
ABSTRACT
Introduction: Collaborative interactions between several diverse biological processes govern the onset and progression of breast cancer. These processes include alterations in cellular metabolism, anti-tumor immune responses, DNA damage repair, proliferation, anti-apoptotic signals, autophagy, epithelial-mesenchymal transition, components of the non-coding genome or onco-mIRs, cancer stem cells and cellular invasiveness. The last two decades have revealed that each of these processes are also directly regulated by a component of the cell cycle apparatus, cyclin D1.
Area covered: The current review is provided to update recent developments in the clinical application of cyclin/CDK inhibitors to breast cancer with a focus on the anti-tumor immune response.
Expert opinion: The cyclin D1 gene encodes the regulatory subunit of a proline-directed serine-threonine kinase that phosphorylates several substrates. CDKs possess phosphorylation site selectivity, with the phosphate-acceptor residue preceding a proline. Several important proteins are substrates including all three retinoblastoma proteins, NRF1, GCN5, and FOXM1. Over 280 cyclin D3/CDK6 substrates have b\een identified. Given the diversity of substrates for cyclin/CDKs, and the altered thresholds for substrate phosphorylation that occurs during the cell cycle, it is exciting that small molecular inhibitors targeting cyclin D/CDK activity have encouraging results in specific tumors.
1. Background
Breast cancer (BrCa) is the most common non-dermatological malignancy in women and represents about 30% of all malignancies diagnosed in women in the USA [Citation1,Citation2]. Breast cancers can be linked to gene mutations inherited from one close relative in more than 5–10% of cases. Historically, breast cancer was considered a disease of the developed World, but almost 50% of breast cancer cases and 58% of deaths occur in less developed Countries. Incidence rates vary greatly worldwide from 19.3 per 100,000 women in Eastern Africa to 89.7 per 100,000 women in Western Europe. The worldwide breast cancer incidence has increased by more than 20% and mortality has increased by 14% since 2008.
In an attempt to reduce mortality from breast cancer, precision medicine approaches have been deployed targeting specific abnormalities identified in the coding region of the genome. Using this approach genetic subtypes of breast cancer have been described in which specific patterns of expression were identified for the coding genome [Citation3]. Growing evidence suggests both the coding and non-coding genome govern the onset and progression of tumorigenesis [Citation4,Citation5] and more recently subtypes of breast cancer have been also demonstrated using patterns of expression for the non-coding genome [Citation6–Citation8].
Using the coding genome, five distinct molecular subtypes were identified referred to as luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-enriched, basal-like, and claudin-low and normal-like [Citation9]. The coding region classification identified potential genetic targets including the estrogen receptor (ERα) and/or progesterone receptor (PR) and Her2. Triple-negative breast cancer (TNBC), which lacks ERα, PR and Her2, is the most deadly form of breast cancer. TNBC is associated in 10- to 15% with mutations of DNA damage repair proteins (BRCA1 BARD1, BRCA1, BRCA2, PALB2, and RAD51D) [Citation10], in 19% with PD-L1 expression [Citation11] and in >95% with CCR5 overexpression [Citation12]. CCR5 expression in TNBC endows the breast cancer cells with characteristics of cancer stems cells [Citation13] and TNBC metastasis can be blocked with CCR5 inhibitors [Citation12], the basis of a current phase 1b/II study (Clinicaltrial.gov, ID#: NCT03838367).
Breast cancer may be characterized based on either the coding [Citation3] or non-coding (Gormley classification, G1-4) [Citation6]. The CCND1 gene, which encodes cyclin D1, has been identified as amplified in 29–58% of breast cancers (Cancer Genome Atlas Network, 2012). In addition, cyclin D1 protein overexpression, through gene amplification, transcriptional or post-transcriptional induction, is found in >50% of breast cancers. Cyclin D1 expression is believed to drive aberrant phosphorylation and inactivation of the retinoblastoma protein (pRB), primarily in luminal A and luminal B where it is associated with increased chromosomal instability [Citation14] ().
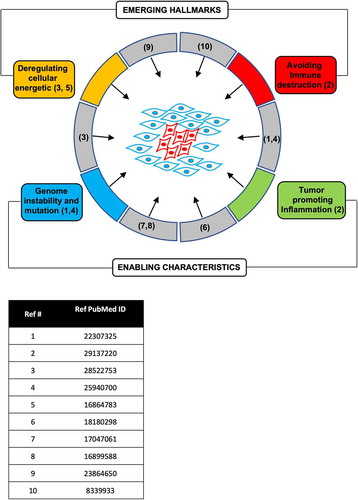
Figure 1. Cyclin D1 participates in both the hallmarks and enabling characteristics of tumorigenesis. The references for studies demonstrating the function of cyclin D1 in the hallmark of cancer and enabling characteristic are shown.
2. The cell-cycle and human breast cancer
Abnormalities of the cell-cycle are a pervasive finding in human breast cancer and other malignancies. A number of recent studies have reviewed the application of the cyclin-dependent kinases (CDK) inhibitor therapy for breast cancer [Citation8,Citation15–Citation17]. This review herein is intended to serve as an update of these studies with a focus on the non-canonical functions of cyclins, including the importance of the anti-tumor immune response, and thereby provide a rationale for additional approaches to further enhance the efficacy of current CDK targeting therapies.
Progression through the cell-cycle occurs in an orderly and precise manner, with descriptive terms related to changes in the cell which are visible by light microscopy, the Gap1 (G1) phase, the S phase, during which time DNA synthesis occur, the G2 phase [Citation8] and cell division or mitosis (the M phase), resulting in the production of two identical daughter cells [Citation18]. As noted above, two distinct processes contribute to cell-cycle progression, firstly changing substrate specificity of the cyclin/CDKs and secondly altered activity of the kinase during the cell cycle. In this regard, the substrates of mitotic kinases are phosphorylated in sequential, kinase-specific waves [Citation19] and phosphorylation of different CDK substrates is temporally ordered in part by a wide dynamic range of differential substrate sensitivity to CDK activity [Citation20].
The history of the current understanding on cyclins in cell cycle progression illustrates the importance of basic science contributions from a number of laboratories around the world [Citation21]. Lee Hartwell’s isolation of temperature-sensitive mutants of budding yeast that could not complete cell division, thus defining cell division cycle, or cdc, genes [Citation22,Citation23] and Mitchison’s work on temperature sensitive budding yeast, in the early 1970s [Citation24], led Paul Nurse, who studied fission yeast, Steve Reed, Kim Nasmyth, Ruderman and others to enter the cell-cycle field [Citation21]. Importantly Nurse discovered wee1, which determined the length of G2, establishing a non-G1 rate-limiting step for progression through the cell cycle [Citation25]. CDC28 one of the ‘start genes’ in budding yeast controlled G1 phase [Citation26] and both Reed and Nasmyth identified approaches to complement CDC28 [Citation27]. The key contributions of many scientists to the field in the 70s and 80s was recently well annotated [Citation21]. The importance of studies on yeast cdc13 [Citation28–Citation30], led ultimately to the complementation experiments and discovery of cyclin D1 by several independent laboratories. In 1989 Andrew Arnold identified a candidate oncogene (PRAD1 or D11S287E) on chromosome 11q13 [Citation31] and in April 1991 published that a human cDNA of this clone, now known as cyclin D1, conveyed the regulatory subunit enzyme activity to phosphorylate histone H1. Shortly thereafter David Beach’s laboratory, who had previously shown cyclin and p34cdc2 are major components of the M-phase specific H1K in sea urchin [Citation32] using a conditionally CLN-deficient yeast strain, identified a human cDNA that complemented the CLN genetic deficiency, designated cyclin D1 (CCND1) [Citation33]. Furthermore, Beach showed that cyclin D1 associated with CDK4, CDK2, CDK5, PCNA and p21 [Citation34]. Independent studies by Charles Sherr’s laboratory showed that colony-stimulating factor-1 induced the abundance of a cyclin-like cDNA (p36CYL) during G1 phase in murine macrophages and that p36CYL was phosphorylated and associated with a polypeptide antigenically related to p34cdc2 suggesting that CYL genes may function during S phase commitment [Citation35].
