
1. Introduction
Cardiovascular diseases (CVD) are among the most common causes of death worldwide. Despite continuous improvement in classical pharmacotherapy, the incidence of CVD is expected to further increase. Thus, novel treatment options are urgently needed to improve patient outcomes. Recent advancements in the molecular understanding of pathomechanisms and disease-contributing factors bear the promise for advanced therapeutics that directly target the molecular or genetic origin of the disease. In combination with the prospective of targeted delivery via gene therapy, curative therapies are finally becoming reality.
One promising molecular target is the small calcium (Ca2+)-binding protein S100A1, which is an important regulator of cardiac performance. S100A1 is a central regulator of Ca2+-driven networks that orchestrates compartmentalized Ca2+ fluxes in the heart [Citation1,Citation2].
In this editorial, we will highlight S100A1’s function and summarize the existing insight into the translational potential of S100A1 for CVD.
2. S100A1: from structure to function
2.1. S100A1: a Ca2+ binding protein
S100A1 belongs to the S100 protein family, which represents the largest group among EF-hand Ca2+-binding proteins exclusively expressed in vertebrates. S100 proteins have been described as central regulators of numerous cellular and molecular processes, often operating in a cell-type-specific manner [Citation3]. On a structural level, S100A1 acts as a symmetric, antiparallel homodimer. Upon Ca2+-binding, the C-terminal extension displays a conformational change, thereby exposing hydrophobic residues that have been implicated in target-protein interaction. Of note, the C-terminal extension displays the most variability between individual S100 proteins, which have been linked to their specific biological activities [Citation3].
2.2. S100A1’s function in the heart
In the heart, S100A1 demonstrates the highest expression in the left ventricle, followed by decreasing concentrations in the right ventricle, right and left atria [Citation4], which correlates with the hemodynamic load of the corresponding cardiac chambers. S100A1 is primarily expressed in cardiomyocytes (CM) [Citation5]. Endothelial cells (EC) express significantly lower S100A1 levels, whereas fibroblasts do not express S100A1 [Citation6,Citation7].
In CM, S100A1 co-localizes with the sarcoplasmic reticulum (SR), the myofilaments and the mitochondria (). S100A1’s interaction with the SR Ca2+ pumps sarcoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and ryanodine receptor 2 (RyR2) regulates their activity, resulting in increased SR Ca2+ release and re-uptake during systole and diastole, respectively. This results in increased Ca2+ cycling and enhancement of excitation-contraction coupling [Citation8–10]. Additionally, in diastole, S100A1 increases RyR2 closure, preventing diastolic Ca2+ leakage [Citation8,Citation11]. At the sarcomeres, S100A1 interacts with titin and attenuates actin-titin interaction, which decreases passive stiffness to facilitate contraction as well as relaxation [Citation12]. In the mitochondria, S100A1 supports adenosine triphosphate (ATP) synthesis and energy provision through its interaction with the ATP-synthase and other mitochondrial proteins [Citation13].
Figure 1. S100A1 is a central regulator of the Ca2+-driven networks in cardiomyocytes. During cardiomyocyte’s excitation-contraction cycle, S100A1’s interaction with the RyR2 and Serca2a increases systolic Ca2+ cycling through the SR, and enhances diastolic RyR2 closure to prevent diastolic SR Ca2+ leakage. S100A1’s interaction with proteins at the myofilaments, such as titin, facilitates contraction as well as relaxation. In the mitochondria, S100A1 interacts with the ATP-synthase, which supports ATP synthesis.
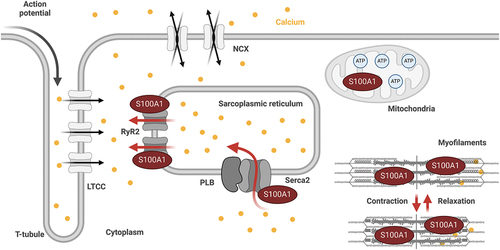
In summary, S100A1’s pleiotropic actions are mediated via direct binding and activity modulation of its target proteins, regulating central Ca2+-driven networks in CM involved in systolic and diastolic performance regulation and energy homeostasis ().
2.3. S100A1 in the vasculature
In ECs, S100A1 mainly co-localizes with the endoplasmic reticulum (ER). Here, it interacts with SERCA2 and the inositol-1,4,5-trisphosphate-receptor (IP3R), regulating ER Ca2+ uptake and release in ECs [Citation6]. Furthermore, S100A1 regulates nitric oxide (NO) production through its interaction with the endothelial nitric oxide synthase (eNOS) and modification of protein kinase C-dependent eNOS phosphorylation, which results in altered NO homeostasis [Citation14].
3. S100A1’s therapeutic use
Expression analysis in failing human hearts unveiled downregulation of S100A1 mRNA and protein levels, sparking the interest in its therapeutic potential [Citation4,Citation15]. Subsequent analyses in a variety of animal heart failure (HF) models corroborated the correlation of attenuated S100A1 expression with disease severity, progression and mortality, and suggested downregulation of S100A1 as part of the fetal gene program [Citation1,Citation4,Citation8,Citation10,Citation16,Citation17]. In support, S100A1 knock-out mice demonstrated accelerated deterioration after chronic stress, whereas S100A1 transgenic mice were protected [Citation10].
