
ABSTRACT
Introduction
Prostaglandin E2 (PGE2) is produced by cyclooxygenases (COX-1/2) and the microsomal prostaglandin E synthase 1 (mPGES-1). PGE2 is pro-inflammatory in diseases such as rheumatoid arthritis, cardiovascular disorders, and cancer. While Nonsteroidal anti-inflammatory drugs (NSAIDs) targeting COX can effectively reduce inflammation, their use is limited by gastrointestinal and cardiovascular side effects resulting from the blockade of all prostanoids. To overcome this limitation, selective inhibition of mPGES-1 is being explored as an alternative therapeutic strategy to inhibit PGE2 production while sparing or even upregulating other prostaglandins. However, the exact timing and location of PGH2 conversion to PGD2, PGI2, TXB2 or PGF2α, and whether it hinders or supports the therapeutic effect of mPGES-1 inhibition, is not fully understood.
Areas covered
The article briefly describes prostanoid history and metabolism with a strong focus on the vascular effects of prostanoids. Recent advances in mPGES-1 inhibitor development and results from pre-clinical and clinical studies are presented. Prostanoid shunting after mPGES-1 inhibition is highlighted and particularly discussed in the context of cardiovascular diseases.
Expert opinion
The newest research demonstrates that inhibition of mPGES-1 is a potent anti-inflammatory treatment strategy and beneficial and safer regarding cardiovascular side effects compared to NSAIDs. Inhibitors of mPGES-1 hold great potential to advance to the clinic and there are ongoing phase-II trials in endometriosis.
GRAPHICAL ABSTRACT
1. Introduction
Since their discovery in the 1930s, prostanoids have been found to have various physiological and pathophysiological functions. These include regulating blood flow, vascular permeability, smooth muscle cell function, renal filtration, gastrointestinal integrity, sleep regulation, platelet function, inflammatory processes, and reproduction [Citation1,Citation2].
Prostanoids are derived from arachidonic acid, which is part of the phospholipid bilayer of cell membranes. When intracellular Ca2+ levels increase, the cytosolic enzyme phospholipase A2 (PLA2) is activated, leading to the hydrolysis of phospholipids. This process generates free arachidonic acid and a lysophospholipid.
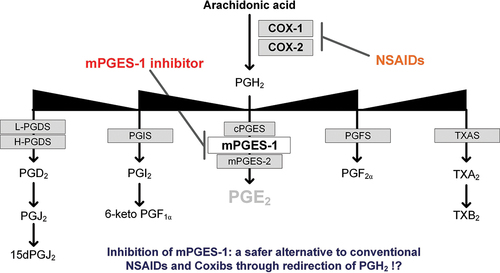
Arachidonic acid is then converted into PGH2 by the enzymes cyclooxygenases (COX-1 and COX-2). Subsequently, PGH2 is further transformed into prostaglandins and thromboxanes, collectively known as prostanoids (PGE2, PGD2, PGI2, PGF2α, and TXA2) (see ). Cyclooxygenases are enzymes bound to the cell membrane that possess cyclooxygenase and peroxidase activities. Initially, arachidonic acid is oxidized with two molecules of oxygen (O2), resulting in the formation of the unstable endoperoxide intermediate PGG2. PGG2 is then reduced to PGH2 [Citation3]. COX-1 and COX-2 are isozymes that differ in their expression patterns and tissue localization. While COX-1 is typically constitutively expressed, COX-2 is also inducible and contributes to prostanoid production during inflammation. PGE2, in particular, is targeted in anti-inflammatory and analgesic treatment strategies. Inflammatory PGE2 is produced from PGH2 by an enzyme called microsomal prostaglandin E synthase 1 (mPGES-1), which was discovered by Jakobsson and colleagues in 1999. MPGES-1 is a microsomal, GSH-dependent, and inducible enzyme [Citation4]. Prostanoids exert their effects through activation of rhodopsin-like seven transmembrane spanning G protein-coupled receptors (GPCRs), acting as endocrine, autocrine, or paracrine signaling molecules. There are nine main prostanoid receptors identified, including DP1 and DP2 (CRTH2) for PGD2, EP1–4 for PGE2, TP for TXA2, IP for PGI2, and FP for PGF2α [Citation5]. The activation of downstream target gene activation results from specific coupling of the prostanoid receptors to different G-proteins (see ). Coupling to the Gq protein, which is the case for EP1, TP and FP activate phospholipase C (PLC) which then cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 consequently activates intracellular Ca2+ levels and DAG activates the protein kinase C (PKC). Gq proteins can also activate Rho kinase. The EP3 as well as the DP2 receptor couple to Gi protein which inhibits adenyl cyclase (AC). This reduces cyclic adenosine monophosphate (cAMP) levels. Coupling to the Gs protein in turn activates AC and stimulates cAMP levels and protein kinase A (PKA). EP2, EP4, DP1 and IP couple to the Gs protein.
Figure 1. Biosynthesis of the primary prostanoids (PGE2, PGD2, PGI2, PGF2α and TXA2) and some of their metabolites. Enzymes are highlighted in gray squares. COX-1 (cyclooxygenase 1), COX-2 (cyclooxygenase 2), mPGES-1 (microsomal prostaglandin E synthase 1), mPGES-1 (microsomal prostaglandin E synthase 2), cPGES (cytosolic prostaglandin E synthase), PGIS (prostacyclin synthase), L-PGDS (lipocalin prostaglandin D synthase), H-PGDS (hematopoietic prostaglandin D synthase), TXS (thromboxane synthase), PGFS (prostaglandin F synthase), MGST3 (microsomal glutathione transferase 3).
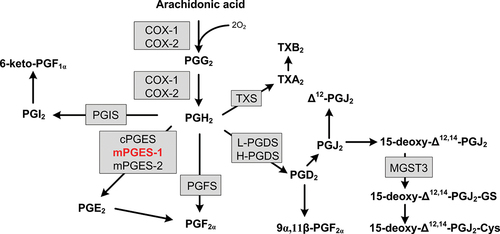
Table 1. VSMC activation by G-protein coupled receptors. All prostanoids can act through the other than their primary receptors, however with lower affinity.