CDKs are a family of conserved serine/threonine protein kinases with a phosphoacceptor site preceding a proline. The binding of CDKs to their cognate regulatory subunit results in the formation of a holoenzyme, which together with additional components, participates in phosphorylation of target substrates governing cell cycle progression [Citation36–Citation44] and other substrates including NRF1 [Citation45], FOXM1 [Citation46] and the basal transcription apparatus [Citation47]. Thirteen CDKs (CDK1 – CDK13) exist in the human species [Citation48,Citation49]. Among the CDKs family, CDK4 and 6 (CDK4/6) together with their D type cyclins regulatory subunit, promote cell cycle progression [Citation50].
The mechanisms by which the mitotic kinases govern progression through the cell cycle was thought to involve phosphorylation of the pRB and related proteins [Citation42]. An increasing array of substrates has added greater complexity to this model [Citation51,Citation52]. Phosphorylation of the substrate pRB, by cyclin D1/CDK4 [Citation53] and other kinases [Citation54] results in the release of E2F/DP proteins (E2F-1 − 5). Thus, in mid/late G1, D-type cyclins and cyclin E, together with cyclin-dependent kinases, pRB, and the related p107 and p130, which then releases E2Fs, which in turn govern the expression of genes promoting cell cycle progression [Citation55]. Such a model suggests that E2F proteins would promote proliferation, however, genetic deletion analysis of E2F function in mice revealed hyper-proliferative phenotypes (reviewed in [Citation56]) suggesting further complexities [Citation8,Citation57].
Despite having high homology with other CDKs, CDK5 is not activated upon binding with a cyclin, however, CDK5 has subsequently been implicated in activating pRb phosphorylation [Citation49,Citation58]. CDK5 has critical functions in both terminally differentiated and proliferating cells and has recently been implicated in diseases, including the development and progression of cancer and neurodegenerative diseases [Citation59]. CDK5 also leads to phosphorylation of the retinoblastoma protein [Citation49,Citation58,Citation60]. CDK5 inhibition is an additional therapeutic target agent to treat cancer [Citation49]. pRB was discovered as a downstream target of CDK5. Expression of CDK5 leads to the phosphorylation of pRB.
CDK5, which has high amino acid sequence homology with other CDKs. Structural studies have shown the similarity of CDK5 (1H4L) with its closest structural homologs (CDK1) (5HQ0) and CDK2 (3QHR) (). CDK5 is upregulated in cancer and tamoxifen reduces both tumor growth and CDK5 activity. Cyclin D1 can attenuate CDK5 kinase activity by competing with p35 for binding with CDK5, thereby forming an inactive complex of cyclin D1 and CDK5 [Citation61–Citation64] (). In targeted drug design focus has been on targeting the ATP binding pocket. In the structural overlay of CDK5 is shown with the inhibitors R-roscovitine (1UNL) and the ATP analogue, [4-Amino-2-[(4-chlorophenyl)amino]-1,3-thiazol-5-yl](3-nitrophenyl)methanone (30OG), with a higher magnification view of the CDK5 active site in which R-roscovitine and the ATP analogue bind ().
Figure 2. Targeting of CDK5. (a). Structural overlay of CDK5 (1H4L) with its closest structural homologs CDK1 and CDK2. (b). Structural overlay of CDK5 with cyclin D1. (c). Structural overlay of CDK5 with the inhibitors R-roscovitine (1UNL) and ATP analogue, {4-Amino-2-[(4-chlorophenyl)amino]-1,3-thiazol-5-yl}(3-nitrophenyl)methanone (30OG). High magnification view is shown of the CDK5 active site in which R-roscovitine and the ATP analog bind.
![Figure 2. Targeting of CDK5. (a). Structural overlay of CDK5 (1H4L) with its closest structural homologs CDK1 and CDK2. (b). Structural overlay of CDK5 with cyclin D1. (c). Structural overlay of CDK5 with the inhibitors R-roscovitine (1UNL) and ATP analogue, {4-Amino-2-[(4-chlorophenyl)amino]-1,3-thiazol-5-yl}(3-nitrophenyl)methanone (30OG). High magnification view is shown of the CDK5 active site in which R-roscovitine and the ATP analog bind.](/cms/asset/fb0bf5ad-675c-4bc8-9cc6-4bf204ec6259/iery_a_1615889_f0002_oc.jpg)
3. Cyclin D1 is sufficient for the hallmarks and enabling characteristics of tumorigenesis
The cell cycle is critical in regulating the balance between the formation of new cells and programmed cell death. The induction and maintenance of tumorigenesis have been proposed to involve a variety of descriptive characteristics or ‘hallmarks’ of cancer (‘proliferative signaling’, ‘resistance to cell death’, ‘replicative immortality’, ‘induction of angiogenesis’, ‘evading growth suppressors’, ‘activating invasion and metastasis’) [Citation65] and ‘enabling capabilities’ (‘avoiding immune destruction’, ‘tumor promoting inflammation’, “deregulated cellular energetics, genomic instability, and mutation) [Citation66] (). It has also been proposed that a dynamic balance between oncogenes and tumor suppressors retrains tumorigenesis [Citation8,Citation48,Citation67–Citation70].
Although cyclin D1 was originally categorized within the presumptive ‘proliferative signaling’ category, the expression of cyclin D1 has been shown to be sufficient for the majority of these ‘hallmarks’ and ‘enabling characteristics’. In this regard cyclin D1 has been shown to induce proliferative signaling [Citation33,Citation71–Citation74], resistance to cell death [Citation71,Citation75], replicative immortality [Citation76], induction of angiogenesis [Citation77], evading growth suppressors, activating invasion and metastasis [Citation61,Citation65] and enabling capabilities (‘avoiding immune destruction’ [Citation63,Citation64]), ‘tumor promoting inflammation’ [Citation78], ‘deregulated cellular energetics’ [Citation45,Citation47,Citation79,Citation80], ‘genomic instability and mutation’ [Citation51,Citation52] (). Although the above models of “cancer hallmarks’ and ‘enabling characteristics’ do not directly embrace the field of cancer stem cells, several studies have shown the importance of cancer stem cells in the onset and progression of cancer [Citation81–Citation83] and the importance of cyclin D1 in expanding the pool of stem [Citation84,Citation85] and/or cancer stem cells [Citation86]. Osteopontin (OPN) was induced by cyclin D1 in fibroblasts, breast epithelial cells and in the murine transgenic mammary gland [Citation78] and OPN was sufficient to induce stem cell expansion [Citation78]. Thus, although cyclin D1 has historically been categorized as a gene product participating in proliferative signaling, given the diverse functions for cyclin D1 in tumorigenesis, it has been reassuring to find that drugs targeting the activity of cyclin D1 have shown promise in patients.