The curative potential of therapeutic S100A1 restoration has since then been demonstrated in comprehensive in vitro approaches and animal models:
Studies in isolated neonatal and adult ventricular rat CM provided the first proof of concept for S100A1’s therapeutic efficacy in vitro. In failing rodent CM, S100A1 re-expression restored contractile performance, enhanced intracellular Ca2+ transients, SR Ca2+ uptake and load, and reduced the β-adrenergic-receptor-triggered (β-AR) SR Ca2+ leak [Citation8,Citation11]. Importantly, S100A1-mediated inotropic functions were additive to β-AR stimulation and occurred independently of cyclic adenosine monophosphate signaling (cAMP) [Citation8]. The anti-arrhythmic and inotropic efficacy of S100A1 gene therapy was further demonstrated in engineered heart tissue (EHTs) [Citation11,Citation18].
The first proof of concept in vivo was performed in a rat HF model: Adenoviral S100A1 gene delivery restored S100A1 protein levels, which improved systolic and diastolic contractile performance as well as β-AR responsiveness [Citation8]. Long-term therapeutic efficacy was subsequently demonstrated using adeno-associated viral (AAV) gene transfer, where restoration of S100A1 protein levels yielded a similar therapeutic profile [Citation11,Citation17].
Successful translation into large animal models was achieved using a porcine post-myocardial infarction (MI) HF model. The pig is well established in heart failure research due to its great anatomical and physiological similarities to humans. S100A1 gene transfer in failing pig hearts yielded long-term improvement of both systolic and diastolic cardiac function as well as attenuated cardiac hypertrophy and reversed remodeling [Citation16]. First insights into the feasibility of human therapy were demonstrated in failing human CM. S100A1 gene transfer into failing human CM in vitro restored S100A1 protein levels in failing CM and corroborated previous findings from animal studies [Citation15].
Additionally to S100A1’s intracellular function, during MI, S100A1 protein can be detected in the circulation in humans as well as in animal models, potentially being released from dying CM. Fibroblasts can take up the secreted S100A1, leading to an immunomodulatory and antifibrotic phenotype through toll-like-receptor-4-dependent signaling mechanisms [Citation7]. Administration of neutralizing S100A1 antibodies during MI lead to enlarged infarct areas and worsened left ventricular function as well as prolonged myocardial inflammation, supporting the hypothesis that extracellular S100A1 exerts anti-inflammatory and protective effects in fibroblasts that contribute to the therapeutic effects of S100A1 by beneficially impacting post-ischemic inflammation and healing [Citation7]. In patients with critical limb ischemia, S100A1 expression is reduced in the gastrocnemius muscle, but strategies to therapeutically enhance S100A1 protein levels in diseased EC have not been tested [Citation14].
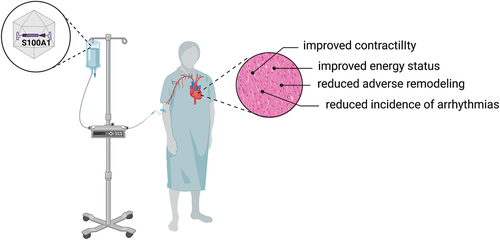
Collectively, these comprehensive proof-of-principle studies provide a strong rationale to advance targeted replacement of S100A1 via gene therapy as a curative therapy option in heart failure patients ().
Figure 2. Conceptual framework of S100A1’s therapeutic actions in the heart. S100A1 therapy could be administered via systemic AAV-gene transfer to heart failure patients. S100A1 has a broad therapeutic profile and results in improved contractility and an improved energy status, as well as reduced adverse remodeling and a reduced incidence of ventricular arrhythmias, making it a promising target for the treatment of heart failure. Created with BioRender.com.
4. Expert opinion
Over the past decades, basic and translational research delivered comprehensive evidence for the therapeutic potential of the Ca2+-binding protein S100A1 in cardiovascular diseases. With multiple intracellular targets, S100A1 is a pleiotropic regulator of cardiomyocyte systolic and diastolic performance and ongoing research will most likely contribute to the identification of further currently unknown regulatory circuits of S100A1 in the future.
Given the gaining momentum for gene-based therapies from the incremental number of clinical trials and the approval of several AAV-based therapeutics, human gene therapy has become a clinical reality. Progress is nevertheless hampered by recently reported severe side effects in clinical studies, that are most likely attributed to broad vector tropism and subsequent off-target effects. Thus, despite the tremendous efforts that have been put into molecular target identification and validation, ongoing research on AAV capsid development needs to be substantiated in order to improve cardiac-specific transduction and safe expression profiles of next generation AAVs.
Regarding the safety profile of S100A1, a moderate S100A1 gene dosage is already sufficient to normalize S100A1 protein expression, yielding significant therapeutic effects. S100A1’s therapeutic window tested to date ranges from expression restoration to local overexpression of ~ 50-fold without occurrence of adverse effects, highlighting its broad therapeutic window. Furthermore, this cAMP-independent, inotropic therapy is not associated with alterations in heart rate, ECG parameters or increased susceptibility to arrhythmias.