1.1. Prostanoids as regulators of vascular tone
PGI2 and TXA2 play a crucial role in regulating vascular tone, thereby controlling blood pressure and blood flow throughout the body. Other prostanoids, such as PGE2, PGD2, and PGF2α, also have an impact on vascular effects (see ). Prostanoids interact directly with intracellular Ca2+ and cAMP levels through their respective receptors, influencing the degree of constriction of a vessel’s smooth muscle cells. The interaction between actin and myosin filaments, which is driven by the influx of Ca2+ from channels at the plasma membrane or from the sarcoplasmic reticulum (SR), is primarily initiated by the activation of Gq-protein coupled receptors (as depicted in ). Gq binding results in the hydrolysis of IP3 and the opening of Ca2+ channels in the SR. Calmodulin in the cytosol binds to Ca2+, activating myosin light chain (MLC) kinase. Phosphorylation of MLC, in turn, facilitates interaction between actin and myosin filaments, leading to constriction [Citation6]. The main Gq-coupled vasoconstrictors include TXA2, endothelin-1, angiotensin-II, vasopressin, and norepinephrine. Changes in intracellular cAMP levels can also regulate the phosphorylation of MLC. Signaling through Gi-coupled receptors results in decreased intracellular cAMP levels and consequent constriction. Alternatively, Gq proteins can activate Rho-kinase, which inhibits MLC dephosphorylation, leading to VSMC constriction [Citation7].
Figure 2. Simplified schematic illustration of VSMC activation by GPCR signaling and NO. Activation of signaling pathways is indicated by ► and inhibition by ┤. Endothelium dependent relaxation can be induced by prostanoids through the activation of Gs coupled receptors on the VSMC. The major endothelium dependent vasodilator is NO (nitric oxide) produced by eNOS (endothelial nitric oxide synthase) which is activated by NO agonists like bradykinin (BK) or acetylcholine (ACh). NO production can be inhibited by the eNOS inhibitor ADMA (asymmetrical dimethylarginine). EDHF (endothelium dependent hyperpolarization factor) includes CYP450 products of arachidonic acid EETs (epoxyeicosatrienoic acids), H2O2 (hydrogen peroxide), potassium ions, gap junctions, and C-type natriuretic peptide. Endothelium dependent contraction of VSMC can be induced through Gi and Gq coupled receptors, activated by for example endothelin-I or prostanoids. Reactive oxygen species such as O2 (superoxide) can react with NO to form OONO− (peroxynitrite), a cytotoxic molecule that reduces NO availability and causes VSMC constriction. SOD (superoxide dismutase) is an antioxidant enzyme and catalyzes the dismutation of superoxide to O2 (oxygen) and H2O2. VSMC constriction can also be induced by other Gq protein coupled receptor agonists such as angiotensin-II, vasopressin and norepinephrine. Additional abbreviations: MLCK (myosin light chain kinase), MLCP (myosin light chain phosphatase), SR (sarcoplasmic reticulum), cGMP (cyclic guanosine monophosphate), cAMP (cyclic adenosine monophosphate), IP3 (inositol triphosphate), PKA (protein kinase A), NO (nitric oxide). Please see text for more detailed descriptions on the depicted pathways.
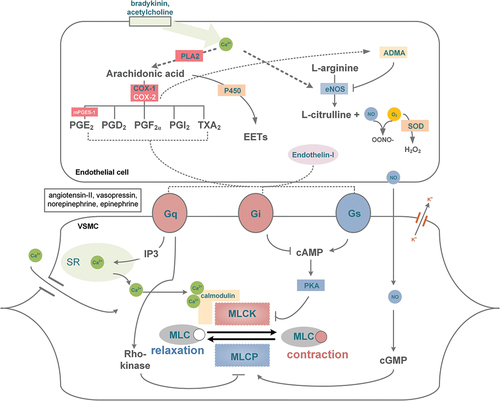
When intracellular Ca2+ levels decrease, MLC phosphorylation also decreases, leading to relaxation of vascular smooth muscle cells (VSMC). Relaxation can also occur through inhibition of MLC phosphorylation or activation of MLC dephosphorylation. Increased cAMP levels, which activate PKA and inhibit MLCK action, mediate the inhibition of MLC phosphorylation. On the other hand, an increase in intracellular cGMP levels leads to MLC dephosphorylation and VSMC relaxation. PGI2, an important endothelium-derived vasodilator, activates IP, a Gs-coupled receptor, and increases intracellular cAMP levels. PGI2 serves as a mediator of inflammation, maintains homeostasis by inhibiting platelet aggregation and vasodilating blood vessels, and is considered the most important cardioprotective prostanoid.
Aside from PGI2, endothelium-dependent relaxation can also be induced by nitric oxide (NO) and endothelium-derived hyperpolarization factor (EDHF). NO increases cGMP levels and induces MLC phosphatase in VSMC. Endothelial NO synthase (eNOS), the enzyme that produces NO in endothelial cells, is constitutively expressed and regulated by intracellular Ca2+ levels. It responds to inflammatory mediators and NO agonists such as bradykinin or acetylcholine [Citation8]. NO production is also regulated through PKA-induced eNOS phosphorylation, which is induced by shear stress. NO diffuses from the endothelial cell into the VSMC, where it binds to guanylyl cyclase which produces cGMP, causing dephosphorylation of MLC, subsequently leading to VSMC relaxation [Citation9]. EDHF causes vascular relaxation independently of NO and PGI2 and is mediated by several potential factors, including CYP450 products of arachidonic acid (epoxyeicosatrienoic acids), H2O2, potassium ions, gap junctions, and C-type natriuretic peptide [Citation10].