4. Role of cyclin D1-CDK4/6 complexes in breast tumorigenesis
Cyclins are regulatory subunits that form holoenzyme complexes with the CDKs [Citation37,Citation41,Citation42,Citation55] in particular CDK4 [Citation87] and CDK6 [Citation88]. Cyclin D1 encodes the regulatory subunit of the holoenzyme that is rate limiting in the proliferation of mammary epithelial cells [Citation89] as well as fibroblasts [Citation74,Citation90] and neurites [Citation91]. The inhibition of cyclin D1 expression leads to cell-cycle arrest, whereas cyclin D1 overexpression promotes G1-S phase progression [Citation74,Citation92,Citation93]. Cyclin D1 was also found to be a rate-limiting factor in the estrogen-induced proliferation of mammary epithelial cells [Citation89]. Cyclin D1 overexpression is sufficient for the induction of mammary tumorigenesis [Citation14,Citation94] with high levels of chromosomal instability (CIN). Furthermore, the rate of mammary tumor onset, and the induction of CIN were identical with a kinase-dead mutant of cyclin D1 [Citation51] suggesting kinase-independent functions may contribute to tumor onset in vivo. In vivo evidence for the requirement of cyclin D1 in mammary tumorigenesis was first identified using cyclin D1 anti-sense plasmids in ErbB2-induced mammary tumors [Citation95]. In subsequent cyclin D1 gene knockout mouse experiments, cyclin D1 was shown to have a pivotal role in the induction of breast tumors by Ras or ErbB2 [Citation96]. Furthermore, cyclin D1 gene deletion evidenced a critical role in skin [Citation97] and gastrointestinal tumorigenesis [Citation98].
Cyclin D1 functions in a variety of cell types that participate in the onset and progression of tumorigenesis, in addition to the mammary epithelial cell, including adipocytes [Citation99], lymphocytes [Citation61], and stromal fibroblasts [Citation78]. Cyclin D1 also affects the development of the mammary gland, in particular affecting the stem cells that may contribute to tumorigenesis [Citation84]. Analysis of cyclin D1 deletion in the mammary gland evidenced an important role for cyclin D1 in the expansion of progenitor cells that gave rise to tumors [Citation84]. Therefore, mammary gland selective cyclin D1 inactivation was conducted in the mature fully developed mammary gland [Citation79]. In contrast, with whole body cyclin D1 gene deletion mice, mammary gland selective cyclin D1 gene inactivation in the adult mammary gland, using inducible anti-sense, revealed a specific role in glucose metabolism. As an in vivo measurement of relative utilization of amino acids from the tricarboxylic acid cycle, the ratio of (glutamate+glutamine)/citrate was assessed using nuclear magnetic resonance. Mammary gland (glutamate+ glutamine)/citrate was increased twofold by endogenous cyclin D1. These studies are consistent with a model in which cyclin D1 determines metabolic substrate prioritization toward amino acid synthesis from the tricarboxylic acid cycle, consistent with the known role for cyclin D1 in DNA synthesis.
5. Cyclin D1 and CDK activity in the anti-tumor immune response
Cyclin D1 has several significant effects on the immune system and the anti-tumor response. First, cyclin D1 is expressed in macrophages and is required for CSF-1 induced guided migration [Citation61,Citation62].
Secondly, cyclin D1 in stromal fibroblasts promotes the secretion of cytokines that govern the expansion of myeloid-derived stem cells (MDSC) [Citation78]. Stromal cyclin D1 had a profound effect on the breast tumor microenvironment increasing the recruitment of F4/80+ and CD11b+ macrophages and increasing angiogenesis [Citation78]. Myeloid-derived suppressor cells (MDSCs) constitute one population of inflammatory cells that suppress the anti-tumor immune response and thereby enhance tumor growth [Citation100]. Cyclin D1 induced the expansion of the MDSC population [Citation78]. Cyclin D1-conditioned medium induced expansion of CD34 positive hematopoietic stem cells (HSCs) and promoted differentiation of CD34 positive hematopoietic stem cells (HSCs) into MDSC [Citation78]. Furthermore, stromal cyclin D1 induces secretion of factors that promotes the expansion of stem cells (breast stem-like cells, embryonic stem cells, and bone marrow-derived stem cells). Stromal cyclin D1 promotes secretion of pro-inflammatory cytokines (CCL2, CCL7, CCL11, CXCL1, CXCL5, CXCL9, CXCL12), CSF (CSF1, GM-CSF1) [Citation78].
Thirdly, cyclin D1/CDK4 induces expression of the immune checkpoint regulatory target PDL-1 [Citation63]. Targeting immune checkpoints such as the one mediated by programmed cell death protein 1 (PD-1) and its ligand PD-L1, have been approved for treating human cancers with durable clinical benefit [Citation101,Citation102]. PD-L1 protein abundance is regulated by cyclin D-CDK4 and the cullin 3-SPOP E3 ligase via proteasome-mediated degradation. Inhibition of CDK4 and CDK6 in vivo increased PD-L1 protein levels [Citation63]. Kaplan-Meier survival curves demonstrated the improved efficacy of combining PD-1 mAb with the CDK4/6 inhibitor, palbociclib in CT26 implanted tumor-bearing mice [Citation63]. Transgenic mice (MMTV-rtTA/tetO-HER2 tumor-bearing mice) tumors treated with abemaciclib and anti-PDL1 combination therapy regressed to a greater degree compared with abemaciclib alone [Citation64].
Fourthly, cyclin D1 triggers anti-tumor immunity [Citation64] via activating endogenous retroviral element expression thereby stimulating type III interferon production and tumor antigen presentation, thereby suppressing regulatory T cell proliferation [Citation64].
Like cyclin D/CDK4, CDK5 has also been implicated in regulating the immune checkpoint. CDK5 has been implicated in upregulation of IFNγ-induced programmed death ligand 1 (PD-L1), which allows certain cells to evade detection by the immune system. Decreased CDK5 expression led to increased expression of the PD-L1 transcriptional repressors IRF2 and IRF2BP and consequent decreased PD-L1 expression [Citation103].
6. Cyclin D1 regulation of the non-coding genome
Micro-RNAs (miRNAs) are 21- to 22-nucleotide-long molecules that regulate a variety of cellular phenotypes by affecting the stability of targeted mRNAs. Yu et al. compared the miRNA expression in cyclin D1-induced mammary tumors and mammary tissues derived from cyclin D1 anti-sense or knockout mice. These studies identified the miR-17/20 cluster as an important regulator of mammary tumorigenesis. miR-17/20 repressed expression of cyclin D1 by targeting the cyclin D1 3′ untranslated region [Citation104]. Cyclin D1 associated in the context of chromatin with the miR-17/20 regulatory region. This was the first study to demonstrate cyclin-dependent regulation of a non-coding RNA [Citation105]. miR-17/20 was shown to regulate the secretion of cytokines and plasminogen activator via the expression of α-enolase and cytokeratin 8. The inhibition of plasminogen activator by miR-17/20 required cyclin D1, thereby defining a mechanism by which cyclin D1 governs a non-coding genome-cytokine loop in breast cancer [Citation105].
