosensory afferents and ventilatory control
Strategies to manipulate ventilatory control system sensitivity (‘loop gain’) by carbonic anhydrase inhibitors are supported in theory and several initial studies. These findings support larger longer-term multicenter studies. A targeted decrease in loop gain may be especially applicable to those OSA patients in whom this factor is the main contributor to their pathophysiology. A mechanistic interpretation is provided based on review of the literature and first principles. Basic science studies also continue to identify other potential targets modulating ventilatory control. Intranasal delivery of agents to stimulate central respiratory activity (e.g. leptin and oxytocin) are inspiring new strategies and avenues of research with translational potential. Target #4: Sleep-Wake Mechanisms. Arousability was the fourth therapeutic target rationalized but was not a focus herein given up-to-date excellent reviews elsewhere.
List of abbreviations
| AHI | = | apnea-hypopnea index |
| CNO | = | clozapine-N-oxide |
| CPAP | = | continuous positive airway pressure |
| GIRK | = | G protein-gated inwardly rectifying K+ |
| IP3 | = | inositol triphosphate |
| Kir | = | inwardly rectifying K+ |
| non-REM | = | non-rapid eye movement |
| NMDA | = | N-methyl-D-aspartate |
| OSA | = | obstructive sleep apnea |
| PCRIT | = | critical closing pressure |
| PIP2 | = | phosphatidylinositol bisphosphate |
| PN | = | nasal pressure |
| REM | = | rapid eye movement |
| RN | = | nasal resistance |
| TASK | = | TWIK-related acid-sensitive K+ |
| TRH | = | thyrotropin-releasing hormon |
| VMAX | = | maximum flow |
Article highlights
Four key targets for obstructive sleep apnea (OSA) pharmacotherapy are identified and mapped to the structural organization of the respiratory control system relevant to OSA pathophysiology.
Rationalized targets for OSA pharmacotherapy are: #1: Pharyngeal Motor Effectors; #2: Upper Airway Sensory Afferents; #3: Chemosensory Afferents and the Ventilatory Control System; and #4: Sleep-Wake Mechanisms.
Among priority categories, the rational targets, strategies, challenges, key assumptions, knowledge gaps, and future directions are identified.
A technological target to accelerate personalized OSA pharmacotherapy is development of a convenient method to accurately quantify the key pathophysiological factors underpinning an individual’s OSA from standard sleep laboratory measurements that can then be simply interpreted and broadly applied using freely available open-source tools.
The current state of the field and anticipated developments supports the proposal that the journey to rational, personalized, and targeted OSA pharmacotherapy is realistically close and achievable in principle, or at least promising.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
A reviewer on this manuscript has disclosed they are chief scientific officer of Apnimed, a company researching on OSA pharmacotherapy. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Young T, Palta M, Dempsey J, et al. The occurrence of sleep-disordered breathing among middle-aged adults. New Engl J Med. 1993;328(17):1230–1235. doi: 10.1056/NEJM199304293281704
- Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002 May 1;165(9):1217–1239. doi: 10.1164/rccm.2109080
- Peppard PE, Young T, Barnet JH, et al. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013 May 1;177(9):1006–1014. doi: 10.1093/aje/kws342
- Benjafield AV, Ayas NT, Eastwood PR, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med. 2019 Aug;7(8):687–698.
- Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005 Nov 10;353(19):2034–2041. doi: 10.1056/NEJMoa043104
- Patil SP, Ayappa IA, Caples SM, et al. Treatment of adult obstructive sleep apnea with positive airway pressure: an american academy of sleep medicine systematic review, meta-analysis, and GRADE assessment. J Clin Sleep Med. 2019 Feb 15;15(2):301–334. doi: 10.5664/jcsm.7638
- Sullivan CE, Issa FG, Berthon-Jones M, et al. Reversal of obstructive sleep apnoea by continuous positive airway pressure applied through the nares. Lancet. 1981 Apr 18;1(8225):862–865.
- Weaver TE, Grunstein RR. Adherence to continuous positive airway pressure therapy: the challenge to effective treatment. Proc Am Thorac Soc. 2008 Feb 15;5(2):173–178. doi: 10.1513/pats.200708-119MG
- McEvoy RD, Antic NA, Heeley E, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016 Sep 8;375(10):919–931. doi: 10.1056/NEJMoa1606599
- Kribbs NB, Pack AI, Kline LR, et al. Objective measurement of patterns of nasal CPAP use by patients with obstructive sleep apnea. Am Rev Respir Dis. 1993 Apr;147(4):887–895.
- Strollo PJ Jr., Soose RJ, Maurer JT, et al. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med. 2014 Jan 9;370(2):139–149. doi: 10.1056/NEJMoa1308659
- Woodson BT, Soose RJ, Gillespie MB, et al. Three-year outcomes of cranial nerve stimulation for obstructive sleep apnea: the STAR trial. Otolaryngol Head Neck Surg. 2016 Jan;154(1):181–188.