2. Exploring alternative strategies to traditional NSAIDs: promising therapeutic approaches with fewer side effects
Traditional NSAIDs, including aspirin, indomethacin, diclofenac, ibuprofen, and naproxen, inhibit both COX enzymes, while COX-2 selective inhibitors, also known as coxibs, such as celecoxib or etoricoxib, primarily act on COX-2. NSAIDs are commonly used as a first-line treatment for various diseases due to their anti-inflammatory, anti-pyretic, and analgesic properties, and are widely available over-the-counter for minor aches and pains, osteoarthritis, rheumatoid arthritis, migraine, and musculoskeletal injuries [Citation11]. However, the use of NSAIDs and coxibs is associated with gastrointestinal and cardiovascular complications when used for prolonged periods. Inhibition of constitutive prostanoid production can lead to an imbalance in prostanoid homeostasis, which alters cell integrity, causing gastrointestinal side effects or reduced renal performance. While selective COX-2 inhibition was initially thought to be a safer alternative to COX-1 inhibition, blocking inducible COX-2 in endothelial cells can lead to severe cardiovascular problems, such as myocardial infarction, pulmonary hypertension, and heart failure [Citation12]. Consequently, rofecoxib and valdecoxib have been withdrawn, and FDA approval for lumiracoxib and etoricoxib has been rejected [Citation13–16], while etoricoxib is approved in Europe and other countries. The underlying molecular mechanism for the cardiovascular side effects of COX-2 inhibition is an imbalance of pro-thrombotic TXA2, mainly produced by platelets via COX-1 in the vasculature, and anti-thrombotic PGI2, mainly produced by vascular cells via COX-2 [Citation17]. Except for low-dose aspirin, NSAIDs are associated with a negative class effect on cardiovascular safety [Citation18]. Therefore, the use of these drugs is concerning, and alternative treatments are needed.
There are several alternative treatment strategies that may have fewer side-effects than traditional NSAIDs. These include dual COX and LOX inhibitors, nitric-oxide (NO)-NSAIDs, and mPGES-1 inhibitors. The combination of COX-1/COX-2 and 5-lipoxygenase (LOX) inhibition is believed to prevent the formation of TXA2 in platelets, downregulate inflammatory prostanoids, and counteract gastric damage by blocking leukotrienes. Licofelone is a COX/LOX inhibitor that has shown promise in clinical trials for treating osteoarthritis with fewer gastrointestinal side effects than traditional NSAIDs [Citation19]. NO-releasing NSAIDs (NO-NSAIDs), have shown promising effects in pre-clinical studies for treating inflammation and hypertension, and naproxcinod (NO-naproxen) is being studied clinically for treating osteoarthritis [Citation20,Citation21]. Selective inhibition of mPGES-1, the synthase of PGE2 downstream of COX, has been proposed as an alternative target to COX. This approach could selectively reduce inflammatory PGE2 without causing undesirable side effects by sparing other prostaglandins, especially PGI2. Thereby, inhibition of mPGES-1 compared to other alternatives holds most promise.
2.1. Anti-inflammatory aspects of mPGES-1 inhibition
Recent advances in mPGES-1 inhibitor characterization and pre-clinical studies demonstrating the anti-inflammatory effects of mPGES-1 inhibition are reviewed by Bergqvist et al. Detailed descriptions of the following examples can be found in [Citation22]. Several mPGES-1 inhibitors (CII, CIII, compound 4b, compound A) have been shown to reduce pouch exudate PGE2 and kidney PGE2 in the air-pouch model of inflammation and to have an anti-pyretic effect in LPS-induced fever [Citation23–26]. The mPGES-1 inhibitor CIII protected the liver and accelerated liver repair compared with vehicle controls in a mouse model of hepatic ischemia reperfusion [Citation27]. The mPGES-1 inhibitor compound 4b was compared with celecoxib during a 14-day observation period regarding its effects on the stomach of mice. Celecoxib caused bleeding ulcers at 50 mg/kg. No adverse effects on the stomach of the mice was observed for the mPGES-1 inhibitor [Citation25]. Inhibition of mPGES-1 has also been reported to reduce tumor growth and block PGE2 formation by cancer-associated fibroblasts in a xenograft neuroblastoma mouse model [Citation28]. The cross-species inhibitors 934, 117, 118, 323 and 323 demonstrated improved selectivity and potency in various pre-clinical assays [Citation29]. The inhibitors blocked human and rat mPGES-1 reversibly and presented bioavailabilities between 10–46%. Besides weak to moderate inhibition of mPGES-2 by all compounds and weak inhibition of COX-2 by 323 no other off target activation was reported. The inhibitors reduced PGE2 levels in A549 cells and whole blood as well as in an air-pouch mouse model. Moreover, the inhibitors reduced paw-swelling in rats and constriction in human resistance arteries ex-vivo [Citation29,Citation30]. The selective human mPGES-1 inhibitor Vipoglanstad (GS-248) reduced acute lung injury and sepsis in a transgenic mouse model [Citation31]. Treatment with Vipoglanstat ameliorated leukocyte infiltration in LPS-induced lung injury and prolonged survival of mice with severe sepsis.
Few mPGES-1 inhibitors have been studied in clinical trials (2013, LY3023703 [Citation32]; 2015, GRC-27864 [Citation33]; 2019, Vipoglanstat [Citation34]; 2022, NS-580). Taken together, the inhibitors decreased LPS-induced PGE2 and increased PGI2 production in blood and/or urine of healthy subjects. The trial for LY3023703 was terminated due to experimentally assessed off-target compound specific liver toxicity observed in two study participants [Citation35]. GRC-27864 was well tolerated by healthy volunteers up to a dose of 1000 mg. Vipoglanstat was recently evaluated in a phase II study (ClinicalTrials.gov Identifier, NCT number: NCT04744207). Vipoglanstat was well tolerated and potently inhibited mPGES-1, but no significant effects were observed on Raynaud’s syndrome symptoms in patients with systemic sclerosis. A phase II investigation of Vipoglanstat in endometriosis is currently being prepared (https://www.gesynta.se/news-media). The mPGES-1 inhibitor NS-580 is currently tested in a phase-II trial for the treatment of endometriosis and chronic pelvic pain syndrome https://rctportal.niph.go.jp/en/detail?trial_id=jRCT2031210685.