Cyclin D1 was subsequently shown to regulate miRNA biogenesis through the transcriptional induction of Dicer [Citation106]. Cyclin D1 and Dicer both maintained heterochromatic histone modification (Tri-m-H3K9). Several functions of Dicer were cyclin D1-dependent. Cyclin D1-mediated cellular proliferation and migration were Dicer-dependent [Citation106]. These findings suggest that cyclin D1 regulates both the expression of individual miRNAs (via binding to their regulatory regions) and can also modulate the processing of other miRNAs via the transcriptional induction of Dicer [Citation106].
A cyclin D1-regulated miRNA signature, was identified in breast cancer cells that included several onco-mirs including miR-193b. When assessed in a superset of 459 breast cancer samples, the cyclin D1-regulated miRNA signature was associated with the Gormley 2 (G2) breast cancer miRNA subset, ERα+ status and activation of the Wnt pathway [Citation6]. Seed elements for cyclin D1-regulated miRNA were identified in 63 genes of the Wnt signaling pathway including DKK [Citation6]. These findings were consistent with studies conducted profiling a large number of different types of cancer cells that identified a distinct class of miRNAs (miR-193b, miR-193a-3p, miR-195-5p, miR-214-5p, miR-890) that target nearly all cyclins/CDKs. These miRNAs were very effective inhibitors of cancer cell proliferation [Citation107].
7. Role of cyclin D1-CDK4/6 in transcription and DNA damage
A substantial body of evidence now supports the model in which cyclin D1 governs gene transcription in the context of chromatin (reviewed in Di Sante et al. [Citation18]). These studies extended earlier findings in which cyclin D1 was shown to interact in a kinase-independent manner with transcriptional coactivators including p300 [Citation108] and more the 30 distinct transcription factors including the ERα [Citation109,Citation110], then DMP1 [Citation111], v-Myb [Citation112] and others (reviewed in cyclins [Citation36,Citation113,Citation114]. In chromatin cyclin, D1 was initially shown to alter recruitment of transcription factors in chromatin and affect local histone acetylation [Citation98]. Subsequently, cyclin D1 was itself shown to be recruited into chromatin using distinct evolving technologies including by ChIP [Citation99,Citation115,Citation116], then ChIP arrays [Citation117] and ChIP Seq [Citation14]. Recruitment into chromatin was independent of the cyclin D1 kinase function [Citation51]. In subsequent studies, other D types cyclins [Citation118,Citation119] and CDK6 was shown to be recruited into chromatin [Citation120,Citation121]. Furthermore, the CDK inhibitors affected phosphorylation of the transcription factors to which CDK bound [Citation122].
Many studies have found proteins with roles in both transcription and DNA repair (), including Topoisomerase IIα [Citation123,Citation124], PARP (Poly(ADP-ribose), TIP60, BRCA1, BRCA2, and the BRCA2 binding protein P/CAF. PARP-1, among other functions, promotes ERα activity [Citation124]. Moreover, cyclin D1 has been shown to regulate the activity of each of these afore-mentioned dual-function proteins. We initially [Citation36,Citation125–Citation129] and other groups subsequently [Citation130], demonstrated that cyclin D1 regulates DNA damage repair (DDR) [Citation129] and binds DDR proteins such as RAD51 [Citation129] and BRCA1 [Citation125]. In addition to binding Rad51, cyclin D1 binds other DNA damage repair proteins [Citation130]. Estrogens can contribute to BrCa by inducing DNA damage [Citation131]. Early BrCa lesions exhibit chromosomal instability with aneuploidy and estrogens lead to double-stranded DNA breaks and genomic instability [Citation132]. Estrogens induce DNA damage via both the production of oxidative metabolites or other oxidative DNA damage [Citation131]. Li et al. showed that cyclin D1 interacts with ERα and regulates a critical role in the E2-dependent DDR in human BrCa cells [Citation128]. Cyclin D1 was recruited to γH2AX foci by E2 and induced RAD51 expression [Citation128] contributing to aberrant growth signaling [Citation36]. The mechanisms governing recruitment of cyclin D1 into chromatin remains to be determined.
8. Stromal fibroblast cyclin D1 and its role in breast cancer
Although not formally included as a either a cancer hallmark, nor an enabling characteristic of tumorigenesis [Citation66], an overwhelming body of evidence has demonstrated the local microenvironment, including the tumor cancer-associated fibroblasts, contribute to the onset and progression of tumorigenesis [Citation133–Citation135]. Cyclin D1 is overexpressed, more than 30-fold in human breast cancer-associated fibroblasts, and reintroduction of cyclin D1 into fibroblasts augmented breast cancer tumor growth in mice [Citation78].
Increased expression of cyclin D1 in the breast cancer stroma is also associated with poor prognosis [Citation78]. When cyclin D1 was expressed in stromal fibroblasts breast epithelial cancer tumor growth was enhanced, apoptosis was restrained, and autophagy was increased [Citation78]. These results demonstrate that stromal cyclin D1 drives tumor microenvironment heterocellular signaling, thereby promoting several key hallmarks of cancer [Citation78].
9. Cyclin D1 regulation in other cancer types
In addition to gene amplification and transcriptional induction of cyclin D1 by Ras and other pathways [Citation136–Citation138], the abundance of cyclin D1 is regulated by posttranslational modification in a cell- type specific manner (reviewed in [Citation139,Citation140]:. Posttranslational modification includes phosphorylation (T286, T288) and ubiquitylation. The ubiquitin hydrolase USP2 [Citation141] and the deubiquitylase USP22, hydrolyzes ubiquitin at a set of specific lysine residues on cyclin D1 (K33, K46, 50, K112/114). Functionally, K33 ubiquitylation is implicated in nuclear localization of cyclin D1, and K112 and K114 ubiquitylation are linked to the interaction of cyclin D1 with partners, including CDK4/6 [Citation142]. Mutation of all cyclin D1 lysines to arginine confers protection from proteasome-dependent degradation, while single mutations only offered a modest increase in the stability of cyclin D1 and the only mutation of K112 has been identified in human breast cancer. K112 affects interactions with a diverse array of proteins including Cdk4/6 and Rho proteins.
Cyclin D1 is polyubiquitinated and subsequently degraded through the 26S proteasome pathway. The formation of polyubiquitin-protein conjugates includes E3, a protein ligase, which attaches ubiquitin to a lysine residue on a target protein. Several distinct ubiquitin-protein E3 ligases have been identified for degrading cyclin D1in distinct cell types [Citation143]. The SCF E3 ubiquitin ligases FBXW8, (Fbw8/Cul7) [Citation144], Fbx4 (SCFFbx4/‹B-crystallin) [Citation145], ß-Trcp [Citation146], APC/C [Citation147] and SCFSkp2 [Citation143] have been shown to regulate cyclin D1 degradation. The F-box protein, SKP2, contributes to the SCFSkp2 complex, which has been shown to ubiquitinate cyclin D1 [Citation143]. A splice variant of Skp2 is retained in the cytoplasm and fails to direct cyclin D1 ubiquitination in the uterine cancer cell line SK-UT [Citation148]. For Fbx4 (SCFFbx4/‹B-crystallin) of interest mutations in Fbx4 have been identified in esophageal squamous cell cancer, associated with increased abundance of cyclin D1 [Citation145]. The kinases governing phosphorylation appear to be cell-type specific, with excellent studies showing the role of GSK3ß [Citation149], Ras/MAPK [Citation144], p38SAPK2 [Citation150], IKKα [Citation146] and ATM/ATR [Citation151]. In lymphoma, cyclin D1 amplification is a frequent cause of overexpression, Eµ-cyclin D1 T-286, but not Eµ-cyclin D1 wt transgenic mice, develop lymphomas [Citation152] and the cyclin D1 phosphorylation site mutant T-286 co-precipitates MEP50 and the methyltransferase PRMT5 [Citation153], consistent with a role for Eµ-cyclin D1 T-286 [Citation152] and PRMT5 in lymphomagenesis [Citation154].