While in animal models, the therapeutic dose can be monitored postmortem through tissue expression analysis, there is currently no biomarker available to monitor expression of therapeutic gene delivery in humans. Thus, future research needs to establish noninvasive biomarkers, that will ultimately allow to assess successful gene delivery to the human heart. In addition, AAV-S100A1 therapeutics could be used to strengthen and advance the concept of engineered cardiac tissue patches in human post-MI therapy.
Besides the continuous efforts to develop S100A1-gene therapy approaches, ongoing research is also focusing on the development of S100A1-derived peptides for the treatment of CVD. S100A1ct, the bioactive c-terminal lead structure derived from the S100A1 protein, is a cell penetrating peptide (CCP). In an ongoing study, we demonstrate that S100A1ct mimics molecular actions of the S100A1 protein, eliciting similar therapeutic actions to its parent protein [Citation19]. Thus, S100A1ct might become an effective systemic treatment option against cardiac contractile dysfunction and tachyarrhythmias of diseased hearts. However, further mechanistic studies of S100A1ct’s interactions with its putative molecular targets are needed to increase the understanding of its molecular mechanism of action and will help to expand the reservoir of S100A1-based therapeutics.
Declaration of interest
P Most and D Kehr hold patents on the therapeutic use of S100A1 in cardiovascular diseases. P Most is the founder of AaviGen GmbH.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Ritterhoff J, Most P. Targeting S100A1 in heart failure. Gene Ther. 2012 Jun;19(6):613–621. doi: 10.1038/gt.2012.8
- Rohde D, Busch M, Volkert A, et al. Cardiomyocytes, endothelial cells and cardiac fibroblasts: S100A1‘s triple action in cardiovascular pathophysiology. Future Cardiol. 2015 May;11(3):309–321.
- Heizmann CW, Fritz G, Schäfer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002 May 1;7(4):d1356–1368. doi: 10.2741/A846
- Remppis A, Greten T, Schäfer BW, et al. Altered expression of the Ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta. 1996 Oct 11;1313(3):253–257. doi: 10.1016/0167-4889(96)00097-3
- Zimmer DB, Song W, Zimmer WE. Isolation of a rat S100 alpha cDNA and distribution of its mRNA in rat tissues. Brain Res Bull. 1991 Aug;27(2):157–162. doi: 10.1016/0361-9230(91)90061-N
- Pleger ST, Harris DM, Shan C, et al. Endothelial S100A1 modulates vascular function via nitric oxide. Circ Res. 2008 Apr 11;102(7):786–794. doi: 10.1161/CIRCRESAHA.108.172031
- Rohde D, Schön C, Boerries M, et al. S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via Toll-like receptor 4. EMBO Mol Med. 2014 Jun;6(6):778–794.
- Most P, Pleger ST, Volkers M, et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004 Dec;114(11):1550–1563.
- Kiewitz R, Acklin C, Schafer BW, et al. Ca2+ -dependent interaction of S100A1 with the sarcoplasmic reticulum Ca2+ -ATPase2a and phospholamban in the human heart. Biochem Biophys Res Commun. 2003 Jun 27;306(2):550–557. doi: 10.1016/S0006-291X(03)00987-2
- Most P, Seifert H, Gao E, et al. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation. 2006 Sep 19;114(12):1258–1268. doi: 10.1161/CIRCULATIONAHA.106.622415
- Ritterhoff J, Volkers M, Seitz A, et al. S100A1 DNA-based inotropic therapy protects against proarrhythmogenic ryanodine receptor 2 dysfunction. Mol Ther. 2015 Aug;23(8):1320–1330.
- Yamasaki R, Berri M, Wu Y, et al. Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J. 2001 Oct;81(4):2297–2313.
- Boerries M, Most P, Gledhill JR, et al. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007 Jun;27(12):4365–4373.
- Most P, Lerchenmüller C, Rengo G, et al. S100A1 deficiency impairs postischemic angiogenesis via compromised proangiogenic endothelial cell function and nitric oxide synthase regulation. Circ Res. 2013 Jan 4;112(1):66–78. doi: 10.1161/CIRCRESAHA.112.275156
- Brinks H, Rohde D, Voelkers M, et al. S100A1 genetically targeted therapy reverses dysfunction of human failing cardiomyocytes. J Am Coll Cardiol. 2011 Aug 23;58(9):966–973. doi: 10.1016/j.jacc.2011.03.054
- Pleger ST, Shan C, Ksienzyk J, et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med. 2011 Jul 20;3(92):92ra64. doi: 10.1126/scitranslmed.3002097
- Pleger ST, Most P, Boucher M, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007 May 15;115(19):2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701
- Remppis A, Pleger ST, Most P, et al. S100A1 gene transfer: a strategy to strengthen engineered cardiac grafts. J Gene Med. 2004 Apr;6(4):387–394.
- Kehr D, Ritterhoff J, Glaser M, et al. S100A1ct: a synthetic peptide derived from human S100A1 protein improves cardiac contractile performance and survival in pre-clinical heart failure models. bioRxiv. 2023; 2023–03.