2.2. Shifting prostanoid balance for potential therapeutic benefits
The feared cardiovascular side effects of NSAIDs are commonly attributed to an imbalance between pro-thrombotic and anti-thrombotic mediators that can lead to thrombotic events, elevated arterial blood pressure, and heart failure. Inhibition of mPGES-1 has been hypothesized to cause similar adverse effects. However, recent studies have indicated a potentially anti-inflammatory and protective cardiovascular profile of mPGES-1 inhibition through a reduction in PGE2 and a shift toward PGI2. Blocking a terminal prostanoid synthase could redirect the substrate PGH2 toward other prostanoid synthases. The upregulation or maintenance of prostanoid levels (PGD2, PGF2α, PGI2, TXA2) upon mPGES-1 inhibition largely depends on the tissue-specific expression of prostanoid synthases in pathological conditions [Citation36]. This shunting phenomenon following mPGES-1 inhibition thereby introduces some uncertainty, raising concerns about potential side effects. However, it equally holds expectations of additional beneficial effects. Tissue- and disease-specific prostanoid shunting upon blockage of mPGES-1 has been observed in various in-vitro and in-vivo assays.
Our research group has recently identified differences in the prostanoid profile of several immune cells, with profound variations in PGH2 redirection after mPGES-1 inhibition [Citation37,Citation38]. Inhibition of mPGES-1 in RAW264.7 cells and mouse bone marrow-derived macrophages (BMDMs) led to shunting toward the PGD2 pathway including pro-resolving 15dPGJ2 and its metabolites [Citation37,Citation38]. In BMDMs, TXB2 and PGF2α were also increased [Citation37,Citation38]. No significant shunting was observed in A549 cells treated with an mPGES-1 inhibitor [Citation37,Citation38]. Enhanced PGI2 production was observed in rheumatoid arthritis synovial fibroblasts (RASFs) treated with an mPGES-1 inhibitor [Citation37,Citation38]. Furthermore, in human arteries stimulated by inflammatory signals ex-vivo, we observed that inhibiting mPGES-1 significantly reduced PGE2 levels while leaving PGI2 levels unchanged or even increased [Citation30]. Conversely, COX-2 inhibition blocked the production of both prostaglandins [Citation30]. Other studies have also reported a notable increase in PGI2 following mPGES-1 inhibition in internal mammary arteries and saphenous veins [Citation39]. Moreover, investigations involving mPGES-1 knockout mice and mPGES-1 inhibitors have revealed effects on lipid mediators beyond prostanoids [Citation40,Citation41].
In an angiotensin-II induced aortic aneurism mouse model, the redirection of PGH2 to PGD2 and PGI2 in urine was associated with a reduction in oxidative stress and a protection from aortic aneurism formation [Citation42]. PGD2 was also increased in bone marrow derived dendritic cells from mPGES-1 KO mice [Citation43], and the PGD2 metabolite 15dPGJ2 was increased in LPS treated mouse peritoneal macrophages lacking mPGES-1 [Citation40]. Mice deficient in mPGES-1 exhibit decreased urinary levels of PGE2, unaltered TXA2 levels, and increased urinary production of PGI2 [Citation44]. In contrast to COX-2 inhibition or deletion, mPGES-1 deletion was found to protect against thrombogenesis and did not cause an increase in blood pressure in mice [Citation44]. Treatment of mPGES-1 KO mice with LPS resulted in reduced platelet activation, aggregation, and release of platelet-derived microparticles compared to WT controls [Citation45]. Depletion and inhibition of mPGES-1 has been found to be protective in models of inflammatory pain, despite enhanced production of the pain mediator PGI2 [Citation46,Citation47]. In rats with post operative pain the mPGES-1 inhibitor UK4b showed equal efficacy compared with opioid treatment [Citation48]. The pain attenuation was associated with reduced PGE2 formation without changing the levels of other prostanoids. This suggests that inhibition of mPGES-1 could be a therapeutic strategy to reduce inflammatory pain.
Ex-vivo inhibition of mPGES-1 with CIII in larger human vessels led to decreased vascular tone via shunting to PGI2 [Citation39]. These results, indicate important vasoactive properties of mPGES-1 inhibitors. We have recently shown that blocking of mPGES-1 in resistance arteries from end-stage kidney disease (ESKD) patients and Non-ESKD controls significantly reduced adrenergic vasoconstriction ex-vivo. In opposite the COX-1/COX-2 inhibitor indomethacin as well as COX-2 selective inhibitors NS-398 and etoricoxib had no effect on NE-induced vasoconstriction [Citation29,Citation30]. In experiments with larger human arteries (saphenous vein and internal mammary artery) and intrarenal arcuate arteries from mice, inhibition of COX-2 caused enhanced constriction [Citation39,Citation49]. Furthermore, mPGES-1 inhibition in resistance arteries led to a significant increase in acetylcholine-induced dilation, suggesting that it may improve blood flow and reduce resistance in-vivo. Blocking PGI2 signaling with an IP receptor antagonist did not restore reduced adrenergic constriction in human resistance arteries, nor did blocking PGE2-EP4 or signaling through PPARγ. Exogenous PGE2 demonstrated a biphasic effect, inducing dilation at nanomolar concentrations and constriction at micromolar concentrations [Citation30]. Whether COX-1 or COX-2 is the primary source of PGI2 in the vasculature is controversial and studies show conflicting results. Deletion of COX-2 in endothelial cells and VSMC has been shown to reduce systemic PGI2 [Citation50,Citation51]. On the other hand, a dominant role of COX-1 in PGI2 production in mouse aorta has been demonstrated [Citation52]. Our laboratory’s immunohistochemistry results showed expression of mPGES-1, COX-1, PGIS, weak expression for COX-2, and receptor expression for PGE2 (EP1–4), thromboxane (TP), and PGI2 (IP) in resistance arteries, supporting the preferential role of COX-1 for PGI2 production [Citation53]. The overall results of ex-vivo vascular studies suggest that inhibition of mPGES-1 has beneficial vasoactive effects, including the promotion of PGI2 signaling and possibly other pathways, such as local reduction of PGE2 and creation of an antioxidant milieu that maintains formation of NO. In human increased PGI2 production in blood and/or urine was observed in phase I or II clinical trials for the mPGES-1 inhibitors LY3023703 [Citation32], GRC-27864 [Citation33] and Vipoglanstat [Citation34].