10. Environmental factors in human breast cancer
Molecular pathological epidemiology (MPE) is an integrative field that utilizes molecular pathology to incorporate interpersonal heterogeneity of a disease process into epidemiology [Citation155]. The MPA approach defines interactive relationships between environmental exposure and disease subtypes in determining disease incidence and mortality and can be used conceptually to integrate the impact of the microbiota as an ongoing exposure [Citation156]. The microbiome influences the onset and progression of cancer [Citation157]. The human gastrointestinal tract is colonized by a complex and diverse community of microorganisms including bacteria, viruses, archaea, and fungi [Citation24]. Recent studies have suggested the microbiota of women with breast cancer differs from that of healthy women, and have proposed potential mechanisms including regulating estrogen-dependent mechanisms [Citation158]. In this regard, alterations in the microbiota/estrobolome (the collection of the enteric bacterial genes whose products metabolize estrogen and its metabolites) can increase circulating estrogens and its metabolites, thereby increasing the risk of breast cancer.
Alternatively, the gut microbiome may affect breast cancer risk through estrogen-independent pathways, both locally and at a distance. In this regard, an enteromammary pathway has been described by which gut bacteria could reach the mammary gland. Furthermore, the gut microbiota differs in composition between healthy and breast cancer patients. The microbiome is present within the normal ductal lobular structure and some studies have identified different flora between individuals. Nipple aspirate fluid collected from breast cancer patients was also shown to have a distinct microbiota profile compared to healthy volunteers. Together these studies suggest an important interaction between the microbiome, food, and lifestyle to molecular pathologies and the influence of these factors on breast cancer and breast cancer therapies warrants consideration.
11. CDK inhibitors for breast cancer
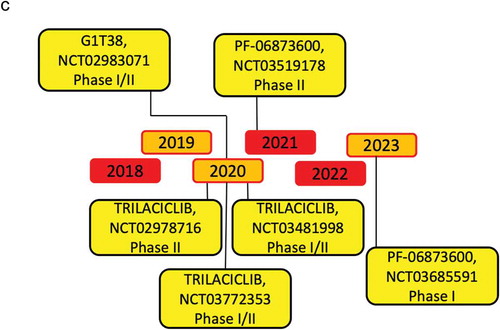
Novel therapies targeting CDK activity have become increasingly precise in targeting cyclin D/CDK activity and have been recently reviewed [Citation159–Citation164]. The effectiveness of CDK inhibitors have been shown in human breast cancer, in which the regulatory subunit, cyclin D1 is overexpressed [Citation15–Citation17]. More than 10 CDK inhibitors have entered clinical trials since 2009, most of which target multiple kinases. Palbociclib (PD0332991), Ribociclib (LEE011), and Abemaciclib (LY2835219) are specific, orally administered, CDK inhibitors with a similar chemical structure () [Citation165,Citation166], that are FDA approved for metastatic breast cancer treatment. They selectively bind to the ATP-binding pocket of CDK4 and CDK6 [Citation165,Citation167–Citation171]. Additional selective CDK inhibitors including G1T38, G1T28 and PF-06873600 are currently under investigation ( and ).
Figure 3. Specific CDK4/6 inhibitors chemical structure.
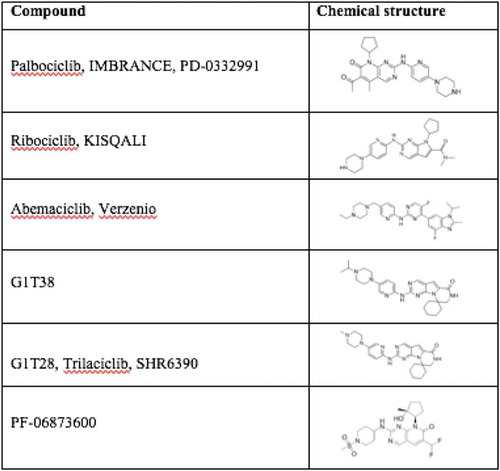
Figure 4. Timeline of the principal CDK4/6 inhibitors clinical trials.
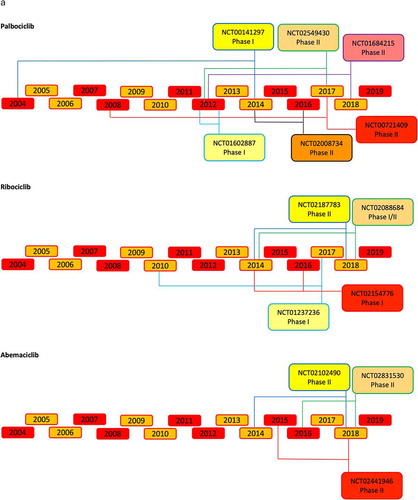
Figure 4. (Continued).
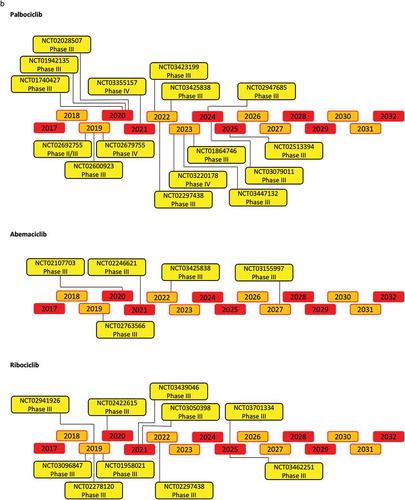
Figure 4. (Continued).
11.1. Pre-clinical studies
The anti-tumor effect of Palbociclib was investigated in neuroblastoma [Citation172], non-small cell lung carcinoma [Citation173] and breast cancer [Citation174]. In-vitro studies demonstrated the effect of palbociclib reduced pRB phosphorylation, and G1 phase cell cycle arrest. Mice carrying human colon cancer cells, treated with palbociclib, showed a reduction of pRB phosphorylation, downregulation of E2F-regulated genes and tumor regression [Citation159,Citation175]. Furthermore, palbociclib sensitized cancer cells to chemotherapy and ionizing radiation [Citation176].
The ribociclib anti-tumor effects have been studied in different types of cancer as well, including breast cancer. Studies on xenograft models determined the efficacy of Ribociclib in many other types of Rb-positive cancers, such as ERα breast cancer [Citation177] and neuroblastoma [Citation172]. These studies showed that ribociclib induces pRB dephosphorylation that leads to G1 arrest in Rb-positive cells [Citation172]. Ribociclib has been administrated in combination with 3-phosphoinositide dependent protein kinase 1 inhibitor (GSK2334470). This study showed ribociclib suppressed proliferation and increase apoptosis in ERα-positive breast cancer cell lines [Citation178].
The anti-tumor effect of abemaciclib has been assessed in several tumors, including breast cancer, and was shown to sensitize cancer cells over-expressing ATP Binding Cassette Subfamily B Member 1 (ABCB1) and ATP Binding Cassette Subfamily G Member 2 (ABCG2), which contribute to multidrug resistance in tumor chemotherapy [Citation179].