- Tong BK, Tran C, Ricciardiello A, et al. CPAP combined with oral appliance therapy reduces CPAP requirements and pharyngeal pressure swings in obstructive sleep apnea. J Appl Physiol. 2020 Nov 1;129(5):1085–1091. doi: 10.1152/japplphysiol.00393.2020
- Edwards BA, Andara C, Landry S, et al. Upper-airway collapsibility and loop gain predict the response to oral appliance therapy in obstructive sleep apnea patients. Am J Respir Crit Care Med. 2016 May 15;194(11):1413–1422.
- Cartwright RD. Effect of sleep position on sleep apnea severity. Sleep. 1984;7(2):110–114. doi: 10.1093/sleep/7.2.110
- Fleury Curado T, Oliven A, Sennes LU, et al. Neurostimulation treatment of OSA. Chest. 2018 Dec;154(6):1435–1447.
- Eckert DJ, White DP, Jordan AS, et al. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013 Oct 15;188(8):996–1004. doi: 10.1164/rccm.201303-0448OC
- Wellman A, Eckert DJ, Jordan AS, et al. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol. 2011 Jun;110(6):1627–1637.
- Younes M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J Appl Physiol. 2008 Nov;105(5):1389–1405. doi: 10.1152/japplphysiol.90408.2008
- Horner RL, Grace KP, Wellman A. A resource of potential drug targets and strategic decision-making for obstructive sleep apnoea pharmacotherapy. Respirology. 2017;22(5):861–873. doi: 10.1111/resp.13079
- Younes M, Loewen AH, Ostrowski M, et al. Genioglossus activity available via non-arousal mechanisms vs. that required for opening the airway in obstructive apnea patients. J Appl Physiol. 2012 Jan;112(2):249–258.
- Younes M. Contributions of upper airway mechanics and control mechanisms to severity of obstructive apnea. Am J Respir Crit Care Med. 2003 Sep 15;168(6):645–658. doi: 10.1164/rccm.200302-201OC
- Lajoie AC, Heinzer R, Series F, et al. Physiology of upper and lower airways. In: Kryger M, Roth T Goldstein C, editors. Principles and practice of sleep medicine 7th ed. Philadelphia PA:Elsevier; 2022. p. 252–259.
- White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005 Dec 1;172(11):1363–1370. doi: 10.1164/rccm.200412-1631SO
- Pham LV, Schwartz AR. The pathogenesis of obstructive sleep apnea. J Thoracic Dis. 2015 Aug;7(8):1358–1372. doi: 10.3978/j.issn.2072-1439.2015.07.28
- Schwartz AR, Smith PL, Wise RA, et al. Induction of upper airway occlusion in sleeping individuals with subatmospheric nasal pressure. J Appl Physiol. 1988 Feb;64(2):535–542.
- Smith PL, Wise RA, Gold AR, et al. Upper airway pressure-flow relationships in obstructive sleep apnea. J Appl Physiol. 1988 Feb;64(2):789–795.
- Patil SP, Schneider H, Marx JJ, et al. Neuromechanical control of upper airway patency during sleep. J Appl Physiol. 2007 Feb;102(2):547–556.
- Horner RL, Innes JA, Holden HB, et al. Afferent pathway(s) for pharyngeal dilator reflex to negative pressure in man: a study using upper airway anaesthesia. J Physiol. 1991;436(1):31–44. doi: 10.1113/jphysiol.1991.sp018537
- Horner RL, Innes JA, Murphy K, et al. Evidence for reflex upper airway dilator muscle activation by sudden negative airway pressure in man. J Physiol. 1991;436(1):15–29. doi: 10.1113/jphysiol.1991.sp018536
- Horner RL. Motor control of the pharyngeal musculature and implications for the pathogenesis of obstructive sleep apnea. Sleep. 1996;19(10):827–853. doi: 10.1093/sleep/19.10.827
- Plowman L, Lauff DC, Berthon-Jones M, et al. Waking and genioglossus muscle responses to upper airway pressure oscillation in sleeping dogs. J Appl Physiol. 1990 Jun;68(6):2564–2573.
- Horner RL, Innes JA, Morrell MJ, et al. The effect of sleep on reflex genioglossus muscle activation by stimuli of negative airway pressure in humans. J Physiol. 1994;476(1):141–151.
- Shea SA, Edwards JK, White DP. Effect of wake-sleep transitions and rapid eye movement sleep on pharyngeal muscle response to negative pressure in humans. J Physiol. 1999;520(3):897–908. doi: 10.1111/j.1469-7793.1999.00897.x
- Stauffer JL, Buick MK, Bixler EO, et al. Morphology of the uvula in obstructive sleep apnea. Am Rev Respir Dis. 1989 Sep;140(3):724–728.
- Friberg D, Gazelius B, Hokfelt T, et al. Abnormal afferent nerve endings in the soft palatal mucosa of sleep apnoics and habitual snorers. Regul Pept. 1997 Jul 23;71(1):29–36. doi: 10.1016/S0167-0115(97)01016-1
- Ryan CF, Lowe AA, Li D, et al. Magnetic resonance imaging of the upper airway in obstructive sleep apnea before and after chronic nasal continuous positive airway pressure therapy. Am Rev Respir Dis. 1991 Oct;144(4):939–944.