2.3. Inhibition of mPGES-1 in models of cardiovascular disease
Several models of cardiovascular disease have demonstrated the beneficial effects of blocking mPGES-1. Besides for example reduction of aneurism formation in hyperlipidemic mice [Citation42], mPGES-1 deletion reduced VSMC proliferation and hyperplasia after vascular injury in mice with femoral artery injury [Citation54]. Endothelial cells lacking the low-density lipoprotein receptor showed increased PGIS and TXS expression after mPGES-1 deletion, and LPS stimulation of mPGES-1-deficient VSMCs and macrophages resulted in a shift toward PGI2 and TXA2 production [Citation55]. Inhibiting mPGES-1 could also protect the heart by sparing the endothelial nitric oxide synthase inhibitor ADMA pathway, as PGI2 and PGE2 can regulate ADMA levels via IP, PPARβ/δ, and EP4 receptors. Compared to COX-2 inhibition, which increased the formation of ADMA in plasma, mPGES-1 KO mice showed increased plasma levels of PGI2 and no effect on ADMA pathway genes [Citation56].
In mice with experimental MI following left coronary artery ligation, global deletion of mPGES-1 did not alter the post-MI cardiac dysfunction observed in wild type animals [Citation57]. However, myeloid-specific deletion of mPGES-1 improved survival after MI without adverse cardiac remodeling [Citation57]. Deletion of mPGES-1 did not increase ischemic myocardial injury 24 hours after coronary artery ligation in mice, unlike wild type mice treated with celecoxib [Citation58]. On the other hand, global deletion of mPGES-1 or its deletion in bone marrow-derived myeloid cells impaired cardiac remodeling after MI in other studies [Citation59,Citation60]. Results from studies using mPGES-1 or COX KO mice and COX inhibitors are conflicting, indicating a need for a better understanding of the link between the prostanoid system and cardiac remodeling after MI.
Several studies suggest a protective role of PGE2 signaling in the adverse effects seen with COX-2 and mPGES-1 depletion in various experimental models, including MI. The PGE2-EP3 pathway has been shown to be important for cardiac recovery after MI in mice [Citation61]. Another study described the protective effect of mPGES-1 through PGE2-EP4 signaling in mice with acute myocardial ischemia-reperfusion injury [Citation62]. A summary of the role and potential of PGE2 in MI/IR injury can be found here [Citation63].
Recent research from our group has shown that chronic administration of the mPGES-1 inhibitor CIII and 118 improved cardiac function in mice after MI compared to vehicle or celecoxib [Citation64,Citation65]. CIII reduced infarct area, increased scar thickness, decreased collagen I/III ratio, decreased fibrosis-related gene expression, and increased capillary density in the ischemic area. Shunting to urinary metabolites of PGI2 after inhibition of mPGES-1 was positively correlated with cardiac function after MI. The PGI2/PGE2 urinary metabolite ratio correlated positively with ejection fraction, fractional shortening, and scar thickness. These findings highlight fundamental differences between experimental enzyme deletion and inhibition and suggest that pharmacological inhibition of mPGES-1 could improve the outcome after MI. Future studies are warranted to validate and expand our understanding of the underlying mechanisms.
3. Expert opinion
NSAIDs are among the most used and recognized medications worldwide. However serious gastrointestinal and especially cardiovascular side-effects associated with their use have led to a number of regulatory actions including the withdrawal of several NSAIDs (e.g. Vioxx in 2004), the introduction of ‘black box’ warnings and the reclassification of the over-the-counter medication diclofenac as prescription only in 2015 (UK). Fears of cardiovascular events caused by NSAIDs have become a public health issue and have generated a ‘guilt by association’ for other anti-inflammatory strategies that target prostanoid signaling pathways, such as inhibition of mPGES-1, which selectively targets PGE2 synthesis downstream of the COX enzymes. However, inhibition of mPGES-1 appears to rebalance the prostanoid profile and reduce the cardiovascular side effects seen with NSAIDs, and is therefore likely superior to COX inhibition, and EP receptor targeting. Another obstacle to the development of mPGES-1 inhibitors has been the fact that inhibitors targeting the human mPGES-1 show little or no effects in rodent models. Only recently cross-species inhibitors have been developed allowing in-depth pre-clinical characterizations. In opposite to the ‘guilt by association,’ recent studies suggest a cardiovascular protective profile of mPGES-1 inhibition through a shift toward PGI2. As has been shown, the redirection of PGH2 to other prostanoids, such as cardioprotective PGI2 or anti-inflammatory PGD2 depends on the cellular composition of the tissue and disease-causing mechanisms, so studies of the larger lipid mediator network are needed for consequence estimation and optimal treatment. Preclinical studies show favorable results with mPGES-1 inhibitors in relieving inflammation and pain, improving vascular function by reducing adrenergic vasoconstriction, protecting against thrombogenesis and protecting against consequences of myocardial infarction. Selective reduction of PGE2, redirection of PGH2 to PGD2 and PGI2 and maintenance of the NO pathway are thought to contribute to the anti-inflammatory and cardioprotective effects observed after inhibition of mPGES-1.
The encouraging results of the use of mPGES-1 inhibitors in a rodent model of myocardial infarction still require further investigation of the underlying mechanisms. These include ablation of prostanoid receptors to identify activated signaling pathways, measurements of plasma ADMA levels at different timepoints as well as the optimal timing of intervention (prophylactic, during acute MI, or post MI). In most of the studies presented, treatment and disease induction collided and a better understanding of whether the inhibitor has similar or improved/worsened effects when used at different phases of the disease model studied could increase clinical relevance.
