
ABSTRACT
Introduction
Drugs targeting mitochondria are emerging as promising antitumor therapeutics in preclinical models. However, a few of these drugs have shown clinical toxicity. Developing mitochondria-targeted modified natural compounds and US FDA-approved drugs with increased therapeutic index in cancer is discussed as an alternative strategy.
Areas covered
Triphenylphosphonium cation (TPP+)-based drugs selectively accumulate in the mitochondria of cancer cells due to their increased negative membrane potential, target the oxidative phosphorylation proteins, inhibit mitochondrial respiration, and inhibit tumor proliferation. TPP+-based drugs exert minimal toxic side effects in rodents and humans. These drugs can sensitize radiation and immunotherapies.
Expert opinion
TPP+-based drugs targeting the tumor mitochondrial electron transport chain are a new class of oxidative phosphorylation inhibitors with varying antiproliferative and antimetastatic potencies. Some of these TPP+-based agents, which are synthesized from naturally occurring molecules and FDA-approved drugs, have been tested in mice and did not show notable toxicity, including neurotoxicity, when used at doses under the maximally tolerated dose. Thus, more effort should be directed toward the clinical translation of TPP+-based OXPHOS-inhibiting drugs in cancer prevention and treatment.
Plain Language Summary
Mitochondria, which are the cell’s powerhouse of energy, are functional in cancer cells. Inhibition of cancer cell respiration is associated with inhibition of cancer cell proliferation. Therefore, mitochondria have become a promising target for developing antitumor drugs to treat cancer. Several classes of drug molecules selectively target cancer cell mitochondria and inhibit mitochondrial respiration or oxidative phosphorylation (OXPHOS). A new class of OXPHOS-targeting drugs is emerging as a potential cancer therapeutic. One of the OXPHOS inhibitor drugs, IACS-010759, developed by investigators at MD Anderson Cancer Center, was tested in patients with acute myeloid leukemia. Patients who were administered the drug developed peripheral neuropathy and other complications (lactic acidosis), resulting in dose reduction. At lower doses, this drug was not effective. Subsequently, the clinical trial was terminated. The investigators then showed the same type of neurotoxicity using a mouse model. These findings were recently published. Thus, there is an urgent need to develop new OXPHOS inhibitors that do not have neurotoxicity in mice or humans.
In this opinion article, we make a case that there are other triphenylphosphonium cation (TPP+)-based mitochondrial OXPHOS inhibitors (inhibiting both complex I and complex III) that are structural modifications of naturally occurring molecules or US FDA-approved drugs. These mitochondria-targeted drugs (MTDs) are as potent as IACS-010759 in cells and in preclinical models. Several TPP+-based MTDs have been tested in mice and did not exert neurotoxicity. TPP+-containing MTDs such as mitochondria-targeted coenzyme Q10 (MitoQ) have been tested in patients with Parkinson’s disease, with no evidence of peripheral neuropathy or other toxicity (e.g., lactic acidosis). Other US FDA-approved drugs (metformin and atovaquone [ATO] or papaverine) are in clinical trials alone or in combination with other standard-of-care treatments (e.g., radiation therapy). We recommend that TPP+-based drugs that have been tested in preclinical models or in humans should undergo clinical trials in patients with cancer.
1. Introduction
Recent reports indicate that some complex I inhibitors (e.g., IACS-010759) induced peripheral neuropathy in phase I clinical trials of patients with relapsed/refractory acute myeloid leukemia and in mice, suggesting that some complex I inhibitors are prone to induce neurotoxicity [Citation1,Citation2]. Developing mitochondria-targeted small-molecule therapeutics like mitochondria-targeted coenzyme Q10 (MitoQ), related analogs, and other triphenylphosphonium cation (TPP+)-based oxidative phosphorylation oxidative phosphorylation (OXPHOS) inhibitory drugs with negligible neurotoxicity in preclinical models and humans, therefore, will be a significant therapeutic advancement in cancer treatment (). Most of these TPP+-based OXPHOS inhibitors are synthesized from naturally occurring molecules or from US Food and Drug Administration (FDA)-approved drugs (). These TPP+-based OXPHOS inhibitors have a great potential to become anticancer agents due to a lack of neurotoxicity in preclinical models and humans.
Figure 1. Chemical structures of MTDs.
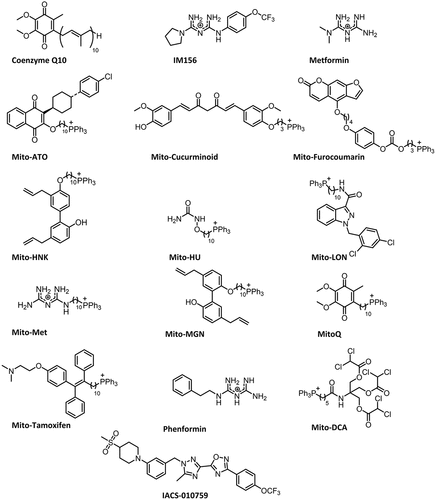
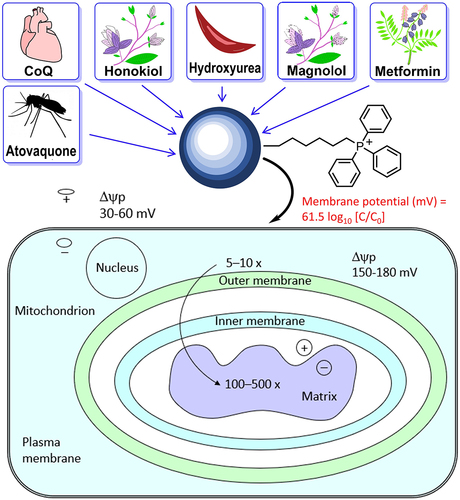
OXPHOS is emerging as a likely druggable target in cancer therapy [Citation3,Citation4]. Increasing evidence supports a significant role for mitochondrial metabolism in promoting cancer development and progression [Citation5–7]. Conjugating delocalized lipophilic cations, such as the TPP+, to compounds of interest is an effective mitochondrial targeting approach [Citation5–7]. The mitochondrial membrane potential in cancer cells (−220 mV) is more hyperpolarized than that in normal cells (−140 mV), and this discrepancy can drive a 100- to 200-fold uptake of cations to cancer cells [Citation5–9] ().
Figure 2. Selective uptake of TPP+-based MTDs into tumor mitochondria. Reprinted (adapted) with permission from Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chemical reviews. 2017;117(15):10043 –10,120. Copyright 2017 American Chemical Society.
The mitochondrial complex III inhibitor, atovaquone (ATO), decreased tumor hypoxia in lung cancer patients and also increased radiosensitivity [Citation10] as result of enhanced tumor oxygenation. Papaverine, a US FDA-approved drug that inhibits mitochondrial respiration, was used as a radiation sensitizer [Citation11]. Mitochondria-targeted atovaquone (Mito-ATO) and other mitochondria-targeted drugs (MTDs) that are much more potent than ATO and the corresponding parent molecule should be more potent hypoxic radiosensitizers. Mitochondrial OXPHOS inhibition enhanced the anti- programmed death-1 (PD-1) antibody immunotherapy in melanoma cancer therapy [Citation12]. lists the chemical structures of MTDs that are currently tested in humans and in preclinical xenograft models. TPP+-containing MTDs were also shown to mitigate the toxic side effects (e.g., cardiotoxicity) of standard-of-care chemotherapeutics such as doxorubicin in preclinical models [Citation13]. lists the potency and toxicity of the selected MTDs in different cancers.
Table 1. Potency and toxicity of selected mitochondria-targeted drugs in different cancers.
2. MitoQ
MitoQ, also referred to as Mito-ubiquinone, is an orally available molecule synthesized from conjugating a lipophilic TPP+ to coenzyme Q via an aliphatic 10-carbon chain. MitoQ is commercially available as a mitochondrial nutrient. Although MitoQ is not yet approved by the US FDA, it is undergoing multiple clinical trials to test its antioxidative mechanism of protection in oxidative pathologies. The antioxidant mechanism of MitoQ likely involves redox cycling between benzoquinone (the oxidized form) and benzoquinol (the reduced form) by complex II or succinate dehydrogenase in the mitochondrial electron transport chain [Citation14]. Mitochondrial reactive oxygen species (often referred to as mtROS) presumably oxidizes Mito-ubiquinol (MitoQH2) back to MitoQ, although the mechanistic details are not known.