- Payne RJ, Kost KM, Frenkiel S, et al. Laryngeal inflammation assessed using the reflux finding score in obstructive sleep apnea. Otolaryngol Head Neck Surg. 2006 May;134(5):836–842.
- Larsson H, Carlsson-Nordlander B, Lindblad LE, et al. Temperature thresholds in the oropharynx of patients with obstructive sleep apnea syndrome. Am Rev Respir Dis. 1992 Nov;146(5 Pt 1):1246–1249.
- Kimoff RJ, Sforza E, Champagne V, et al. Upper airway sensation in snoring and obstructive sleep apnea. Am J Respir Crit Care Med. 2001 Jul 15;164(2):250–255. doi: 10.1164/ajrccm.164.2.2010012
- Nguyen AT, Jobin V, Payne R, et al. Laryngeal and velopharyngeal sensory impairment in obstructive sleep apnea. Sleep. 2005 May 1;28(5):585–593.
- Taranto-Montemurro L, Messineo L, Wellman A. Targeting endotypic traits with medications for the pharmacological treatment of obstructive sleep apnea. A review of the current literature. J Clin Med. 2019 Nov 2;8(11):1846.
- Dahan A, van der Schrier R, Smith T, et al. Averting opioid-induced respiratory depression without affecting analgesia. Anesthesiology. 2018 May;128(5):1027–1037.
- Buckler KJ. TASK-like potassium channels and oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2007 Jul 1;157(1):55–64. doi: 10.1016/j.resp.2007.02.013
- Schmickl CN, Landry S, Orr JE, et al. Effects of acetazolamide on control of breathing in sleep apnea patients: mechanistic insights using meta-analyses and physiological model simulations. Physiol Rep. 2021 Oct;9(20):e15071. doi: 10.14814/phy2.15071
- Edwards BA, Sands SA, Eckert DJ, et al. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012 Mar 1;590(5):1199–1211. doi: 10.1113/jphysiol.2011.223925
- Hedner J, Stenlof K, Zou D, et al. A randomized controlled clinical trial exploring safety and tolerability of sulthiame in sleep apnea. Am J Respir Crit Care Med. 2022 Jun 15;205(12):1461–1469. doi: 10.1164/rccm.202109-2043OC
- Saboisky JP, Chamberlin NL, Malhotra A. Potential therapeutic targets in obstructive sleep apnoea. Expert Opin Ther Targets. 2009 Jul;13(7):795–809. doi: 10.1517/14728220903005608
- Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med. 2004 Mar 1;169(5):623–633. doi: 10.1164/rccm.200307-1023OC
- Eckert DJ, Younes MK. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol. 2014 Feb 1;116(3):302–313. doi: 10.1152/japplphysiol.00649.2013
- Hudgel DW, Patel SR, Ahasic AM, et al. The role of weight management in the treatment of adult obstructive sleep apnea. an official american thoracic society clinical practice guideline. Am J Respir Crit Care Med. 2018 Sep 15;198(6):e70–e87. doi: 10.1164/rccm.201807-1326ST
- Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022 Jul 21;387(3):205–216. doi: 10.1056/NEJMoa2206038
- Blackman A, Foster GD, Zammit G, et al. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: the SCALE sleep apnea randomized clinical trial. Int J Obes (Lond). 2016 Aug;40(8):1310–1319.
- Messineo L, Eckert DJ, Taranto-Montemurro L, et al. Ventilatory drive withdrawal rather than reduced genioglossus compensation as a mechanism of obstructive sleep apnea in REM sleep. Am J Respir Crit Care Med. 2022 Jan 15;205(2):219–232. doi: 10.1164/rccm.202101-0237OC
- Gell LK, Vena D, Alex RM, et al. Neural ventilatory drive decline as a predominant mechanism of obstructive sleep apnoea events. Thorax. 2022 Jul;77(7):707–716.
- Horner RL, Malhotra A. Control of breathing and upper airways during sleep. In: Broaddus C, Mason R Ernst J, editors. Murray & nadel’s textbook of respiratory medicine 6th ed. St. Louis MO:Elsevier, Saunders; 2016. p. 1511–1526.
- White DP. Advanced concepts in the pathophysiology of obstructive sleep apnea. Adv Otorhinolaryngol. 2017;80:7–16.