In conclusion, there is a great need for clinical research of mPGES-1 inhibitors in chronic inflammatory diseases such as rheumatoid arthritis, spondyloarthritis, and osteoarthritis, as well as diseases with a strong component of vasculopathy and in cancer. The cardioprotective effects observed in experimental models warrant phase II trials in MI, possibly also as prophylactic treatment in ‘at risk’ individuals for cardiovascular disease such as those with hypertension, diabetes mellitus or metabolic syndrome. It is encouraging that phase II studies are ongoing and planned in endometriosis, a complex female disease with components of hormonal regulation, angiogenesis, cell proliferation, inflammation and pain. The authors believe that additional mechanistic studies of mPGES-1 inhibitors are unlikely to further predict outcome in humans. Therefore, multiple phase II studies should be conducted, possibly in an academic setting led by leading scientists in the respective fields, which usually reduce study costs. There are several examples where the indication of a developed drug was planned or predicted for one disease but ultimately proved to be better in another disease. Secukinumab is one such interesting example, which went from an indication for rheumatoid arthritis to psoriasis arthritis and spondyloarthritis.
Overall, the results of recent pre-clinical and clinical investigations suggest that inhibition of mPGES-1 is a promising anti-inflammatory therapy with the potential to prevent and ameliorate inflammation and pain, cancer, cardiovascular diseases and other inflammatory conditions with cardiovascular comorbidities.
Abbreviations
| ADMA | = | asymmetric dimethyl arginine |
| COX | = | cyclooxygenase |
| EC | = | endothelial cell |
| ESKD | = | end-stage kidney disease |
| LPS | = | lipopolysaccharide |
| MI | = | myocardial infraction |
| MI/IR | = | myocardial infarction ischemia reperfusion |
| mPGES-1 | = | microsomal prostaglandin E synthase 1 |
| NO | = | nitric oxide |
| Non-ESKD | = | non end-stage kidney disease |
| NSAIDs | = | non-steroidal anti-inflammatory drugs |
| PPAR | = | peroxisome proliferator activated receptor |
| PGI2 | = | prostaglandin I 2 (prostacyclin) |
| PGE2 | = | prostaglandin E 2 |
| TXB2 | = | thromboxane B 2 |
| VSMC | = | vascular smooth muscle cells |
Article highlights
Inhibition of mPGES-1 affects PGE2 and other prostanoids specific for the cell type and disease condition.
Inhibition of mPGES-1 is anti-inflammatory and vasoactive.
Inhibition of mPGES-1 may activate anti-inflammatory and cardioprotective pathways encouraging implications for the treatment of inflammation without cardiovascular side effects.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
This review is partly based on the PhD thesis of Julia Steinmetz-Späh.
Additional information
Funding
References
- V Euler US. Über die Spezifische Blutdrucksenkende Substanz des Menschlichen Prostata-und Samenblasensekretes. Klinische Wochenschrift. 1935 Aug 01;14(33):1182–1183.
- Goldblatt MW. Properties of human seminal plasma. Journal Of Physiology. 1935;84(2):208–218. doi: 10.1113/jphysiol.1935.sp003269
- Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996 Dec 27;271(52):33157–33160. doi: 10.1074/jbc.271.52.33157
- Jakobsson PJ, Thorén S, Morgenstern R, et al. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999 Jun;96(13):7220–7225. doi: 10.1073/pnas.96.13.7220
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004 Aug;103(2):147–166. doi: 10.1016/j.pharmthera.2004.06.003
- Conti MA, Adelstein RS. The relationship between calmodulin binding and phosphorylation of smooth muscle myosin kinase by the catalytic subunit of 3‘: 5’ cAMP-dependent protein kinase. J Biol Chem. 1981 Apr 10;256(7):3178–3181. doi: 10.1016/S0021-9258(19)69586-4
- Ahmed S, Warren DT. Vascular smooth muscle cell contractile function and mechanotransduction. Vessel Plus. 2018;2:36. doi: 10.20517/2574-1209.2018.51
- Chen K, Pittman RN, Popel AS. Nitric oxide in the vasculature: where does it come from and where does it go? A quantitative perspective. Antioxid Redox Signal. 2008 Jul;10(7):1185–1198. doi: 10.1089/ars.2007.1959
- Blatter LA, Wier WG. Nitric oxide decreases [Ca2+]i in vascular smooth muscle by inhibition of the calcium current. Cell Calcium. 1994 Feb 01;15(2):122–131.
- Luksha L, Agewall S, Kublickiene K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis. 2009 Feb;202(2):330–344. doi: 10.1016/j.atherosclerosis.2008.06.008
- Crofford LJ. Use of NSAIDs in treating patients with arthritis. Arthritis Res Ther. 2013;15(3):S2. doi: 10.1186/ar4174
- Wang M, FitzGerald GA. The cardiovascular biology of microsomal prostaglandin E synthase-1. Trends Cardiovasc Med. 2010;20(6):189–195. doi: 10.1016/j.tcm.2011.04.002
- Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005 Mar 17;352(11):1092–1102. doi: 10.1056/NEJMoa050493
- John S, Martin J. Report of John S. Martin Jr. To the special committee of the board of directors of Merck & Co., Inc. Concerning the Conduct of Senior Management in the Development and Marketing of Vioxx. 2006.