2.1. Potency
Recent in vitro and in vivo studies show that MitoQ inhibits breast cancer and pancreatic cancer metastases, and the MitoQ effect was attributed to its ability to inhibit mitochondrial superoxide [Citation15,Citation16]. More importantly, MitoQ administration prevented the recurrence of breast cancer in mice xenografted with human cancer cells [Citation17].
Recent studies show that at submicromolar concentrations, both MitoQ and dimethoxy MitoQ (DM-MitoQ) potently inhibit mitochondrial respiration and tumor cell proliferation. Recently, MitoQ and its redox-crippled analog, DM-MitoQ, were shown to target and inactivate the mitochondrial chaperone protein, tumor necrosis factor receptor-associated protein 1 (Trap1), that preferentially inhibits cancer cells including the triple-negative breast cancer cells [Citation18]. MitoQ and DM-MitoQ inhibit Trap1, a mitochondrial heat shock protein 90. Trap1 also regulates mitochondrial respiration, acting as a switch between OXPHOS and glycolysis [Citation19,Citation20]. The investigators concluded that the antioxidant mechanism of MitoQ was not responsible for its enhanced cytotoxicity in breast cancer, as both MitoQ and redox-inactive analogs also induced a similar cytotoxic response [Citation20]. However, results from emerging studies suggest that MitoQ and DM-MitoQ potently inhibit complex I-induced oxygen consumption and proliferation of breast cancer and glioma cells, and that inhibition of OXPHOS by MitoQ and DM-MitoQ is primarily responsible for the antiproliferative effects in breast cancer and glioma cells [Citation21].
2.2. Toxicity
MitoQ was shown to be well tolerated without any perceptible neurotoxicity in Parkinson’s patients [Citation22]. Administration of MitoQ for a 12-week duration reversed the cardiotoxicity of doxorubicin in rats [Citation23]. Reports suggest that MitoQ was able to ameliorate peripheral neuropathy [Citation24]. Supraphysiological levels of MitoQ (i.e., three orders of magnitude higher than its effective concentrations in cancer cells) caused nephrotoxicity [Citation25].
New findings using redox-crippled MitoQ revealed that the antiproliferative and antitumor mechanism of MitoQ involves selective inhibition of OXPHOS in tumor cells.
3. Mito-HNK
Mitochondria-targeted honokiol (Mito-HNK) was synthesized by attaching a TPP+ moiety via an alkyl side chain to honokiol (HNK) [Citation26]. HNK is a key bioactive molecule in magnolia bark extracts that has been used for centuries in Asian countries for treating various inflammatory disorders. Also, it is a widely used nutritional supplement. HNK itself is a mitochondrial complex I inhibitor used in chemoprevention research [Citation26].
3.1. Lack of in vivo toxicity in mice
Mito-HNK was tested in an eight-week toxicology study in A/J mice [Citation26]. Neurons and muscle cells typically have a much higher plasma membrane potential that could enhance Mito-HNK uptake [Citation26]. Thus, a subchronic toxicity screen was used to monitor the central nervous system and neuromuscular effects during Mito-HNK treatment. Mice were treated orally with vehicle control and Mito-HNK at various doses (up to 20 times the effective dose) for five days per week for eight weeks. No histopathological changes were observed in neural or skeletal muscles. Even at 20-fold higher than the effective dose, Mito-HNK did not affect the motor function. Both body weight and rectal temperature remained constant during eight weeks of treatment with Mito-HNK. Detailed pharmacokinetic/pharmacodynamic studies are currently ongoing in mouse models of lung cancer as part of a cancer preclinical drug development program.
3.2. In vitro and in vivo potency: inhibition of lung cancer metastasis to the brain
Mito-HNK inhibits mitochondrial complex I-induced oxygen consumption, induces superoxide and hydrogen peroxide formation and activation of adenosine monophosphate-activated protein kinase (AMPK), inhibits signal transducer and activator of transcription 3 (STAT3) phosphorylation, and inhibits proliferation of cancer cells [Citation26]. Further, Mito-HNK inhibits lung cancer progression and prevents metastasis of lung cancer cells to lymph nodes and to the brain [Citation26]. From a mechanistic standpoint, the antitumor and antimetastatic effects were shown to be mediated by the STAT3 pathway. Knockdown of STAT3 abrogated both the antiproliferative and antimetastatic effects of Mito-HNK [Citation26]. Mito-HNK inhibits STAT3 phosphorylation irrespective of the epidermal growth factor receptor mutation status in lung cancer cells [Citation26]. Reports indicate that a decrease in the core body temperature and death result from the excessive inhibition of OXPHOS [Citation46]. Mito-HNK did not elicit these effects [Citation26]. Currently, treatment of lung cancer metastasis does not start until after diagnosis. Mito-HNK treatment inhibits metastasis of primary lung cancer to the brain. Based on the lack of in vivo toxicity, including neurotoxicity, in mice, we suggest that Mito-HNK is a promising antitumor candidate drug for preventing primary and metastatic lung cancer.
4. Mito-MGN
Magnolol is present in abundance in magnolia extract, a traditional herbal medicine used effectively for centuries in East Asia to treat inflammatory diseases. Mitochondrial-targeted magnolol (Mito-MGN) belongs to a new class of mitochondria-targeted polyphenolic drugs. Mito-MGN is synthesized by conjugating a TPP+ moiety via an alkyl side chain to magnolol [Citation27]. Currently, there are no effective drugs that treat melanoma, an aggressive form of skin cancer. B-Raf serine/threonine kinase inhibitor antiglycolytic drugs induce a rapid onset of drug resistance. B-Raf serine/threonine kinase inhibitors cause metabolic reprogramming from a glycolytic phenotype to an OXPHOS phenotype that is attributed to resistance against antiglycolytic kinase inhibiting drugs (e.g., vemurafenib). The increased dependence on OXPHOS for energy makes OXPHOS a vulnerable target in drug-resistant melanoma cells. Increased mitochondrial biogenesis and upregulated OXPHOS genes are associated with enhanced mitochondrial respiration in drug-resistant melanoma cells. Mito-MGN inhibited mitochondrial complex I-induced oxygen consumption, AKT-forkhead box O signaling, blocked cell cycle progression, and melanoma cell proliferation [Citation27,Citation28]. Mito-MGN was shown to potently inhibit melanoma cell proliferation and tumor growth in murine melanoma xenografts [Citation27,Citation28].
4.1. Toxicity
We have not done any detailed toxicology studies (e.g., like those that were done for Mito-HNK) for Mito-MGN. However, mice were treated three times a week with 1 mg of Mito-MGN administered intratumorally [Citation27]. Key metabolites of cardiac, hepatic, and renal function were measured in the serum. There were no significant differences in the serum levels of alanine transaminase (ALT), blood urea nitrogen, albumin, glucose, and protein in control and Mito-MGN-treated mice. Aspartate aminotransferase (AST) levels were considerably elevated upon treatment with Mito-MGN.
4.2. In vitro and in vivo potency
Mito-MGN potently inhibits mitochondrial complex I-induced mitochondrial respiration, blocks cell cycle progression, and inhibits proliferation of melanoma cells, primarily through downregulation of mammalian target of rapamycin (mTOR)/protein kinase B (AKT) signaling and mitophagy [Citation27].
Mito-MGN induced AMPK–threonine 172 phosphorylation, activating AMPK signaling, mitophagy, and energy-related proteins in melanoma cells. Mito-MGN treatment was equally effective in inhibiting drug-resistant melanoma cells (with enhanced OXPHOS) [Citation27].
Mito-MGN inhibited tumor progression in an immune-competent mouse xenograft model [Citation28]. Also, Mito-MGN remodeled the tumor microenvironment (TME) in a mouse melanoma model. Mito-MGN induced infiltration of T cells, decreased myeloid-derived suppressor cells (MDSCs), and decreased tumor-associated macrophages in melanoma tumors [Citation28]. The antitumor effect of Mito-MGN is inhibited by immune depletion [Citation28]. We conclude that the antitumor immunity effect of mitochondria-targeted polyphenolics is an exciting area of therapeutic drug targeting and TME remodeling.