- White DP. Pharmacologic approaches to the treatment of obstructive sleep apnea. Sleep Med Clin. 2016 Jun;11(2):203–212. doi: 10.1016/j.jsmc.2016.01.007
- Kim LJ, Freire C, Fleury Curado T, et al. The role of animal models in developing pharmacotherapy for obstructive sleep apnea. J Clin Med. 2019 Nov 22;8(12):2049. doi: 10.3390/jcm8122049
- Finnsson E, Olafsdottir GH, Loftsdottir DL, et al. A scalable method of determining physiological endotypes of sleep apnea from a polysomnographic sleep study. Sleep. 2021 Jan 21;44(1). doi: 10.1093/sleep/zsaa168
- Sands SA, Edwards BA, Terrill PI, et al. Phenotyping pharyngeal pathophysiology using polysomnography in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2018 May 1;197(9):1187–1197. doi: 10.1164/rccm.201707-1435OC
- Younes M, Schwab R. Con: can physiological risk factors for obstructive sleep apnea be determined by analysis of data obtained from routine polysomnography? Sleep. 2023 May 10;46(5). doi: 10.1093/sleep/zsac158
- Sands SA, Edwards BA. Pro: can physiological risk factors for obstructive sleep apnea be determined by analysis of data obtained from routine polysomnography? Sleep. 2023 May 10;46(5). doi: 10.1093/sleep/zsac310
- Alex RM, Sofer T, Azarbarzin A, et al. Within-night repeatability and long-term consistency of sleep apnea endotypes: the multi-ethnic study of atherosclerosis and osteoporotic fractures in men study. Sleep. 2022 Sep 8;45(9). doi: 10.1093/sleep/zsac129
- Lim DC, Mazzotti DR, Sutherland K, et al. Reinventing polysomnography in the age of precision medicine. Sleep Med Rev. 2020 Aug;52:101313.
- Taranto-Montemurro L, Messineo L, Sands SA, et al. The combination of atomoxetine and oxybutynin greatly reduces obstructive sleep apnea severity: A randomized, placebo-controlled, double-blind crossover trial. Am J Respir Crit Care Med. 2019 Nov 5;199:1267–1276.
- Wadman M. Drug pair shows promise for treating sleep apnea. Science. 2018 Sep 21;361(6408):1174–1175. doi: 10.1126/science.361.6408.1174
- Fleury Curado T, Berger S, Polotsky VY. Pharmacotherapy of obstructive sleep apnea: is salvation just around a corner? Am J Respir Crit Care Med. 2019 May 15;199(10):1186–1187. doi: 10.1164/rccm.201811-2135ED
- Lim R, Messineo L, Grunstein RR, et al. The noradrenergic agent reboxetine plus the antimuscarinic hyoscine butylbromide reduces sleep apnoea severity: a double-blind, placebo-controlled, randomised crossover trial. J Physiol. 2021 Jun 26;599(17):4183–4195. doi: 10.1113/JP281912
- O’Halloran KD. One step closer to pharmacotherapy for sleep apnoea. J Physiol. 2021 Jul 23;599(17):4015–4016. doi: 10.1113/JP282061
- Aishah A, Lim R, Sands SA, et al. Different antimuscarinics when combined with atomoxetine have differential effects on obstructive sleep apnea severity. J Appl Physiol. 2021 May 1;130(5):1373–1382.
- Schweitzer PK, Maynard JP, Wylie PE, et al. Efficacy of atomoxetine plus oxybutynin in the treatment of obstructive sleep apnea with moderate pharyngeal collapsibility. Sleep Breath. 2022 May;13:1–9.
- Messineo L, Taranto-Montemurro L, Calianese N, et al. Atomoxetine and fesoterodine combination improves obstructive sleep apnoea severity in patients with milder upper airway collapsibility. Respirology. 2022 Nov;27(11):975–982.
- Rosenberg R, Abaluck B, Thein S. Combination of atomoxetine with the novel antimuscarinic aroxybutynin improves mild to moderate OSA. J Clin Sleep Med. 2022 Dec 1;18(12):2837–2844. doi: 10.5664/jcsm.10250
- Kinouchi T, Terada J, Sakao S, et al. Effects of the combination of atomoxetine and oxybutynin in Japanese patients with obstructive sleep apnoea: A randomized controlled crossover trial. Respirology. 2022 Oct 2;28(3):273–280.
- Perger E, Taranto Montemurro L, Rosa D, et al. Reboxetine plus oxybutynin for obstructed sleep apnea treatment a 1-week randomized, placebo-controlled, double-blind crossover trial. Chest. 2022 Sep 17;161(1):237–247.
- Taranto-Montemurro L, Messineo L, Azarbarzin A, et al. Effects of the combination of atomoxetine and oxybutynin on OSA endotypic traits. Chest. 2020 Jun;157(6):1626–1636.
- Taranto-Montemurro L, Sands SA, Edwards BA, et al. Desipramine improves upper airway collapsibility and reduces OSA severity in patients with minimal muscle compensation. Eur Respir J. 2016 Nov;48(5):1340–1350.
- Taranto-Montemurro L, Edwards BA, Sands SA, et al. Desipramine increases genioglossus activity and reduces upper airway collapsibility during non-REM sleep in healthy subjects. Am J Respir Crit Care Med. 2016 Mar 11;194(7):878–885. doi: 10.1164/rccm.201511-2172OC
- Lim R, Carberry JC, Wellman A, et al. Reboxetine and hyoscine butylbromide improve upper airway function during nonrapid eye movement and suppress rapid eye movement sleep in healthy individuals. Sleep. 2019 Apr 1;42(4). doi: 10.1093/sleep/zsy261
- Aishah A, Loffler KA, Toson B, et al. One month dosing of atomoxetine plus oxybutynin in obstructive sleep apnea: a randomized, placebo-controlled trial. Ann Am Thoracic Soc. 2023 Apr;20(4):584–595.