- Geusens P, Lems W. Efficacy and tolerability of lumiracoxib, a highly selective cyclo-oxygenase-2 (COX2) inhibitor, in the management of pain and osteoarthritis. Ther Clin Risk Manag. 2008;4(2):337–344. doi: 10.2147/TCRM.S1209
- El-Malah AA, Gineinah MM, Deb PK, et al. Selective COX-2 inhibitors: road from success to controversy and the quest for repurposing. Pharmaceuticals (Basel). 2022 Jul 3;15(7):827. doi: 10.3390/ph15070827
- Dogné JM, Supuran CT, Pratico D. Adverse cardiovascular effects of the coxibs. J Med Chem. 2005 Apr 7;48(7):2251–2257. doi: 10.1021/jm0402059
- Mitchell JA, Kirkby NS. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacol. 2019 04;176(8):1038–1050. doi: 10.1111/bph.14167
- Vinod K. Licofelone in osteoarthritis: is this the awaited drug? a systematic review. Int J Basic Clin Pharmacol. 2021 May;10(5):564. doi: 10.18203/2319-2003ijbcp20211654
- Muscará MN, McKnight W, Del Soldato P, et al. Effect of a nitric oxide-releasing naproxen derivative on hypertension and gastric damage induced by chronic nitric oxide inhibition in the rat. Life Sci. 1998;62(15):L235–40. doi: 10.1016/S0024-3205(98)00072-1
- Wallace JL, Ignarro LJ, Fiorucci S. Potential cardioprotective actions of no-releasing aspirin. Nat Rev Drug Discov. 2002 May;1(5):375–382. doi: 10.1038/nrd794
- Bergqvist F, Morgenstern R, Jakobsson PJ. A review on mPGES-1 inhibitors: from preclinical studies to clinical applications. Prostaglandins Other Lipid Mediat. 2019 Nov;147:106383. doi: 10.1016/j.prostaglandins.2019.106383
- Leclerc P, Pawelzik SC, Idborg H, et al. Characterization of a new mPGES-1 inhibitor in rat models of inflammation. Prostaglandins Other Lipid Mediat. 2013 Apr;102-103:1–12.
- Leclerc P, Idborg H, Spahiu L, et al. Characterization of a human and murine mPGES-1 inhibitor and comparison to mPGES-1 genetic deletion in mouse models of inflammation. Prostaglandins Other Lipid Mediat. 2013 Dec;107:26–34.
- Ding K, Zhou Z, Hou S, et al. Structure-based discovery of mPGES-1 inhibitors suitable for preclinical testing in wild-type mice as a new generation of anti-inflammatory drugs. Sci Rep. 2018 Mar;8(1):5205. doi: 10.1038/s41598-018-23482-4
- Sugita R, Kuwabara H, Sugimoto K, et al. A novel selective prostaglandin E2 synthesis inhibitor relieves pyrexia and chronic inflammation in rats. Inflammation. 2016 Apr;39(2):907–915. doi: 10.1007/s10753-016-0323-5
- Nishizawa N, Ito Y, Eshima K, et al. Inhibition of microsomal prostaglandin E synthase-1 facilitates liver repair after hepatic injury in mice. J Hepatol. 2018 Jul;69(1):110–120. doi: 10.1016/j.jhep.2018.02.009
- Kock A, Larsson K, Bergqvist F, et al. Inhibition of microsomal prostaglandin E synthase-1 in cancer-associated fibroblasts suppresses neuroblastoma tumor growth. EBioMedicine. 2018 Jun;32:84–92.
- Larsson K, Steinmetz J, Bergqvist F, et al. Biological characterization of new inhibitors of microsomal PGE synthase-1 in preclinical models of inflammation and vascular tone. Br J Pharmacol. 2019 Aug;176(24):4625–4638. doi: 10.1111/bph.14827
- Steinmetz-Späh J, Arefin S, Larsson K, et al. Effects of microsomal prostaglandin E synthase-1 (mPGES-1) inhibition on resistance artery tone in patients with end stage kidney disease. Br J Pharmacol. 2021 Nov 11;179(7):1433–1449. doi: 10.1111/bph.15729
- Gurusamy M, Nasseri S, Rampa DR, et al. Inhibition of microsomal prostaglandin E synthase-1 ameliorates acute lung injury in mice. J Transl Med. 2021 Aug 9;19(1):340. doi: 10.1186/s12967-021-03016-9
- Jin Y, Smith CL, Hu L, et al. Pharmacodynamic comparison of LY3023703, a novel microsomal prostaglandin e synthase 1 inhibitor, with celecoxib. Clin Pharmacol Ther. 2016 Mar;99(3):274–284. doi: 10.1002/cpt.260
- Sant S, Tandon M, Menon V, et al. GRC 27864, novel, microsomal prostaglandin E synthase-1 enzyme inhibitor: phase 1 study to evaluate safety, PK and biomarkers in healthy, adult subjects. Osteoarthritis Cartilage. 2018;26:S351–S352. doi: 10.1016/j.joca.2018.02.698
- Edenius C, Ekström G, Kolmert J, et al. SAT035 inhibition of microsomal prostaglandin E synthase-1 (mPGES-1) by GS-248 reduces prostaglandin E2 biosynthesis while increasing prostacyclin in human subjects. Ann Rheumatic Dis. 2020;79(Suppl 1):1103. doi: 10.1136/annrheumdis-2020-eular.5503
- Norman BH, Fisher MJ, Schiffler MA, et al. Identification and mitigation of reactive metabolites of 2-aminoimidazole-containing microsomal prostaglandin E synthase-1 inhibitors terminated due to clinical drug-induced liver injury. J Med Chem. 2018 03;61(5):2041–2051. doi: 10.1021/acs.jmedchem.7b01806
- Koeberle A, Laufer SA, Werz O. Design and development of microsomal prostaglandin E2 synthase-1 inhibitors: challenges and future directions. J Med Chem. 2016 07;59(13):5970–5986. doi: 10.1021/acs.jmedchem.5b01750
- Liu J, Peng B, Steinmetz-Späh J, et al. Microsomal prostaglandin E synthase-1 inhibition promotes shunting in arachidonic acid metabolism during inflammatory responses in vitro. Prostaglandins Other Lipid Mediat. 2023 Apr 23;167:106738. doi: 10.1016/j.prostaglandins.2023.106738
- Steinmetz-Späh J, Liu J, Singh R, et al. Biosynthesis of prostaglandin 15dPGJ2 -glutathione and 15dPGJ2-cysteine conjugates in macrophages and mast cells via MGST3. J Lipid Res. 2022;63(12):100310. Nov 09(published online ahead of print):100310. doi: 10.1016/j.jlr.2022.100310
- Ozen G, Gomez I, Daci A, et al. Inhibition of microsomal PGE synthase-1 reduces human vascular tone by increasing PGI2: a safer alternative to COX-2 inhibition. Br J Pharmacol. 2017;174(22):4087–4098. doi: 10.1111/bph.13939
- Idborg H, Olsson P, Leclerc P, et al. Effects of mPGES-1 deletion on eicosanoid and fatty acid profiles in mice. Prostaglandins Other Lipid Mediat. 2013 Dec;107:18–25.