5. Mito-ATO
ATO is a US FDA-approved drug for use in combination with proguanil for the prevention and treatment of malaria, a US FDA-approved alternative for the prevention or treatment of Pneumocystis pneumonia, and an alternative for treating Toxoplasma in combination with sulfadiazine [Citation47–55]. ATO is an inhibitor of OXPHOS and does not cause serious side effects when given at 3- to 42-fold the normal dose [Citation50–53]. ATO has been shown to inhibit the proliferation of cancer cells by targeting complex III and to exhibit antitumor activity in animal models of acute myelogenous leukemia, acute lymphocytic leukemia, and glioblastoma [Citation3,Citation56–59]. Inhibition of tumor growth by ATO was attributed to its direct effects on cancer cells and its ability to perturb immune cell recognition and/or alter the host’s TME [Citation3,Citation29,Citation30,Citation56–59].
We developed Mito-ATO by attaching the bulky TPP+ group to ATO via a long alkyl chain, which separates TPP+ from ATO’s structure and increases its lipophilicity and mitochondrial uptake in immunosuppressive cells in the TME [Citation30]. We showed that among the different Mito-ATO analogs tested, Mito10-ATO showed maximal potency to inhibit complex I- and III-induced mitochondrial oxygen consumption [Citation30].
5.1. Toxicity
Acute and subchronic toxicology of Mito-ATO has not been done extensively. Intratumoral delivery of Mito10-ATO showed no alterations in body weights, AST levels, or ALT levels in mice [Citation29].
5.2. In vitro and in vivo potency
We and others have shown that MTDs can target subtypes of immune cells in the TME [Citation5–7,Citation60–63]. We also explored the effects of Mito10-ATO on immune cells within the TME in mouse tumor models [Citation29]. Using an in situ vaccination approach, Mito10-ATO was injected locally into primary tumors, which triggered a potent T cell immune response that attacked both the local tumor and metastatic cancer cells. The amount of Mito10-ATO leaking into the circulation (~0.8% of the injected amount) was unable to induce the observed antitumor effects; therefore, the potent antitumor effects on the nontreated side tumor or metastatic tumor cells appear to be mediated by a systemic adaptive T cell tumor-specific immune response, including an increase in cytotoxic CD4+, T cells triggered by intratumoral Mito10-ATO injection of the treated side tumor [Citation29]. Flow cytometry analysis found that Mito10-ATO treatment decreased intratumoral granulocytic MDSCs and regulatory T cells (Tregs) and increased effector CD4+ T cells [Citation29]. Single-cell RNA sequencing showed that the reduction of granulocytic MDSCs and Tregs were linked to Mito10-ATO’s inhibition of OXPHOS (by suppression of mitochondrial complexes) and glycolysis in granulocytic MDSCs and Tregs, leading to the death of these cells via both extrinsic and intrinsic (mitochondrial) pathways of apoptosis [Citation29].
We demonstrated for the first time that injection of the novel OXPHOS inhibitor Mito10-ATO into primary tumors triggers a potent T cell immune responses locally and in distant sites of tumor metastasis by reversing immunosuppression in the TME [Citation5,Citation30].
MTDs that undergo redox activation to generate a huge flux of reactive oxygen species can inhibit metastasizing cancer cells via a prooxidant mechanism. Redox-active MTDs can exert antitumor effects via an OXPHOS-inhibiting mechanism and a pro-oxidant promoting mechanism [Citation64,Citation65]. Mito-ATO and MitoQ may also be considered as redox-active MTDs by virtue of the futile redox-cycling mechanism of the semiquinone in the presence of oxygen. Findings from our studies suggest that Mito-ATO is one of the most potent OXPHOS-inhibiting drugs that primarily targets mitochondrial complex III in cancer cells.
6. Mito-LND
Mito-lonidamine (Mito-LND) is lonidamine (LND) conjugated to TPP+ via an alkyl side chain linker [Citation31]. LND, a derivative of indole-3-carboxylic acid, is an antiglycolytic chemotherapeutic drug that has undergone Phase II and Phase III trials targeting lung cancer. It was shown to be safe with limited efficacy. Mito-LND is one of the least toxic MTDs developed in our laboratory. TPP+ modification converted a minimally effective antiglycolytic drug into a significantly more potent MTD. Mito-LND is nearly 100-fold more potent than LND as an OXPHOS inhibitor in lung cancer cells [Citation31]. Mito-LND induced mitophagy and autophagy in cancer cells [Citation31].
6.1. Toxicity
Mito-LND is distinctly different from LND with regard to target of inhibition. LND targets hexokinase and missense constraint whereas Mito-LND targets complex I and II in the mitochondrial electron transport chain. Mito-LND caused no toxicity in mice, even when administered at 50 times the effective cancer inhibitory dose for eight weeks [Citation31]. No adverse effects were noticed in tissues by histopathology or liver function tests. We used a Modified Irwin Screen, a comprehensive observational battery test employing 35 distinct measurements to assess sensorimotor, neurological, and autonomic nervous system function. No neurotoxicity was observed in Mito-LND treated mice.
6.2. In vitro and in vivo potency
Mito-LND was effective at low micromolar concentrations in inhibiting lung cancer cell proliferation. The half-maximal inhibitory concentration (IC50) of Mito-LND was 200–300-fold lower than the IC50 of LND for inhibiting the proliferation of lung cancer cells. Mito-LND downregulates the autophagy-linked energy-sensing mTOR/AKT signaling pathway in cancer cells [Citation31]. Mito-LND inhibits primary lung tumors and suppresses lung cancer brain metastases in mouse models of lung cancer. Mito-LND was more effective than LND. We conclude that Mito-LND is one of the least toxic and most potent MTDs for inhibiting lung tumor brain metastasis.
7. Mitochondria-targeted biguanides
7.1. Metformin
Metformin, a US FDA-approved drug, which has been used in diabetic patients (in daily doses of 2–3 g) for more than 50 years, has an excellent safety profile. However, cancer researchers have suggested that more potent biguanides should be used in cancer treatment because of the poor bioavailability and low plasma levels of metformin in diabetic cancer patients treated with metformin [Citation66]. The bioavailability of metformin is relatively poor. Metformin is undergoing several clinical trials in the field of cancer [Citation67]. Although interest in metformin therapy to treat cancer is slowly decreasing, this can be attributed in part to a lack of attention to patient selection, mechanism of action, and appropriate combinational modality. Metformin’s ability to reshape through the TME in preclinical models shows promise in combinational therapies involving immunotherapy.
7.2. Phenformin
Phenformin, a more lipophilic phenyl group substituted analog of metformin, was withdrawn because of a high risk of lactic acidosis [Citation68]. Clearly, lactic acidosis is one of the expected side effects associated with OXPHOS inhibitors that induce a compensatory increase in glycolysis in response to metabolic adaptation. Repurposing of phenformin in cancer therapy was recently reviewed [Citation69]. At therapeutic doses, phenformin suppressed Hedgehog-dependent tumor growth via a complex I-independent mechanism [Citation70]. Phenformin inhibited mitochondrial glycerophosphate dehydrogenase, leading to elevated levels of intracellular nicotinamide adenine dinucleotide hydride (NADH) and the association between corepressors CtBP2 and Gli1. Unlike metformin, Phenformin activates AMPK signaling in ornithine transcarbamylase-deficient tumor cells. Phenformin suppressed the growth of hepatocellular carcinoma in conjunction with mTOR inhibitors [Citation71]. Phenformin was used in conjunction with gemcitabine to eradicate high OXPHOS pancreatic cancer cells [Citation72].
7.3. IM156
IM156, also known as Lixumistat, is an orally active analog of metformin that inhibits OXPHOS more potently than metformin in tumor cells [Citation33]. The first-in-human study of IM156 in patients with advanced solid tumors and lymphoma was recently published (Immunomet Therapeutics, Inc.) [Citation33]. IMI56 inhibits mitochondrial protein complex I that that oxidizes NADH in the mitochondrial electron transport chain, leading to adenosine triphosphate formation. In contrast with metformin that requires active transporters such as organic cation transporter 1, IM156, being more hydrophobic, diffuses across the cell membrane. It was concluded that IM156 should be used in combination with other targeted antitumor therapies and/or in selected cancer patients and not as a single agent due to limited clinical activity in an unselected population of cancer patients [Citation33].