- Grace KP, Hughes SW, Horner RL. Identification of the mechanism mediating genioglossus muscle suppression in REM sleep. Am J Respir Crit Care Med. 2013 Dec 6;187(3):311–319. doi: 10.1164/rccm.201209-1654OC
- Veasey S, White DP. Obstructive sleep apnea pharmacotherapy: one step closer. Am J Respir Crit Care Med. 2013 Feb 1;187(3):226–227. doi: 10.1164/rccm.201211-2047ED
- Fenik VB, Davies RO, Kubin L. Noradrenergic, serotonergic and GABAergic antagonists injected together into the XII nucleus abolish the REM sleep-like depression of hypoglossal motoneuronal activity. J Sleep Res. 2005 Dec;14(4):419–429. doi: 10.1111/j.1365-2869.2005.00461.x
- Chan E, Steenland HW, Liu H, et al. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med. 2006;174:1264–1273.
- Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med. 2005;172:1322–1330.
- Song G, Poon CS. Alpha2-adrenergic blockade rescues hypoglossal motor defense against obstructive sleep apnea. JCI Insight. 2017 Feb 23;2(4):e91456.
- Fenik VB, Rukhadze I. Activity of pontine A7 noradrenergic neurons is suppressed during REM sleep. J Appl Physiol Applied Physiology (Bethesda, Md: 1985). 2022 May 26;133(1):130–143. doi: 10.1152/japplphysiol.00771.2021
- Niakani S, Liu H, Liu WY, et al. Differential pharmacological and sex-specific effects of antimuscarinic agents at the hypoglossal motor nucleus in vivo in rats. Sci Rep. 2022 Sep 1;12(1):14896. doi: 10.1038/s41598-022-19233-1
- Altree TJ, Aishah A, Loffler KA, et al. The norepinephrine reuptake inhibitor reboxetine alone reduces obstructive sleep apnea severity: a double-blind, placebo-controlled, randomized crossover trial. J Clin Sleep Med. 2023 Jan 1;19(1):85–96. doi: 10.5664/jcsm.10256
- Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1(8):876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981
- Taranto-Montemurro L, Pho H, White DP. Development of a combination of noradrenergic and antimuscarinic drugs for the treatment of obstructive sleep apnea: challenges and progress. Front Sleep. 2023;2:1148282. doi: 10.3389/frsle.2023.1148282
- Zhu L, Chamberlin NL, Arrigoni E. Muscarinic inhibition of hypoglossal motoneurons: possible implications for upper airway muscle hypotonia during REM sleep. J Neurosci. 2019 Oct 2;39(40):7910–7919. doi: 10.1523/JNEUROSCI.0461-19.2019
- Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007 Jan 11;445(7124):168–176. doi: 10.1038/nature05453
- Bayliss DA, Viana F, Kanter RK, et al. Early postnatal development of thyrotropin-releasing hormone (TRH) expression, TRH receptor binding, and TRH responses in neurons of rat brainstem. J Neurosci. 1994;14(2):821–833. doi: 10.1523/JNEUROSCI.14-02-00821.1994
- Rekling JC, Funk GD, Bayliss DA, et al. Synaptic control of motoneuronal excitability. Physiol Rev. 2000 Apr;80(2):767–852.
- Johansson O, Hokfelt T, Pernow B, et al. Immunohistochemical support for three putative transmitters in one neuron: coexistence of 5-hydroxytryptamine, substance P- and thyrotropin releasing hormone-like immunoreactivity in medullary neurons projecting to the spinal cord. Neuroscience. 1981;6(10):1857–1881. doi: 10.1016/0306-4522(81)90028-2
- Khomane KS, Meena CL, Jain R, et al. Novel thyrotropin-releasing hormone analogs: a patent review. Expert Opin Ther Patents. 2011 Nov;21(11):1673–1691.
- Bayliss DA, Viana F, Berger AJ. Effects of thyrotropin-releasing hormone on rat motoneurons are mediated by G proteins. Brain Res. 1994;668(1–2):220–229. doi: 10.1016/0006-8993(94)90527-4
- Kinoshita K, Yamamura M, Sugihara J, et al. Taltirelin hydrate (TA‐0910): an orally active thyrotropin‐releasing hormone mimetic agent with multiple actions. CNS Drug Rev. 1998;1:25–41.