11.2. Clinical pharmacology
The selective CDK4/6 inhibitors have been further tested in clinical trials. The updated list of completed and open clinical trials in breast cancer are shown in Supplemental and Supplemental . The first phase I clinical study assessed the pharmacokinetics of palbociclib in patients with pRB-positive solid tumors [Citation180]. To assess both dose-limiting toxicity (DLT) and maximum tolerated dose (MTD), palbociclib was administered once daily for 21 of 28 days. This phase I study (NCT00141297) tested oral palbociclib in six dose-escalation cohorts. A daily dose of 125 mg over a period of 3 weeks, followed by 1 week off, was established to be the maximum tolerated dose (MTD) [Citation180]. In the phase II clinical trial (NCT01237236) of ribociclib, the drug was administrated in an escalating dose from 50 mg to 120 mg daily. The MTD was 900 mg/day over a 3 weeks’ period with 1 week off [Citation181]. In the phase I trial (NCT01394016) of abemaciclib the MTD resulted was 200 mg daily [Citation182].
Table 1. Completed clinical trials.
Table 2. Ongoing clinical trials per each CDK inhibitor.
11.3. Toxicities
Each of the three CDK inhibitors induce dose-related neutropenia, with rare neutropenic fevers. Although abemaciclib is associated with less neutropenia, monitoring is required with the potential need to reduce the dose. Palbociclib and ribociclib are administered on a 3-week-on, 1-week-off schedule, a schedule necessitated by the greater neutropenia seen with these agents compared with abemaciclib. Ribociclib has been associated with QT prolongation, leading to a requirement for electrocardiogram monitoring. Gastrointestinal toxicity, in the form of diarrhea, is more common with abemaciclib. Diarrhea is dose-dependent and in the MONARCH 2 trial led to a decrease in the dose of abemaciclib from 200 mg to 150 mg. This diarrhea occurs early is readily manageable with conventional antidiarrheal agents, and in the MONARCH 2 trial and led to drug discontinuation in only 1.6% of patients.
Neutropenia was the primary adverse event that determined DLT for palbociclib. Other reported adverse events included fatigue, diarrhea, nausea, dyspnea, and arthralgia. During the Phase II and III trials a dose of 125 mg daily of palbociclib was well tolerated and the toxicity was consistent between the trials [Citation180]. These adverse events caused an increased monitoring of the blood components during the treatment [Citation181]. Abemaciclib showed a higher rate of diarrhea and fatigue as adverse events [Citation182].
No biomarkers reliably define patients who will, or will not, benefit from the addition of a CDK4/6 inhibitor to their endocrine therapy. Adding CDK4/6 inhibitor to endocrine therapy prolongs PFS compared to endocrine therapy alone as first-line treatment in advanced breast cancer. The magnitude of PFS benefit is ethnicity-dependent but there were no interethnic differences in relative treatment-related toxicities [Citation183]. Recently several biomarkers studies have been published with regards to the use of circulating tumor DNA (ctDNA) analysis performed either at baseline, shortly after treatment or at progression in the context of the PALOMA-3 study [Citation184,Citation185]. Longitudinal detection and persistence of PI3KCA, ESR1, and Rb1 seems to predict resistance to the combination of palbociclib and fulvestrant in patients with endocrine-resistant disease [Citation186].
11.4. CDK4/6 and CDK5 as therapeutic targets for breast cancer
Multiple studies have shown that the addition of CDK4/6 inhibitors confers prolonged progression-free survival (PFS) in the context of patients receiving endocrine therapy. In this regard, improvement has been observed in combination with aromatase inhibitors [Citation169,Citation171,Citation187], and fulvestrant [Citation167,Citation188,Citation189] for palbociclib, ribociclib, and abemaciclib therapy. In the Palbociclib: Ongoing Trials in the Management of Breast Cancer [PALOMA]–1), palbociclib resulted in a progression-free survival benefit in patients with previously untreated, ERα+, HER2− advanced breast cancer [Citation190]. The subsequent randomized, phase 3 trial, PALOMA-2, showed that as first-line therapy for ERα+ HER2− advanced breast cancer, palbociclib prolonged progression-free survival when given in combination with letrozole [Citation171],. In the PALOMA-3 phase 3 trial, treatment with palbociclib and fulvestrant, prolonged progression-free survival among patients with ERα+ positive, HER2− advanced breast cancer who had disease progression after previous endocrine therapy compared with placebo and fulvestrant [Citation191]. This was the first study with a CDK 4/6 inhibitor to report mature data for OS analysis and demonstrating that, in patients with endocrine-sensitive disease the use of combination regimen is associated with approximately 10 months improvement in OS [Citation191].
Abemaciclib received FDA approval for the frontline treatment of postmenopausal women with ERα-positive, HER2− advanced or metastatic breast cancer, based on results from the phase III MONARCH 3 trial, in which the addition of abemaciclib to anastrozole or letrozole reduced the risk of progression or death by 46% compared with a nonsteroidal aromatase inhibitor alone. Abemaciclib has previously received FDA approval as monotherapy, based on the MONARCH 1 trial, which demonstrated an objective response rate of 19.7%, with a median duration of response of 8.6 months. Importantly these studie showed benefit independent of hormonal therapy.
Ribociclib has, unlike its competitors, been specifically studied in premenopausal women in the frontline setting (in the MONALEESA-7 trial), and as in postmenopausal women, the results suggest an impressive improvement in PFS, with similar toxicity to that seen in the postmenopausal setting.
CDK5 has been considered as a therapeutic target for breast cancer because of the evidence above that CDK5 participates in augmenting pRB phosphorylation and the participation of CDK5 in the anti-tumor immune response [Citation49]. Ahn and coworkers have reported three CDK5/p25 inhibitors able to reduce tau phosphorylation at Ser396 and Ser404, namely Bellidin (14, IC50 = 0.2 μM, ATP competitive), diazaphenanthrene 15 (IC50 = 2.0 μM, substrate competitive), and 4-aminothiazole 16a(IC50 = 17 μM, ATP competitive) [Citation192,Citation193].
11.5. Therapy resistance
Although the rationale for CDK inhibitors was based on a model in which pRB is the key substrate, subsequent studies revealed the biology is more complex. It has also been proposed that durable CDK inhibition [Citation114] reflects the induction of a senescence phenotype [Citation194]. Knockdown of pRB induced only partial resistance to CDK inhibition by palbociclib [Citation195–Citation197]. The combination of high p16ink4a expression with loss of RB expression has emerged as one of the more commonly used approaches to exclude or include patients on clinical trials with CDK4/6 inhibitors. In preclinical models with genetic loss of p16ink4a there is evidence for acquired resistance to CDK4/6 inhibitors [Citation198,Citation199]. However, in cancers that lose CDKN2A there is little evidence for a direct correlation between response and loss of the tumor suppressor [Citation182]. pRB inactivation in breast cancer is primarily due to overexpression or amplification of the cyclin D1 gene (CCND1) which is observed in as many as 50% of breast cancers, wherein it is believed to drive aberrant phosphorylation/inactivation of RB protein [Citation200]. Inactivation of the CDK-inhibitor p16ink4a (CDKN2A) contributes to the deregulation of RB phosphorylation, however, increased p16ink4a is often found in human breast cancer and deletion of p16inka4a/ARF did not affect MMTV-Erbb2 induced mammary tumors [Citation201]. Expression rates of p16 and pRB differ according to the breast cancer molecular subgroup [Citation202]. High p16INK4a mRNA expression is associated with high tumor grade, is silenced in a fraction of breast cancer cases [Citation203], Tumor samples overexpressing p16INK4a were predominantly ERα negative [Citation204]. reduced expression of p16ink4a due to de novo INK4a methylation occurred in 24/120 in one study [Citation204]. Lastly, loss of heterozygosity at the RB gene (RB1) locus has been defined in 20–30% of breast cancer and histological loss of RB protein has been documented with varying frequency up to 20% with a higher frequency in high-grade tumors [Citation205].