7.4. Mito-Mets
Mitochondria-targeted metformins (Mito-Mets) are a novel class of compounds synthesized by attaching a TPP+ moiety or a substituted TPP+ moiety via an alkyl side chain of varying lengths to metformin [Citation35–37]. Mito10-Met exhibits a 1,000-fold higher potency than metformin in inhibiting mitochondrial respiration, AMPK activation, and suppression of forkhead box protein M1 in cancer cells, and therefore, could provide significant therapeutic advantage in patients with advanced pancreatic cancer [Citation35]. Mito-Mets have been used as a potent sensitizer in radiation therapy due to their ability to inhibit OXPHOS and decrease tumor hypoxia.
7.4.1. Toxicity
Serum from mice dosed with Mito10-Met was collected, and hepatic and nephrotoxicity assessed using standard AST, ALT, alkaline phosphatase, and blood urea nitrogen assays, respectively. Neither metformin nor Mito10-Met showed any significant off-target toxicity in vivo [Citation35]. After two weeks of administration of Mito10-Met in mice, there was an increased accumulation of Mito-Met10 in liver, kidney, spleen, and tumor tissues.
7.4.2. In vitro and in vivo potency
The antiproliferative effects of Mito-Mets increased with increasing alkyl side chain lengths (100-fold to 1,000-fold). The lead compound, Mito10-Met, activated AMPK phosphorylation at a much lower concentration than metformin in pancreatic and colon cancer cells [Citation35,Citation36]. Mito10-Met abrogated pancreatic cancer growth in preclinical mouse models more potently than metformin.
We conclude that Mito-Mets could serve as an effective antitumor drug for treating pancreatic, hepatic, and renal cancers in combination with radiation or other treatment modalities.
8. Mito-HUs
Hydroxyurea (HU) is a US FDA-approved drug for treating sickle cell disease. It has also been used as an antitumor drug alone or in combination with other chemotherapeutics or radiation. Because it inhibits the enzyme ribonucleotide reductase responsible for DNA synthesis, HU is also used to treat myeloproliferative diseases. Mitochondria-targeted hydroxyurea (Mito-HU) was synthesized by substituting the hydroxyl group in HU with a TPP+ attached to an aliphatic side chain of different lengths [Citation38]. HU is very hydrophilic, so a considerably longer side chain length (n = 14–20) was needed to enhance the hydrophobicities of Mito-HU.
8.1. Toxicity
Most of the studies with Mito-HU analogs were performed in colon cancer and immune cells. The IC50 values of the Mito-HUs on the inhibition of cell proliferation in HCT116 colon cancer cells indicate that with increasing alkyl side chain length, Mito-HUs became increasingly potent. No obvious cytotoxicity was observed [Citation38]. Mito-HUs may have the potential to induce reprogramming of immune cells in the TME [Citation38]. Studies using Mito-HUs in in vivo immune competent and immune deficient mouse models have not been done.
8.2. In vitro and in vivo potency
Elongating the alkyl side chain length in the Mito-HU class of compounds increased its hydrophobicity, as well as its OXPHOS inhibiting and antiproliferating potencies in several cancer cells [Citation38]. This chain length dependent potency of Mito-HU compounds was also reflected in the immunomodulatory effects. The more hydrophobic Mito-HUs (e.g., Mito14-HU, Mito18-HU) more strongly inhibited the MDSCs and suppressive neutrophils while stimulating T cell response [Citation38]. We conclude that Mito-HUs represent a new class of MTDs exhibiting antimetastatic characteristics.
9. MTDs developed in other laboratories
Brief descriptions of some MTDs developed by other investigators are provided in the following sections.
9.1. Mito-Tamoxifen
Tamoxifen is an estrogen-modulating drug used to treat women and men with advanced breast cancer. This drug was banned due to its serious side effects, including a small increased risk of uterine cancer and other problems (e.g., pulmonary embolism, stroke). The mechanism of action of Tamoxifen involves inhibition of estrogen uptake through the estrogen receptors. TPP+ conjugation to Tamoxifen resulted in an MTD, Mito-Tamoxifen, that is undergoing clinical trials for treatment of metastatic solid tumors [Citation39,Citation40]. The investigators concluded that mitochondrial electron transport chain is a suitable therapeutic target in human epidermal growth factor receptor 2 overexpressing breast cancer. Selective disruption of respiratory supercomplexes is a new strategy in cancer treatment for several MTDs [Citation40,Citation73,Citation74].
9.2. Mito-Curcuminoids
Mitochondria-targeted curcumin analogs (Mito-Curcuminoids) were developed as antitumor agents that disrupt thioredoxin redox enzyme [Citation41,Citation42]. This strategy may overcome the limitation of poor bioavailability of curcumin.
9.3. Mito-Furocoumarins
Mitochondrial ion channels have been targeted in cancer therapy, especially the mitochondrial potassium-selective channel. Mitochondria-targeted furocoumarins (Mito-Furocoumarins) effectively block the mitochondrial potassium channel, enhancing apoptosis of tumor cells [Citation43].
9.4. Mito-DCA
Mitochondria-targeted dichloroacetic acid (Mito-DCA) is a tumor-selective metabolic inhibitor and is synthesized by conjugating TPP+ to the parent small molecule, dichloroacetic acid [Citation44]. Mito-DCA suppressed lactic acid production in tumor cells, which led to reprogramming of the immune system and activation of tumor cell-toxic T cells [Citation44]. Mito-DCA did not affect normal cells; it only affected tumor cells with dysfunctional mitochondria [Citation44].
10. Relative inhibitory effects of MTDs: hydrophobicity differences
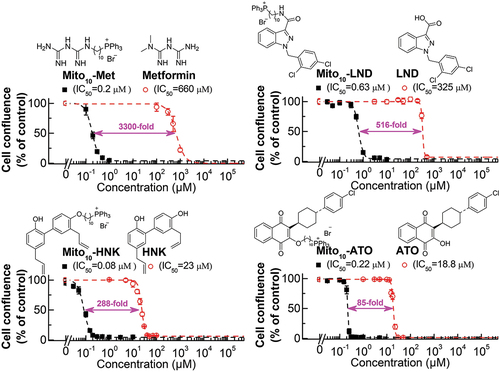
We previously reported that increasing the aliphatic chain length in TPP+-conjugated molecules greatly enhanced the antiproliferative potencies in tumor cells [Citation35]. The fold difference between the parent compound and the TPP+-modified compound (with 10 carbons in the linker side chain) is dependent on the parent compound, especially its hydrophobicity. shows the dose response characteristics of metformin, LND, HNK, and ATO and the TPP+-modified analogs in human pancreatic cancer (MiaPaCa-2) cells [Citation26,Citation35,Citation75]. The difference in the antiproliferative effect between ATO and Mito-ATO is 85-fold, whereas the difference between metformin and Mito10-Met is 3,300-fold. Although many factors are responsible for the fold difference between the TPP+-modified drug and the unmodified drug, the hydrophobicity of the parent drug is a major factor. If the parent compound is very hydrophilic, TPP+ modification will likely induce a greater fold difference in antiproliferative effect and inhibition of mitochondrial respiration in tumor cells [Citation38] due to the more negative mitochondrial membrane potential of tumor cells as compared with normal cells [Citation9,Citation76–79].
Figure 3. Comparisons of MTDs and the corresponding parent compounds on cell proliferation inhibitions in human pancreatic cancer (MiaPaca-2) cells. The effects of MTDs and their parental compounds on the proliferation of MiaPaCa-2 cells were monitored in the IncuCyte Live-cell Analysis system. The IC50 values were determined at the point at which control cells reached ~ 90% confluence. Relative cell confluence (control is taken as 100%) is plotted against concentration. Dashed lines represent the fitting curves used to determine the IC50 values as indicated. The fold of differences as indicated were calculated by the potency difference of the IC50 values between each mitochondria-targeted drug and its parental compound. The IC50 values of Mito-Met and metformin were published previously in Cancer Research (Cheng G et al., cancer Res, 2016). The IC50 values of Mito-ATO and ATO were published in Scientific Reports (Cheng G et al., Scientific Reports, 2020; Cheng G et al., Scientific Reports, 2022). This figure is re-used under CC by 4.0 from Cheng G, Hardy M, Kalyanaraman B. Antiproliferative effects of mitochondria-targeted N-acetylcysteine and analogs in cancer cells. Scientific Reports. 2023;13:7254.