- Liu WY, Ladha R, Liu H, et al. Thyrotropin-releasing hormone analog as a stable upper airway-preferring respiratory stimulant with arousal properties. J Appl Physiol Of Applied Physiology (Bethesda, Md: 1985). 2022 Nov 1;133(5):1067–1080. doi: 10.1152/japplphysiol.00414.2022
- Liu WY, Liu H, Aggarwal J, et al. Differential activating effects of thyrotropin-releasing hormone and its analog taltirelin on motor output to the tongue musculature in vivo. Sleep. 2020 Sep 14;43(9):zsaa053. doi: 10.1093/sleep/zsaa053
- Messineo L, Carter SG, Taranto-Montemurro L, et al. Addition of zolpidem to combination therapy with atomoxetine-oxybutynin increases sleep efficiency and the respiratory arousal threshold in obstructive sleep apnoea: A randomized trial. Respirology. 2021 Jun 23;26(9):878–886. doi: 10.1111/resp.14110
- Messineo L, Eckert DJ, Lim R, et al. Zolpidem increases sleep efficiency and the respiratory arousal threshold without changing sleep apnoea severity and pharyngeal muscle activity. J Physiol. 2020 Oct;598(20):4681–4692.
- Smales ET, Edwards BA, Deyoung PN, et al. Trazodone effects on obstructive sleep apnea and non-REM arousal threshold. Ann Am Thoracic Soc. 2015 May;12(5):758–764.
- Heinzer RC, White DP, Jordan AS, et al. Trazodone increases arousal threshold in obstructive sleep apnoea. Eur Respir J. 2008 Jun;31(6):1308–1312.
- Veasey SC, Fenik P, Panckeri K, et al. The effects of trazodone with L-tryptophan on sleep-disordered breathing in the English bulldog. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1659–1667.
- Eckert DJ, Malhotra A, Wellman A, et al. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep. 2014 Apr 1;37(4):811–819. doi: 10.5665/sleep.3596
- AbdelFattah MR, Jung SW, Greenspan MA, et al. Efficacy of antidepressants in the treatment of obstructive sleep apnea compared to placebo. A systematic review with meta-analyses. Sleep Breath. 2020 Jun;24(2):443–453.
- Chen CY, Chen CL, Yu CC. Trazodone improves obstructive sleep apnea after ischemic stroke: a randomized, double-blind, placebo-controlled, crossover pilot study. J Neurol. 2021 Aug;268(8):2951–2960. doi: 10.1007/s00415-021-10480-2
- Algera MH, Cotten JF, van Velzen M, et al. Respiratory effects of thyrotropin-releasing hormone and its analogue taltirelin on opioid-induced respiratory depression. Br J Anaesth. 2022 Jul;129(1):e4–e6.
- Algera MH, Cotten JF, van Velzen M, et al. Are thyrotropin-releasing hormone (TRH) and analog taltirelin viable reversal agents of opioid-induced respiratory depression? Pharmacol Res Perspect. 2022 Jun;10(3):e00974.
- Boghosian JD, Luethy A, Cotten JF. Intravenous and intratracheal thyrotropin releasing hormone and its analog taltirelin reverse opioid-induced respiratory depression in isoflurane anesthetized rats. J Pharmacol Exp Ther. 2018 Jul;366(1):105–112. doi: 10.1124/jpet.118.248377
- Altree TJ, Eckert DJ. Obstructive sleep apnea endotypes and their postoperative relevance. Int Anesthesiol Clin. 2022 Apr 1;60(2):1–7.
- Altree TJ, Chung F, Chan MTV, et al. Vulnerability to postoperative complications in obstructive sleep apnea: importance of phenotypes. Anesth Analg. 2021 May 1;132(5):1328–1337. doi: 10.1213/ANE.0000000000005390
- Töpert C, Döring F, Wischmeyer E, et al. Kir2.4: a novel K+ inward rectifier channel associated with motoneurons of cranial nerve nuclei. J Neurosci. 1998 Jun 1;18(11):4096–4105. doi: 10.1523/JNEUROSCI.18-11-04096.1998
- Grace KP, Hughes SW, Horner RL. Identification of a pharmacological target for genioglossus reactivation throughout sleep. Sleep. 2014 Jan;37(1):41–50. doi: 10.5665/sleep.3304
- Grace KP, Hughes SW, Shahabi S, et al. K+ channel modulation causes genioglossus inhibition in REM sleep and is a strategy for reactivation. Respir Physiol Neurobiol. 2013 Sep 15;188(3):277–288. doi: 10.1016/j.resp.2013.07.011
- Horner RL, Hughes SW, Malhotra A. State-dependent and reflex drives to the upper airway: basic physiology with clinical implications. J Appl Physiol Applied Physiology (Bethesda, Md: 1985). 2014 Feb 1;116(3):325–336. doi: 10.1152/japplphysiol.00531.2013
- Weaver CD, Denton JS. Next-generation inward rectifier potassium channel modulators: discovery and molecular pharmacology. Am J Physiol Cell Physiol. 2021 Jun 1;320(6):C1125–C1140. doi: 10.1152/ajpcell.00548.2020
- Fleury Curado T, Fishbein K, Pho H, et al. Chemogenetic stimulation of the hypoglossal neurons improves upper airway patency. Sci Rep. 2017 Mar 10;7(1):44392. doi: 10.1038/srep44392
- Horton GA, Fraigne JJ, Torontali ZA, et al. Activation of the hypoglossal to tongue musculature motor pathway by remote control. Sci Rep. 2017 Apr 6;7(1):45860. doi: 10.1038/srep45860
- Fleury Curado T, Pho H, Freire C, et al. Designer Receptors exclusively activated by designer drugs approach to treatment of sleep-disordered breathing. Am J Respir Crit Care Med. 2021 Jan 1;203(1):102–110. doi: 10.1164/rccm.202002-0321OC
- Fleury Curado TA, Pho H, Dergacheva O, et al. Silencing of hypoglossal motoneurons leads to sleep disordered breathing in lean mice. Front Neurol. 2018;9:962. doi: 10.3389/fneur.2018.00962
- Roth BL. Dreadds for neuroscientists. Neuron. 2016 Feb 17;89(4):683–694. doi: 10.1016/j.neuron.2016.01.040
- Rogan SC, Roth BL, Morrow AL. Remote control of neuronal signaling. Pharmacol Rev. 2011 Jun;63(2):291–315. doi: 10.1124/pr.110.003020
- Urban DJ, Roth BL. Dreadds (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol. 2015;55(1):399–417. doi: 10.1146/annurev-pharmtox-010814-124803
- Taranto-Montemurro L, Sands SA, Azarbarzin A, et al. Effect of 4-aminopyridine on genioglossus muscle activity during sleep in healthy adults. Ann Am Thoracic Soc. 2017 Jul;14(7):1177–1183.
- Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011 Sep 22;477(7365):495–498. doi: 10.1038/nature10370
- Soom M, Schonherr R, Kubo Y, et al. Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett. 2001 Feb 9;490(1–2):49–53. doi: 10.1016/S0014-5793(01)02136-6
- Issa SS, Shaimardanova AA, Solovyeva VV, et al. Various AAV serotypes and their applications in gene therapy: an overview. Cells. 2023 Mar 1;12:(5).
- Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019 May;18(5):358–378. doi: 10.1038/s41573-019-0012-9
- Saper CB, Scammell TE. Emerging therapeutics in sleep. Ann Neurol. 2013 Sep;74(3):435–440. doi: 10.1002/ana.24000
- Doyle BM, Singer ML, Fleury-Curado T, et al. Gene delivery to the hypoglossal motor system: preclinical studies and translational potential. Gene Ther. 2021 Feb 11;28(7–8):402–412. doi: 10.1038/s41434-021-00225-1
- de Beeck S O, Wellman A, Dieltjens M, et al. Endotypic mechanisms of successful hypoglossal nerve stimulation for obstructive sleep apnea. Am J Respir Crit Care Med. 2021 Mar 15;203(6):746–755.
- Talley EM, Lei Q, Sirois JE, et al. TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron. 2000;25(2):399–410.
- Steenland HW, Liu H, Horner RL. Endogenous glutamatergic control of rhythmically active mammalian respiratory motoneurons in vivo. J Neurosci. 2008 Jul 2;28(27):6826–6835. doi: 10.1523/JNEUROSCI.1019-08.2008
- Gurges P, Liu H, Horner RL. Modulation of TASK-1/3 channels at the hypoglossal motoneuron pool and effects on tongue motor output and responses to excitatory inputs in vivo: implications for strategies for obstructive sleep apnea pharmacotherapy. Sleep. 2021 Jan 21;44(1). doi: 10.1093/sleep/zsaa144
- Sormann J, Schewe M, Proks P, et al. Gain-of-function mutations in KCNK3 cause a developmental disorder with sleep apnea. Nature Genet. 2022 Oct;54(10):1534–1543.
- Simonson TS, Moya EA, Malhotra A. Genomic insights into TASK-1 reveal functional roles in sleep apnea. Nature Genet. 2022 Oct;54(10):1451–1452. doi: 10.1038/s41588-022-01195-9
- Rodstrom KEJ, Kiper AK, Zhang W, et al. A lower X-gate in TASK channels traps inhibitors within the vestibule. Nature. 2020 Jun;582(7812):443–447.
- Kiper AK, Rinne S, Rolfes C, et al. Kv1.5 blockers preferentially inhibit TASK-1 channels: TASK-1 as a target against atrial fibrillation and obstructive sleep apnea? Pflugers Arch - Eur J Physiol. 2015 May;467(5):1081–1090.
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000 May 15;525(Pt 1):135–142.
- Mulkey DK, Talley EM, Stornetta RL, et al. TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J Neurosci. 2007 Dec 19;27(51):14049–14058. doi: 10.1523/JNEUROSCI.4254-07.2007
- Kim D, Cavanaugh EJ, Kim I, et al. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J Physiol. 2009 Jun 15;587(Pt 12):2963–2975.
- Olschewski A, Veale EL, Nagy BM, et al. TASK-1 (KCNK3) channels in the lung: from cell biology to clinical implications. Eur Respir J. 2017 Nov;50(5):1700754.