Acquired resistance to CDK4/6 inhibition has implicated deregulated cyclin E expression in some [Citation198,Citation206], but not all studies [Citation182]. TP53 mutation was associated with poor clinical response to the CDK4/6 inhibitor abemaciclib in breast cancer patients [Citation182] however this finding could be due to the association of TP53 mutations with triple-negative disease in the cohort analyzed [Citation114]. Suppression of MDM2 was associated with improved response to CDK4/6 inhibition in a small collection of liposarcomas treated with palbociclib [Citation207]. Therapy that interfering with CDK6 activity may also be associated with a higher risk of acquiring TP53 mutations because inhibition of CDK6 kinase activity may provoke the outgrowth of p53-mutant clones from premalignant cells [Citation122]. CDK6 expression levels correlate with the p53 pathway status in murine and human tumors and CDK6 binds to the promoters of genes governing the p53 antagonists Prmt5, Ppm1d, and Mdm4 [Citation122].
Approaches to overcoming CDKI resistance were recently reviewed [Citation114]. Additional secondary changes that occur upon treatment with CDK inhibitors that may contribute to both therapeutic efficacy and resistance include altered regulation of the Akt/mTORC pathway, and altered gene expression a function of non-canonical signaling by cyclins [Citation36,Citation113,Citation114]. Upstream mitogenic signaling is activated by CDK4/6 inhibition resulting in increased expression of cyclin D1. This, in turn, is associated with upregulation of Akt signaling [Citation178], suggesting a role for PI3K/Akt inhibitors to overcome CDKI resistance [Citation208]. Preclinical studies have suggested that resistance to CDK4/6 inhibitor can emerge through the selection for acquired mutation of RB1, amplification of cyclin E, amplification of CDK6, or suppression of CDK2 inhibitors (e.g. p27kip1 or p21cip1). Resistant cells coordinately upregulated expression of cyclins A, E and D1, activated phospho-CDK2 and phospho-S477/T479 AKT. Additional findings in CDKI resistant cells, suggesting potential targets to overcome resistance include amplification of cdk6 [Citation209], increased Cdk4 phosphorylation status (CDK4 T172 phosphorylation) [Citation210]. In addition, Increased androgen receptor (AR) function has been described, suggesting a role for androgen antagonists [Citation211]. In identifying potential sensitizers, CDK4/6 Inhibitors were shown to sensitize PIK3CA mutant breast cancer to PI3K inhibitors [Citation212]. Cyclin D1 overexpression, known to be associated with mitochondrial dysfunction (inhibition) [Citation45] in breast cancer cells, was shown to correlate with dependence upon glutamine in an esophageal cancer cell line and CB-839 (glutaminase 1 inhibitor) plus metformin/phenformin reduced growth of palbociclib resistant esophageal cancer cell lines https://doi.org/10.1038/s41467-019-09179-w. Collectively these studies suggest the potential to overcome CDK inhibitor resistance with inhibitors of immune checkpoints, Akt, AR and glutaminase.
12. Expert opinion
Therapeutic targeting of CDKs for breast cancer has demonstrated improvement in outcomes. New CDK inhibitors with reduced toxicities are being explored. Although the CDKs have diverse substrates, cyclin D1 has been shown to be sufficient for the majority of the ‘hallmarks’ and ‘enabling characteristics’ of tumorigenesis, perhaps providing an explanation of the efficacy for these therapies. Of concern, patients continue to perish while CDK activity, assessed by kinase activity, is inhibited in their tumors These findings suggest additional molecular mechanisms continue to drive therapeutic resistance, and or tumor growth through CDK-independent mechanisms. These mechanisms may relate to the growing number of kinase-independent functions of cyclins, including ERα agonist function, activation of chromosomal instability and recruitment of other enzyme including protein methylases [Citation213].
There are several recent findings with important implications for targeting cyclin D1/CDK activity in breast cancer. Firstly, the findings that cyclin D1/CDK govern aspects of the anti-tumor immune response [Citation61–Citation64] suggest the opportunity to explore combination therapy between CDK inhibitors and checkpoint inhibitors. Secondly, the side effects of bone marrow suppression, as a limiting toxicity, warrant the development of additional CDK inhibitors with distinct toxicity profiles. In preliminary preclinical studies, GiT38 appears to have less neutropenia [Citation214]. Thirdly, it will be important to develop efficient predictors of response to CDKs inhibitors with ctDNA appearing promising. Fourthly, because distinct domains of cyclin D1 conduct transcriptional activities that drive the process of mammary tumorigenesis [Citation113], including chromosomal instability [Citation14] in a kinase-independent manner [Citation51,Citation52,Citation215], the notion of targeting additional domains of cyclin D1 appears warranted. A growing body of evidence has demonstrated that cyclin D drives transcriptional programs in breast cancer, lymphoma [Citation118] and neural cells [Citation119].
Finally, and most importantly, women continue to die from metastatic breast cancer, including patients being treated with CDK inhibitors, despite the ability of the inhibitors to reduce CDK activity. Given the diverse additional functions of cyclin D1 to drive tumorigenesis, it will important to assess additional complementary approaches to inactivate the non-canonical functions of cyclin D1.
Article highlights
The cell cycle regulatory complex ‘cyclin D1-CDK4/6’ participates in the onset and maintenance of breast tumorigenesis.
Cyclin D1 is sufficient for the hallmarks and enabling characteristics of tumorigenesis
Breast cancer clinical studies have demonstrated the efficacy of targeting cyclin D/CDK activity, in human breast cancer. (an updated list and description of completed and ongoing clinical trials is provided)
Many functions of cyclin D1 are Cdk-independent, including the induction of chromosomal instability which may promote tumor heterogeneity.
Cyclin D1 and CDK inhibitors modulate anti-tumor immunity.
Declaration of interest
R Pestell is the founder of LightSeed LLC, has issued patents in cancer treatments, and is both a shareholder and employee of CytoDyn Inc. M Cristofanilli has acted as a consultant for Novartis, Merus, and CytoDyn, and has received honoraria from Pfizer. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Supplemental Material
Download MS Word (78.7 KB)Supplementary material
Supplemental data can be accessed here.
Additional information
Funding
Related Research Data
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017 Jan;67(1):7–30.
- Gilabert M, Bertucci F, Esterni B, et al. Capecitabine after anthracycline and taxane exposure in HER2-negative metastatic breast cancer patients: response, survival and prognostic factors. Anticancer Res. 2011 Mar;31(3):1079–1086.
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012 Oct 4;490(7418):61–70.
- Di Leva G, Cheung DG, Croce CM. miRNA clusters as therapeutic targets for hormone-resistant breast cancer. Expert Rev Endocrinol Metab. 2015;10(6):607–617.