11. OXPHOS inhibitors, MDSCs, and metastatic cancer
The metabolic reprogramming (enhanced OXPHOS) that occurs in metastatic cancer cells likely plays a major role in metastatic cancer cell survival and progression [Citation80,Citation81]. Reports indicate that an OXPHOS inhibitor, IACS-010759, inhibits melanoma brain metastasis [Citation32]. The mitochondrial complex I inhibitor also inhibits MDSCs in the metastatic TME. TPP+-conjugated OXPHOS inhibitors of mitochondrial complex I and complex III – Mito-MGN, Mito-ATO, and Mito-HU – are potentially suitable antimetastatic drugs [Citation27,Citation30]. It was reported that brain metastases from patients with melanoma displayed a considerable degree of immunosuppression and increased expression of genes related to OXPHOS. IACS-010759, a reported mitochondrial complex I inhibitor, blocks metastasis formation in mouse models [Citation32]. Mito-HNK, Mito-ATO, and Mito-LND are potent OXPHOS inhibitors and inhibit lung cancer metastasis to the brain in mouse models [Citation30,Citation31].
Drug resistance to kinase inhibitors is attributed to metabolic reprogramming from glycolysis to mitochondrial oxidative metabolism. Bioenergetic mapping results showed that tumor cells with enhanced mitochondrial OXPHOS were more sensitive to TPP+-based MTDs and other OXPHOS inhibitors [Citation82]. Enhanced OXPHOS reprograms mitochondrial energy to promote triple-negative breast cancer metastasis [Citation83,Citation84]. Metastatic breast cancer cells exhibit enhanced mitochondrial OXPHOS activity [Citation85]. Mitochondrial OXPHOS genes were transcriptionally upregulated in primary breast cancers. Breast cancer metastases have higher tricarboxylic acid cycle flux compared with primary tumors [Citation86]. Melanoma brain metastases have enhanced OXPHOS genes [Citation32,Citation87,Citation88]. The progression of breast cancer to brain metastasis is a major cause of fatalities. Similar to Mito-HNK and Mito-LND [Citation26,Citation31], MitoQ crosses the blood-brain barrier [Citation89,Citation90]. This is significant for MTDs as antitumor drugs in metastatic cancers.
12. OXPHOS inhibitors, tumor hypoxia, and biomarkers in radiation therapy
Radiation is not very effective in killing hypoxic tumor cells. OXPHOS inhibitors are effective hypoxic radiation sensitizers [Citation11,Citation36,Citation91,Citation92]. The enhanced susceptibility to radiation results from inhibiting mitochondrial respiration. Inhibition of mitochondrial respiration effectively increases the oxygen concentration, decreases tumor hypoxia, and inhibits hypoxic gene expression in lung cancer patients [Citation93]. Tumor hypoxia is recognized as a potential barrier to other cancer treatment modalities such as chemotherapy, radiation therapy, and immunotherapy [Citation3]. In addition to a reduction in tumor hypoxia (measured by positron emission tomography–computed tomography), another major pharmacodynamic endpoint of ATO treatment in cancer patients was shown to be downregulation in hypoxia-regulated genes in ATO-treated tumors [Citation93].
13. Molecular targets of MTDs
Previously, we used low-temperature electron paramagnetic resonance to determine the mitochondrial redox changes in pancreatic cancer cells treated with TPP+-containing MTDs [Citation94]. Based on the electron paramagnetic resonance spectral changes of mitochondrial complex I iron-sulfur (FeS) clusters, [2Fe2S]+ and [4Fe-4S]+, we surmised that TPP+-containing MTDs (mitochondrial complex I inhibitors) bind closer to the NADH-dehydrogenase site in the mitochondrial complex I dictated by the NAD+/NADH couple [Citation95].
Mito10-ATO, formed by attachment of TPP+ to ATO, inhibits both complex I- and complex III-induced oxygen consumption [Citation30]. Similar to ATO, Mito10-ATO likely targets the Q0 site of the cytochrome bc1 complex (ubiquinol cytochrome c oxidoreductase or complex III) [Citation30]. However, increasing the alkyl side chain length in Mito-ATO (e.g., Mito12-ATO or Mito16-ATO) inhibited only the complex I- and not complex III-induced oxygen consumption, suggesting that the side chain length and hydrophobicity are critical for stabilization at the Q0 site of complex III [Citation30]. Additional structural studies are needed to provide further insights on the molecular binding of Mito-ATO analogs in mitochondria.
Cryo-electron microscopy studies recently provided new insights on the binding and bonding characteristics of metformin, a weakly cationic MTD, and the more hydrophobic analogs of metformin (phenformin and IM1092) in the Q-channel of mammalian respiratory complex I [Citation96]. Hydrophobicity differences are primarily responsible for the observed differences in the potency of the unsubstituted and halogen-containing phenyl group in biguanides [Citation96]. Going forward, cryo-electron microscopy structural studies should be performed with TPP+ containing MTDs.
14. Conclusion
In this opinion article, we reviewed a new class of TPP+-based drugs as potential OXPHOS-targeted cancer therapeutics. For the most part, these drugs are modifications of naturally occurring molecules or US FDA-approved drugs. Several TPP+-based drugs lack the neurotoxicity associated with an OXPHOS drug whose clinical trial was recently terminated. We make a case here that TPP+-based drugs free of neurotoxicity and other toxicities (e.g., lactic acidosis) in preclinical models should be tested in cancer patients in future clinical trials.
15. Expert opinion
Most of the publications on mitochondrial inhibitors in cancer are centered on a few drugs (e.g., metformin, IACS-010759, ATO). Metformin is a US FDA-approved antidiabetic drug. Its bioavailability is poor, and it is a weak mitochondrial complex I inhibitor. In contrast, Mito-Met is nearly 1,000-times more potent than metformin with respect to its inhibition of cancer cell respiration and proliferation. ATO is a mitochondrial complex III inhibitor that has been repurposed in cancer research. Mito10-ATO is a much more potent mitochondria-targeted complex III inhibitor of cancer cells. The OXPHOS-inhibiting drug, IACS-010759, exhibited neurotoxicity in a preclinical model and in cancer patients. The MTDs discussed here are structurally modified, naturally occurring compounds or US FDA-approved drugs with an established safety profile. MTDs target proteins in the electron transport chain in the mitochondria of cancer cells and inhibit mitochondrial respiration.
The mitochondrial electron transport chain is a vulnerable target in some drug-resistant cancer cells and cancer stem cells. These cells have increased OXPHOS genes and are more susceptible to MTDs. Mito-HNK is not toxic at 20 times the effective dose. There was no perceived neurotoxicity in Mito-HNK treated mice.
There is a direct correlation between hydrophobicity and enhanced antiproliferative effect in cancer cells treated with MTDs. Hydrophobicity is altered with different chain lengths and substituted phenyl groups (−CF3) in MTDs.
TPP+-based MTDs (e.g., MitoQ) have been tested in humans with no signs of peripheral neuropathy. Recent studies show that MitoQ inhibits metastasis of breast cancer in mice.
Mitochondrial complex I inhibitors are synthetically lethal in PTEN-deficient metastatic prostate cancer cells [Citation97]. The scope of the antitumor mechanism of MTDs can be greatly expanded in the context of synthetic lethality.
Enhanced expression and activity of OXPHOS genes and OXPHOS activity were suggested as a suitable biomarker for selecting cancer patients for targeted therapy with MTDs in combination with a standard-of-care antitumor therapy or therapeutic agents [Citation72,Citation98–100]. To overcome chemoresistance due to high OXPHOS, future clinical trials should use MTDs that are highly potent, have the least toxicity, and are used to treat other conditions in humans and/or MTDs that have been tested extensively for various toxicities (including neurotoxicity) in preclinical models.
Cancer stem cells, which are resistant to chemotherapy, exhibit enhanced mitochondrial OXPHOS activity and are vulnerable to drugs containing TPP+ [Citation101].