- Bohnen MS, Roman-Campos D, Terrenoire C, et al. The impact of heterozygous KCNK3 mutations associated with pulmonary arterial hypertension on channel function and pharmacological recovery. J Am Heart Assoc. 2017 Sep 9;6(9). doi: 10.1161/JAHA.117.006465
- Osman AM, Mukherjee S, Altree TJ, et al. Topical Potassium channel blockage improves pharyngeal collapsibility: a translational, placebo-controlled trial. Chest. 2023 Nov 24;163(4):953–965.
- Wirth KJ, Steinmeyer K, Ruetten H. Sensitization of upper airway mechanoreceptors as a new pharmacologic principle to treat obstructive sleep apnea: investigations with AVE0118 in anesthetized pigs. Sleep. 2013 May 1;36(5):699–708. doi: 10.5665/sleep.2630
- Hillman D. A new pharmacological treatment to treat obstructive sleep apnea? Sleep. 2013 May 1;36(5):635–636. doi: 10.5665/sleep.2614
- Wirth KJ, Paehler T, Rosenstein B, et al. Atrial effects of the novel K(+)-channel-blocker AVE0118 in anesthetized pigs. Cardiovasc Res. 2003 Nov 1;60(2):298–306.
- Gaisl T, Turnbull CD, Weimann G, et al. BAY 2253651 for the treatment of obstructive sleep apnoea: a multicentre, double-blind, randomised controlled trial (SANDMAN). Eur Respir J. 2021 Nov;58(5):2101937.
- Genta PR, Taranto-Montemurro L. Task accomplished: promising effects of a new topical potassium channel antagonist in OSA. Chest. 2023 Apr;163(4):749–750. doi: 10.1016/j.chest.2023.01.004
- Hedner J, Stenlof K, Zou D, et al. Reply to Chen et al. Am J Respir Crit Care Med. 2022 Oct 15;206(8):1051. doi: 10.1164/rccm.202207-1275LE
- Whyte KF, Gould GA, Airlie MA, et al. Role of protriptyline and acetazolamide in the sleep apnea/hypopnea syndrome. Sleep. 1988 Oct;11(5):463–472.
- Tojima H, Kunitomo F, Kimura H, et al. Effects of acetazolamide in patients with the sleep apnoea syndrome. Thorax. 1988 Feb;43(2):113–119.
- Orr JE, Malhotra A, Sands SA. Pathogenesis of central and complex sleep apnoea. Respirology. 2017 Jan;22(1):43–52. doi: 10.1111/resp.12927
- Dempsey JA, Veasey SC, Morgan BJ, et al. Pathophysiology of sleep apnea. Physiol Rev. 2010 Jan;90(1):47–112.
- Younes M, Ostrowski M, Thompson W, et al. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001 Apr;163(5):1181–1190.
- Berger S, Pho H, Fleury-Curado T, et al. Intranasal leptin relieves sleep-disordered breathing in mice with diet-induced obesity. Am J Respir Crit Care Med. 2019 Mar 15;199(6):773–783. doi: 10.1164/rccm.201805-0879OC
- MSM I, Mokhlesi B. Activating leptin receptors in the central nervous system using intranasal leptin. A Novel therapeutic target for sleep-disordered breathing. Am J Respir Crit Care Med. 2019 Mar 15;199(6):689–691.
- Pho H, Berger S, Freire C, et al. Leptin receptor expression in the dorsomedial hypothalamus stimulates breathing during NREM sleep in db/db mice. Sleep. 2021 Jun 11;44(6). doi: 10.1093/sleep/zsab046
- Amorim MR, Dergacheva O, Fleury-Curado T, et al. The effect of DREADD activation of leptin receptor positive neurons in the nucleus of the solitary tract on sleep disordered breathing. Int J Mol Sci. 2021 Jun 23;22(13):6742. doi: 10.3390/ijms22136742
- Freire C, Pho H, Kim LJ, et al. Intranasal leptin prevents opioid-induced sleep-disordered breathing in obese mice. Am J Respir Cell Mol Biol. 2020 Oct;63(4):502–509.
- Dergacheva O, Polotsky VY, Mendelowitz D. Oxytocin mediated excitation of hypoglossal motoneurons: implications for treating obstructive sleep apnea. Sleep. 2023 Apr 12;46(4). doi: 10.1093/sleep/zsad009
- Jain V, Marbach J, Kimbro S, et al. Benefits of oxytocin administration in obstructive sleep apnea. Am J Physiol Lung Cell Mol Physiol. 2017 Nov 1;313(5):L825–L833. doi: 10.1152/ajplung.00206.2017
- Jain V, Kimbro S, Kowalik G, et al. Intranasal oxytocin increases respiratory rate and reduces obstructive event duration and oxygen desaturation in obstructive sleep apnea patients: a randomized double blinded placebo controlled study. Sleep Med. 2020 Oct;74:242–247.
- Osman AM, Altree TJ, Eckert DJ. The scent of love is in the air(way): a potential drug target for sleep apnea? Sleep. 2023 Apr 12;46(4). doi: 10.1093/sleep/zsad019