- Bergqvist F, Ossipova E, Idborg H, et al. Inhibition of mPGES-1 or COX-2 results in different proteomic and lipidomic profiles in A549 lung cancer cells. Front Pharmacol. 2019;10:636. doi: 10.3389/fphar.2019.00636
- Wang M, Lee E, Song W, et al. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008 Mar;117(10):1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398
- Monrad SU, Kojima F, Kapoor M, et al. Genetic deletion of mPGES-1 abolishes PGE2 production in murine dendritic cells and alters the cytokine profile, but does not affect maturation or migration. Prostaglandins Leukot Essent Fatty Acids. 2011 Mar;84(3–4):113–121. doi: 10.1016/j.plefa.2010.10.003
- Cheng Y, Wang M, Yu Y, et al. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006 May;116(5):1391–1399. doi: 10.1172/JCI27540
- Raouf J, Mobarrez F, Larsson K, et al. Deletion of mPGES-1 affects platelet functions in mice. Clin Sci. 2016;130(24):2295–2303. doi: 10.1042/CS20160463
- Kamei D, Yamakawa K, Takegoshi Y, et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004 Aug 6;279(32):33684–33695. doi: 10.1074/jbc.M400199200
- Xu D, Rowland SE, Clark P, et al. MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008 Sep;326(3):754–763. doi: 10.1124/jpet.108.138776
- Stewart MJ, Weaver LM, Ding K, et al. Analgesic effects of a highly selective mPGES-1 inhibitor. Sci Rep. 2023 Feb 27;13(1):3326. doi: 10.1038/s41598-023-30164-3
- Kirkby NS, Sampaio W, Etelvino G, et al. Cyclooxygenase-2 selectively controls renal blood flow through a novel PPARβ/δ-dependent vasodilator pathway. Hypertension. 2018;71(2):297–305. doi: 10.1161/HYPERTENSIONAHA.117.09906
- Yu Y, Ricciotti E, Scalia R, et al. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci, trans med. 2012;4(132):ra13254–ra13254. doi: 10.1126/scitranslmed.3003787
- Tang SY, Monslow J, Todd L, et al. Cyclooxygenase-2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation. 2014 Apr 29;129(17):1761–1769. doi: 10.1161/CIRCULATIONAHA.113.007913
- Kirkby NS, Lundberg MH, Harrington LS, et al. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci USA. 2012;109(43):17597–17602. doi: 10.1073/pnas.1209192109
- Norel X, Walch L, Fau - Gascard J-P, et al. Prostacyclin release and receptor activation: differential control of human pulmonary venous and arterial tone. Br J Pharmacol. 2004;142(4):788–796. (0007-1188 (Print)). doi: 10.1038/sj.bjp.0705843
- Wang M, Ihida-Stansbury K, Kothapalli D, et al. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011 Feb;123(6):631–639. doi: 10.1161/CIRCULATIONAHA.110.973685
- Wang M, Zukas AM, Hui Y, et al. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. 2006 Sep;103(39):14507–14512. doi: 10.1073/pnas.0606586103
- Kirkby NS, Raouf J, Ahmetaj-Shala B, et al. Mechanistic definition of the cardiovascular mPGES-1/COX-2/ADMA axis. Cardiovasc Res. 2020 Oct;116(12):1972–1980. doi: 10.1093/cvr/cvz290
- Chen L, Yang G, Jiang T, et al. Myeloid cell mPges-1 deletion attenuates mortality without affecting remodeling after acute myocardial infarction in mice. J Pharmacol Exp Ther. 2019 Jul;370(1):18–24. doi: 10.1124/jpet.118.256057
- Wu D, Mennerich D, Arndt K, et al. Comparison of microsomal prostaglandin E synthase-1 deletion and COX-2 inhibition in acute cardiac ischemia in mice. Prostaglandins Other Lipid Mediat. 2009 Nov;90(1–2):21–25. doi: 10.1016/j.prostaglandins.2009.06.006
- Degousee N, Fazel S, Angoulvant D, et al. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation. 2008 Apr 1;117(13):1701–1710. doi: 10.1161/CIRCULATIONAHA.107.749739
- Degousee N, Simpson J, Fazel S, et al. Lack of microsomal prostaglandin E(2) synthase-1 in bone marrow-derived myeloid cells impairs left ventricular function and increases mortality after acute myocardial infarction. Circulation. 2012 Jun 12;125(23):2904–2913. doi: 10.1161/CIRCULATIONAHA.112.099754
- Tang J, Shen Y, Chen G, et al. Activation of E-prostanoid 3 receptor in macrophages facilitates cardiac healing after myocardial infarction. Nat Commun. 2017 Mar 03;8(1):14656. doi: 10.1038/ncomms14656
- Zhu L, Xu C, Huo X, et al. The cyclooxygenase-1/mPGES-1/endothelial prostaglandin EP4 receptor pathway constrains myocardial ischemia-reperfusion injury. Nat Commun. 2019 Apr 23;10(1):1888. doi: 10.1038/s41467-019-09492-4
- Zhu L, Zhang Y, Guo Z, et al. Cardiovascular biology of prostanoids and drug discovery. Arteriosclerosis Thrombosis Vasc Biol. 2020 Jun 01;40(6):1454–1463. doi: 10.1161/ATVBAHA.119.313234
- Zhang Y, Steinmetz-Späh J, Idborg H, et al. Microsomal prostaglandin E synthase-1 inhibition prevents adverse cardiac remodelling after myocardial infarction in mice. Br J Pharmacol. 2023 Feb 14;180(15):1981–1998. doi: 10.1111/bph.16061
- Steinmetz-Späh J. Studies on anti-inflammatory and vasoactive effects of mPGES-1 inhibition [elektronisk resurs].