Mito-ATO induced metabolic reprogramming in the TME [Citation29]. Recent results show that MDSCs and Tregs are novel targets in Mito-ATO [Citation30]. Mito-ATO decreased MDSCs and Tregs and increased tumor-infiltrating cytotoxic T cells. Mito-ATO improved the efficacy of PD-1 blockade immunotherapy [Citation29]. The combinatorial antitumor effect of MTDs such as Mito-ATO and analogs in immuno-oncology is an exciting development. Preclinical data suggest a rational basis for future clinical evaluation of MTDs such as Mito-ATO or its analogs in combination with PD-1 blockade and other immune checkpoint inhibitors [Citation29].
Article highlights
Review of potent, tumor-cell-selective, and low-toxicity drugs
Translatable to the clinic
Ability to inhibit metastases of primary tumors
Potential synergy with radiation and immunotherapies
Abbreviations list
| AKT | = | protein kinase B |
| ALT | = | alanine transaminase |
| AMPK | = | adenosine monophosphate-activated protein kinase |
| AST | = | aspartate aminotransferase |
| ATO | = | atovaquone |
| DM-MitoQ | = | dimethoxy MitoQ |
| EPR | = | electron paramagnetic resonance |
| FeS | = | iron-sulfur |
| FDA | = | Food and Drug Administration |
| IC50 | = | half-maximal inhibitory concentration |
| HNK | = | honokiol |
| HU | = | hydroxyurea |
| LND | = | lonidamine |
| MAPKi | = | mitogen-activated protein kinases inhibitor |
| MBM | = | mouse brain membrane |
| MDSC | = | myeloid-derived suppressor cell |
| METC | = | mitochondrial electron transport chain |
| Mito-ATO | = | mitochondria-targeted atovaquone |
| Mito-Curcuminoids | = | mitochondria-targeted curcumin analogs |
| Mito-DCA | = | mitochondria-targeted dichloroacetic acid |
| Mito-Furocoumarins | = | mitochondria-targeted furocoumarin analogs |
| Mito-HNK | = | mitochondria-targeted honokiol |
| Mito-HU | = | mitochondria-targeted hydroxyurea |
| Mito-LND | = | mitochondria-targeted lonidamine |
| Mito-Mets | = | mitochondria-targeted metformin analogs |
| Mito-MGN | = | mitochondrial-targeted magnolol |
| MitoQ | = | mitochondria-targeted coenzyme Q10 |
| MitoQH2 | = | mito-ubiquinol |
| Mito-Tamoxifen | = | mitochondria-targeted tamoxifen |
| MTD | = | mitochondria-targeted drug |
| mTOR | = | mammalian target of rapamycin |
| mtROS | = | mitochondrial reactive oxygen species |
| NAD+ | = | nicotinamide adenine dinucleotide |
| NADH | = | nicotinamide adenine dinucleotide hydride |
| OXPHOS | = | oxidative phosphorylation |
| PD-1 | = | programmed death-1 |
| STAT3 | = | signal transducer and activator of transcription 3 |
| TME | = | tumor microenvironment |
| TPP+ | = | triphenylphosphonium cation |
| Trap1 | = | tumor necrosis factor receptor-associated protein 1 |
| Tregs | = | regulatory T cells |
| US FDA | = | US Food and Drug Administration |
Declaration of interest
Balaraman Kalyanaraman is an inventor of US Patent No. 9,956,233, ‘Neuroprotection by mitochondria-targeted metformin.’
Balaraman Kalyanaraman and Micael Hardy are inventors of US Patent No. 11,274,114, ‘Modified mito-metformin compounds and methods of synthesis and use thereof;’ US Patent No. 10,836,782, ‘Mito-honokiol compounds and methods of synthesis and use thereof;’ and US Patent No. 11,352,382, ‘Mito-lonidamine, compositions and methods of use.’
Balaraman Kalyanaraman, Gang Cheng, and Micael Hardy are inventors of US Patent No. 11,083,739 and US Patent No. 11,612,610, both titled ‘Mito-magnolol compounds and methods of synthesis and use thereof,’ and WO2021081500A1, ‘Mitochondria-targeted atovaquone: a more potent and more effective antitumor, antimicrobial, and antimalarial drug,’ which is under consideration by the US Patent and Trademark Office.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Thank you to Lydia Washechek for preparing and proofreading the manuscript.
Additional information
Funding
References
- Yap TA, Daver N, Mahendra M, et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: phase I trials. Nat Med. 2023;29(1):115–126.
- Zhang X, Dang CV. Time to hit pause on mitochondria-targeting cancer therapies. Nat Med. 2023;29(1):29–30.
- Ashton TM, McKenna WG, Kunz-Schughart LA, et al. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018 Jun 1;24(11):2482–2490. doi: 10.1158/1078-0432.CCR-17-3070
- Xu Y, Xue D, Bankhead A 3rd, et al. Why all the Fuss about oxidative phosphorylation (OXPHOS)? J Med Chem. 2020 Dec 10;63(23):14276–14307. doi: 10.1021/acs.jmedchem.0c01013
- Huang M, Myers CR, Wang Y, et al. Mitochondria as a novel target for cancer chemoprevention: emergence of mitochondrial-targeting agents. Cancer Prev Res (Phila). 2021 Mar;14(3):285–306. doi: 10.1158/1940-6207.CAPR-20-0425
- Kalyanaraman B. Teaching the basics of repurposing mitochondria-targeted drugs: from Parkinson’s disease to cancer and back to Parkinson’s disease. Redox Biol. 2020 Aug 3;36:101665. doi: 10.1016/j.redox.2020.101665
- Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015 Sep 15;43(3):435–449. doi: 10.1016/j.immuni.2015.09.001
- Smith RAJ, Porteous CM, Gane AM, et al. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5407–5412. doi: 10.1073/pnas.0931245100
- Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018 Dec;17(12):865–886. doi: 10.1038/nrd.2018.174
- Ashton TM, Fokas E, Kunz-Schughart LA, et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat Commun. 2016 Jul 25;7(1):12308. doi: 10.1038/ncomms12308
- Benej M, Hong X, Vibhute S, et al. Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc Natl Acad Sci U S A. 2018 Oct 16;115(42):10756–10761. doi: 10.1073/pnas.1808945115
- Nishida M, Yamashita N, Ogawa T, et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes. J Immunother Cancer. 2021 Sep;9(9):e002954. doi: 10.1136/jitc-2021-002954
- Chandran K, Aggarwal D, Migrino RQ, et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J. 2009 Feb 18;96(4):1388–1398. doi: 10.1016/j.bpj.2008.10.042
- Graham D, Huynh NN, Hamilton CA, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009 Aug;54(2):322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351
- Capeloa T, Krzystyniak J, d’Hose D, et al. MitoQ inhibits human breast cancer cell migration, invasion and Clonogenicity. Cancers (Basel). 2022 Mar 16;14(6):1516. doi: 10.3390/cancers14061516
- Capeloa T, Van de Velde JA, d’Hose D, et al. Inhibition of mitochondrial redox signaling with MitoQ prevents metastasis of human pancreatic cancer in mice. Cancers (Basel). 2022 Oct 7;14(19):4918. doi: 10.3390/cancers14194918
- Capeloa T, Krzystyniak J, Rodriguez AC, et al. MitoQ prevents human breast cancer recurrence and lung metastasis in mice. Cancers (Basel). 2022 Mar 15;14(6):1488. doi: 10.3390/cancers14061488
- Yoon NG, Lee H, Kim SY, et al. Mitoquinone inactivates mitochondrial chaperone TRAP1 by blocking the client binding site. J Am Chem Soc. 2021 Dec 1;143(47):19684–19696. doi: 10.1021/jacs.1c07099
- Yoshida S, Tsutsumi S, Muhlebach G, et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci U S A. 2013 Apr 23;110(17):E1604–12. doi: 10.1073/pnas.1220659110
- Rasola A, Neckers L, Picard D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. 2014 Aug;24(8):455–463. doi: 10.1016/j.tcb.2014.03.005
- Cheng G, Karoui H, Hardy M, et al. Redox-crippled MitoQ potently inhibits breast cancer and glioma cell proliferation: a negative control for verifying the antioxidant mechanism of MitoQ in cancer and other oxidative pathologies. Free Radic Biol Med. 2023 Aug 20;205:175–187. doi: 10.1016/j.freeradbiomed.2023.06.009
- Smith RAJ, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ [conference paper]. Ann N Y Acad Sci. 2010;1201:96–103.
- Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010 May 11;107(19):8788–8793. doi: 10.1073/pnas.1003428107
- Fink B, Coppey L, Davidson E, et al. Effect of mitoquinone (Mito-Q) on neuropathic endpoints in an obese and type 2 diabetic rat model. Free Radic Res. 2020 May;54(5):311–318. doi: 10.1080/10715762.2020.1754409
- Gottwald EM, Duss M, Bugarski M, et al. The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol Rep. 2018 Apr;6(7):e13667. doi: 10.14814/phy2.13667
- Pan J, Lee Y, Cheng G, et al. Mitochondria-targeted honokiol confers a striking inhibitory effect on lung cancer via inhibiting complex I activity. iScience. 2018 May 25;3:192–207. doi: 10.1016/j.isci.2018.04.013
- Cheng G, Hardy M, Zielonka J, et al. Mitochondria-targeted magnolol inhibits OXPHOS, proliferation, and tumor growth via modulation of energetics and autophagy in melanoma cells. Cancer Treat Res Commun. 2020 Sep 17;25:100210.
- AbuEid M, McAllister DM, McOlash L, et al. Synchronous effects of targeted mitochondrial complex I inhibitors on tumor and immune cells abrogate melanoma progression. iScience. 2021 Jun 25;24(6):102653. doi: 10.1016/j.isci.2021.102653
- Huang M, Xiong D, Pan J, et al. Prevention of tumor growth and dissemination by in situ vaccination with Mitochondria-Targeted Atovaquone. Adv Sci. 2022;9(12):2101267. doi: 10.1002/advs.202101267
- Cheng G, Hardy M, Topchyan P, et al. Potent inhibition of tumour cell proliferation and immunoregulatory function by mitochondria-targeted atovaquone Sci Rep. 2020 Oct 21;10(1):17872. doi: 10.1038/s41598-020-74808-0
- Cheng G, Zhang Q, Pan J, et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat Commun. 2019;10(1):2205. doi: 10.1038/s41467-019-10042-1
- Fischer GM, Jalali A, Kircher DA, et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 2019 May;9(5):628–645. doi: 10.1158/2159-8290.CD-18-1489
- Janku F, Beom SH, Moon YW, et al. First-in-human study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2022 Oct;40(5):1001–1010. doi: 10.1007/s10637-022-01277-9
- Izreig S, Gariepy A, Kaymak I, et al. Repression of LKB1 by miR-17∼92 sensitizes MYC-Dependent lymphoma to biguanide treatment. Cell Rep Med. 2020 May 19;1(2):100014. doi: 10.1016/j.xcrm.2020.100014
- Cheng G, Zielonka J, Ouari O, et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. 2016 Jul 1;76(13):3904–3915. doi: 10.1158/0008-5472.CAN-15-2534
- Boyle KA, Van Wickle J, Hill RB, et al. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J Biol Chem. 2018 Sep 21;293(38):14891–14904. doi: 10.1074/jbc.RA117.001469
- AbuEid M, Keyes RF, McAllister D, et al. Fluorinated triphenylphosphonium analogs improve cell selectivity and in vivo detection of mito-metformin. iScience. 2022 Dec 22;25(12):105670. doi: 10.1016/j.isci.2022.105670
- Cheng G, Hardy M, Topchyan P, et al. Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects. iScience. iScience. 2021 Jun 25;24(6):102673. doi: 10.1016/j.isci.2021.102673
- Bielcikova Z, Stursa J, Krizova L, et al. Mitochondrially targeted tamoxifen in patients with metastatic solid tumours: an open-label, phase I/Ib single-centre trial. EClinicalMedicine. 2023 Mar;57:101873. doi: 10.1016/j.eclinm.2023.101873
- Rohlenova K, Sachaphibulkij K, Stursa J, et al. Selective disruption of respiratory supercomplexes as a new strategy to suppress Her2(high) breast cancer. Antioxid Redox Signal. 2017 Jan 10;26(2):84–103. doi: 10.1089/ars.2016.6677
- Reddy CA, Somepalli V, Golakoti T, et al. Mitochondrial-targeted curcuminoids: a strategy to enhance bioavailability and anticancer efficacy of curcumin. PLoS One. 2014;9(3):e89351. doi: 10.1371/journal.pone.0089351
- Jayakumar S, Patwardhan RS, Pal D, et al. Mitochondrial targeted curcumin exhibits anticancer effects through disruption of mitochondrial redox and modulation of TrxR2 activity. Free Radic Biol Med. 2017 Dec;113:530–538.
- Mattarei A, Romio M, Managò A, et al. Novel mitochondria-targeted furocoumarin derivatives as possible anti-cancer agents. Front Oncol. 2018;8:122. doi: 10.3389/fonc.2018.00122
- Pathak RK, Marrache S, Harn DA, et al. Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate [article]. ACS Chem Biol. 2014 May 16;9(5):1178–1187. doi: 10.1021/cb400944y
- Kolb D, Kolishetti N, Surnar B, et al. Metabolic modulation of the tumor microenvironment leads to multiple checkpoint inhibition and immune cell infiltration. ACS Nano. 2020;14(9):11055–11066. doi: 10.1021/acsnano.9b10037
- Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018 Jul;24(7):1036–1046. doi: 10.1038/s41591-018-0052-4
- Srivastava IK, Rottenberg H, Vaidya AB. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem. 1997 Feb 14;272(7):3961–3966. doi: 10.1074/jbc.272.7.3961
- Radloff PD, Philipps J, Nkeyi M, et al. Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet. 1996 Jun 1;347(9014):1511–1514. doi: 10.1016/S0140-6736(96)90671-6
- Hughes W, Leoung G, Kramer F, et al. Comparison of atovaquone (566C80) with trimethoprim-sulfamethoxazole to treat pneumocystis carinii pneumonia in patients with AIDS. N Engl J Med. 1993 May 27;328(21):1521–1527. doi: 10.1056/NEJM199305273282103
- Freeman CD, Klutman NE, Lamp KC, et al. Relative bioavailability of atovaquone suspension when administered with an enteral nutrition supplement. Ann Pharmacother. 1998 Oct;32(10):1004–1007. doi: 10.1345/aph.17464
- Darade A, Pathak S, Sharma S, et al. Atovaquone oral bioavailability enhancement using electrospraying technology. Eur J Pharm Sci. 2018 Jan 1;111:195–204. doi: 10.1016/j.ejps.2017.09.051
- Dixon R, Pozniak AL, Watt HM, et al. Single-dose and steady-state pharmacokinetics of a novel microfluidized suspension of atovaquone in human immunodeficiency virus-seropositive patients. Antimicrob Agents Chemother. 1996 Mar;40(3):556–560. doi: 10.1128/AAC.40.3.556
- Nixon GL, Moss DM, Shone AE, et al. Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother. 2013;68(5):977–985. doi: 10.1093/jac/dks504
- Fry M, Pudney M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4’-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem Pharmacol. 1992 Apr 1;43(7):1545–1553. doi: 10.1016/0006-2952(92)90213-3
- Mather MW, Darrouzet E, Valkova-Valchanova M, et al. Uncovering the molecular mode of action of the antimalarial drug atovaquone using a bacterial system. J Biol Chem. 2005 Jul 22;280(29):27458–27465. doi: 10.1074/jbc.M502319200
- Fiorillo M, Lamb R, Tanowitz HB, et al. Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016 Jun 7;7(23):34084–34099. doi: 10.18632/oncotarget.9122
- Xiang M, Kim H, Ho VT, et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood. 2016 Oct 6;128(14):1845–1853. doi: 10.1182/blood-2015-07-660506
- Gupta N, Srivastava SK. Atovaquone: an antiprotozoal drug suppresses primary and resistant breast tumor growth by inhibiting HER2/β-catenin signaling. Mol Cancer Ther. 2019 Oct;18(10):1708–1720. doi: 10.1158/1535-7163.MCT-18-1286
- Tian S, Chen H, Tan W. Targeting mitochondrial respiration as a therapeutic strategy for cervical cancer. Biochem Biophys Res Commun. 2018 May 23;499(4):1019–1024. doi: 10.1016/j.bbrc.2018.04.042
- Loftus RM, Finlay DK. Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem. 2016 Jan 1;291(1):1–10. doi: 10.1074/jbc.R115.693903
- Andrejeva G, Rathmell JC. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017 Jul 5;26(1):49–70. doi: 10.1016/j.cmet.2017.06.004
- Li X, Wenes M, Romero P, et al. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019 Jul;16(7):425–441. doi: 10.1038/s41571-019-0203-7
- Gaber T, Strehl C, Buttgereit F. Metabolic regulation of inflammation. Nat Rev Rheumatol. 2017 May;13(5):267–279. doi: 10.1038/nrrheum.2017.37
- Tasdogan A, Ubellacker JM, Morrison SJ. Redox regulation in cancer cells during metastasis. Cancer Discov. 2021 Nov;11(11):2682–2692. doi: 10.1158/2159-8290.CD-21-0558
- Tasdogan A, Faubert B, Ramesh V, et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature. 2020 Jan;577(7788):115–120. doi: 10.1038/s41586-019-1847-2
- Burton LP, Deng G, Yanes CD, et al. Novel metformin analogues for treatment of pancreatic cancer. J Endocr Soc. 2021;5:1027–1028. doi: 10.1210/jendso/bvab048.2103
- Lord SR, Harris AL. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br J Cancer. 2023 Apr;128(6):958–966. doi: 10.1038/s41416-023-02204-2
- McGuinness ME, Talbert RL. Phenformin-induced lactic acidosis: a forgotten adverse drug reaction. Ann Pharmacother. 1993 Oct;27(10):1183–1187. doi: 10.1177/106002809302701004
- Zhao H, Swanson KD, Zheng B. Therapeutic repurposing of biguanides in cancer. Trends Cancer. 2021 Aug;7(8):714–730. doi: 10.1016/j.trecan.2021.03.001
- Di Magno L, Manni S, Di Pastena F, et al. Phenformin inhibits hedgehog-dependent tumor growth through a complex I-Independent redox/corepressor module. Cell Rep. 2020 Feb 11;30(6):1735–1752.e7. doi: 10.1016/j.celrep.2020.01.024
- Veiga SR, Ge X, Mercer CA, et al. Phenformin-induced mitochondrial dysfunction sensitizes hepatocellular carcinoma for dual inhibition of mTOR. Clin Cancer Res. 2018 Aug 1;24(15):3767–3780. doi: 10.1158/1078-0432.CCR-18-0177
- Masoud R, Reyes-Castellanos G, Lac S, et al. Targeting mitochondrial complex I overcomes chemoresistance in high OXPHOS pancreatic cancer. Cell Rep Med. 2020 Nov 17;1(8):100143. doi: 10.1016/j.xcrm.2020.100143
- Hollinshead KER, Parker SJ, Eapen VV, et al. Respiratory supercomplexes promote mitochondrial efficiency and growth in severely hypoxic pancreatic cancer. Cell Rep. 2020 Oct 6;33(1):108231. doi: 10.1016/j.celrep.2020.108231
- Stemberkova-Hubackova S, Zobalova R, Dubisova M, et al. Simultaneous targeting of mitochondrial metabolism and immune checkpoints as a new strategy for renal cancer therapy. Clin Transl Med. 2022 Mar;12(3):e645. doi: 10.1002/ctm2.645
- Boulware DR, Pullen MF, Bangdiwala AS, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for covid-19. N Engl J Med. 2020 Aug 6;383(6):517–525. doi: 10.1056/NEJMoa2016638
- Crunkhorn S. Targeting the mitochondria to block tumour growth. Nat Rev Drug Discov. 2021 Feb;20(2):97. doi: 10.1038/d41573-021-00001-1
- Zorova LD, Popkov VA, Plotnikov EY, et al. Mitochondrial membrane potential. Anal Biochem. 2018 Jul 1;552:50–59. doi: 10.1016/j.ab.2017.07.009
- Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 2017 Jun 27;117(15):10043–10120. doi: 10.1021/acs.chemrev.7b00042
- Dong L, Neuzil J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019 Oct 25;39(1):1–3. doi: 10.1186/s40880-019-0412-6
- Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020 Apr 10;368(6487). doi: 10.1126/science.aaw5473
- Gouirand V, Guillaumond F, Vasseur S. Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front Oncol. 2018;8:117. doi: 10.3389/fonc.2018.00117
- Cheng G, Zielonka J, McAllister D, et al. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br J Cancer. 2014 Jul 8;111(1):85–93. doi: 10.1038/bjc.2014.272
- LeBleu VS, O’Connell JT, Gonzalez Herrera KN, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014 Oct;16(10):992-1003, 1–15. doi: 10.1038/ncb3039
- Park JH, Vithayathil S, Kumar S, et al. Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016 Mar 8;14(9):2154–2165. doi: 10.1016/j.celrep.2016.02.004
- Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012 Apr 1;11(7):1445–1454. doi: 10.4161/cc.19841
- Bartman CR, Weilandt DR, Shen Y, et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature. 2023 Feb;614(7947):349–357. doi: 10.1038/s41586-022-05661-6
- Najjar YG, Menk AV, Sander C, et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight. 2019 Mar 7;4(5). doi: 10.1172/jci.insight.124989
- Jaiswal AR, Liu AJ, Pudakalakatti S, et al. Melanoma evolves complete immunotherapy resistance through the acquisition of a hypermetabolic phenotype. Cancer Immunol Res. 2020 Nov;8(11):1365–1380. doi: 10.1158/2326-6066.CIR-19-0005
- McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011 Nov 2;31(44):15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656.
- d’Hose D, Mathieu B, Mignion L, et al. EPR investigations to study the impact of Mito-metformin on the mitochondrial function of prostate cancer cells. Molecules. 2022 Sep 10;27(18). doi: 10.3390/molecules27185872
- Kalyanaraman B, Cheng G, Hardy M, et al. Mitochondria-targeted metformins: anti-tumour and redox signalling mechanisms. Interface Focus. 2017 Apr 6;7(2):20160109. doi: 10.1098/rsfs.2016.0109
- Skwarski M, McGowan DR, Belcher E, et al. Mitochondrial inhibitor atovaquone increases tumor oxygenation and inhibits hypoxic gene expression in patients with non-small cell lung cancer. Clin Cancer Res. 2021 May 1;27(9):2459–2469. doi: 10.1158/1078-0432.CCR-20-4128
- Kalyanaraman B, Cheng G, Zielonka J, et al. Low-temperature EPR spectroscopy as a probe-Free technique for monitoring oxidants formed in tumor cells and tissues: implications in drug resistance and OXPHOS-Targeted therapies. Cell Biochem Biophys. 2019 Mar;77(1):89–98. doi: 10.1007/s12013-018-0858-1
- Cheng G, Zielonka M, Dranka B, et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: potentials, pitfalls, and the future. J Biol Chem. 2018 Jun 29;293(26):10363–10380. doi: 10.1074/jbc.RA118.003044
- Bridges HR, Fedor JG, Blaza JN, et al. Structure of inhibitor-bound mammalian complex I. Nat Commun. 2020 Oct 16;11(1):5261. doi: 10.1038/s41467-020-18950-3
- Naguib A, Mathew G, Reczek CR, et al. Mitochondrial complex I inhibitors expose a vulnerability for selective killing of Pten-null cells. Cell Rep. 2018 Apr 3;23(1):58–67. doi: 10.1016/j.celrep.2018.03.032
- Weinberg SE, Singer BD, Steinert EM, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019 Jan;565(7740):495–499. doi: 10.1038/s41586-018-0846-z
- Zhao Z, Mei Y, Wang Z, et al. The effect of oxidative phosphorylation on cancer drug resistance. Cancers (Basel). 2022 Dec 22;15(1):62. doi: 10.3390/cancers15010062
- Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020 Sep 1;32(3):341–352. doi: 10.1016/j.cmet.2020.06.019
- De Francesco EM, Ózsvári B, Sotgia F, et al. Dodecyl-TPP targets mitochondria and potently eradicates Cancer Stem Cells (CSCs): synergy with FDA-Approved drugs and natural compounds (vitamin C and berberine). Front Oncol. 2019;9:615. doi: 10.3389/fonc.2019.00615