- Yu Z, Pestell RG. Small non-coding RNAs govern mammary gland tumorigenesis. J Mammary Gland Biol Neoplasia. 2012 Mar;17(1):59–64.
- Wang G, Gormley M, Qiao J, et al. Cyclin D1-mediated microRNA expression signature predicts breast cancer outcome. Theranostics. 2018;8(8):2251–2263.
- Zhang Y, Xu B, Zhang XP. Effects of miRNAs on functions of breast cancer stem cells and treatment of breast cancer. Onco Targets Ther. 2018;11:4263–4270.
- Xu H, Yu S, Liu Q, et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol. 2017 Apr 24;10(1):97.
- Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000 Aug 17;406(6797):747–752.
- Shimelis H, LaDuca H, Hu C, et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J Natl Cancer Inst. 2018 Aug;110(8):855-862-.
- Mittendorf EA, Philips AV, Meric-Bernstam F, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014 Apr;2(4):361–370.
- Velasco-Velazquez M, Jiao X, De La Fuente M, et al. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012 Aug 1;72(15):3839–3850.
- Jiao X, Velasco-Velazquez MA, Wang M, et al. CCR5 governs DNA damage repair and breast cancer stem cell expansion. Cancer Res. 2018 Apr 1;78(7):1657–1671.
- Casimiro MC, Crosariol M, Loro E, et al. ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. J Clin Invest. 2012 Mar;122(3):833–843.
- Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015 Feb;14(2):130–146.
- Shah AN, Cristofanilli M. The growing role of CDK4/6 inhibitors in treating hormone receptor-positive advanced breast cancer. Curr Treat Options Oncol. 2017 Jan;18(1):6.
- Sablin MP, Ricci F, Loirat D, et al. [Cell cycle inhibitors in endocrine receptor positive breast cancer]. Bull Cancer. 2017 Feb;104(2):114–122.
- Di Sante G, Casimiro MC, Li Z, et al. D-type cyclins and gene transcription. In: Hinds PW, Brown NE, editors. D-Type Cyclins Cancer. Cham (Switzerland): Springer International Publishing; 2018. p. 61–90.
- Swaffer MP, Jones AW, Flynn HR, et al. Quantitative phosphoproteomics reveals the signaling dynamics of cell-cycle kinases in the fission yeast schizosaccharomyces pombe. Cell Rep. 2018 Jul 10;24(2):503–514.
- Swaffer MP, Jones AW, Flynn HR, et al. CDK substrate phosphorylation and ordering the cell cycle. Cell. 2016 Dec 15;167(7):1750–1761 e16.
- Nurse P. A journey in science: cell-cycle control. Mol Med. 2017 Jun;22:112–119.
- Hartwell LH. Genetic control of the cell division cycle in yeast. II. Genes controlling DNA replication and its initiation. J Mol Biol. 1971 Jul 14;59(1):183–194.
- Hartwell LH, Mortimer RK, Culotti J, et al. Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics. 1973 Jun;74(2):267–286.
- Mitchison JM. The biology of the cell cycle. Cambridge: Cambridge University Press; 1971.
- Nurse P. Genetic control of cell size at cell division in yeast. Nature. 1975 Aug 14;256(5518):547–551.
- Hartwell LH. Saccharomyces cerevisiae cell cycle. Bacteriol Rev. 1974 Jun;38(2):164–198.
- Nasmyth KA, Reed SI. Isolation of genes by complementation in yeast: molecular cloning of a cell-cycle gene. Proc Natl Acad Sci U S A. 1980 Apr;77(4):2119–2123.
- Alfa CE, Booher R, Beach D, et al. Fission yeast cyclin: subcellular localisation and cell cycle regulation. J Cell Sci Suppl. 1989;12:9–19.
- Booher RN, Alfa CE, Hyams JS, et al. The fission yeast cdc2/cdc13/suc1 protein kinase: regulation of catalytic activity and nuclear localization. Cell. 1989 Aug 11;58(3):485–497.
- Hagan I, Hayles J, Nurse P. Cloning and sequencing of the cyclin-related cdc13+ gene and a cytological study of its role in fission yeast mitosis. J Cell Sci. 1988 Dec;91(Pt 4):587–595.
- Arnold A, Kim HG, Gaz RD, et al. Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest. 1989 Jun;83(6):2034–2040.
- Meijer L, Arion D, Golsteyn R, et al. Cyclin is a component of the sea urchin egg M-phase specific histone H1 kinase. Embo J. 1989 8;Aug(8):2275–2282.
- Xiong Y, Connolly T, Futcher B, et al. Human D-type cyclin. Cell. 1991;65(4):691–699.
- Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992 Oct 30;71(3):505–514.
- Matsushime H, Roussel MF, Ashmun RA, et al. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–713.
- Di Sante G, Di Rocco A, Pupo C, et al. Hormone-induced DNA damage response and repair mediated by cyclin D1 in breast and prostate cancer. Oncotarget. 2017 Oct 10;8(47):81803–81812.
- Pestell RG, Albanese C, Reutens AT, et al. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev. 1999;20(4):501–534.
- Lee YM, Sicinski P. Targeting cyclins and cyclin-dependent kinases in cancer: lessons from mice, hopes for therapeutic applications in human. Cell Cycle. 2006 Sep;5(18):2110–2114.
- Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev. 2007 Feb;17(1):60–65.
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004 Nov 15;18(22):2699–2711.
- Hunter T, Pines J. Cyclins and cancer II: cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330.
- Casimiro MC, Velasco-Velázquez M, Aguirre-Alvarado C, et al. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin Investig Drugs. 2014 Mar;23(3):295–304.
- Fu M, Wang C, Li Z, et al. Minireview: cyclin D1: normal and abnormal functions. Endocrinology. 2004 Dec;145(12):5439–5447.
- Wang C, Li Z, Lu Y, et al. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A. 2006 Aug 1;103(31):11567–11572.
- Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011 Nov 15;20(5):620–634.
- Lee Y, Dominy JE, Choi YJ, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014 Jun 26;510(7506):547–551.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009 Mar;9(3):153–166.
- Shupp A, Casimiro MC, Pestell RG. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget. 2017 Mar 7;8(10):17373–17382.
- Udvardy A. The role of controlled proteolysis in cell-cycle regulation. Eur J Biochem. 1996 Sep 1;240(2):307–313.
- Casimiro MC, Di Sante G, Crosariol M, et al. Kinase-independent role of cyclin D1 in chromosomal instability and mammary tumorigenesis. Oncotarget. 2015 Apr 20;6(11):8525–8538.
- Casimiro MC, Arnold A, Pestell RG. Kinase independent oncogenic cyclin D1. Aging (Albany NY). 2015. 7;Jul(7):455–456.
- Kato J, Matsushime H, Hiebert SW, et al. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993 Mar;7(3):331–342.
- Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18(2):753–761.
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677.
- Weinberg RA. E2F and cell proliferation: A world turned upside down. Cell. 1996;85:457–459.
- Johnson J, Thijssen B, McDermott U, et al. Targeting the RB-E2F pathway in breast cancer. Oncogene. 2016 Sep 15;35(37):4829–4835.
- Pozo K, Castro-Rivera E, Tan C, et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell. 2013 Oct 14;24(4):499–511.
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122.
- Bhandari D, Lopez-Sanchez I, To A, et al. Cyclin-dependent kinase 5 activates guanine nucleotide exchange factor GIV/Girdin to orchestrate migration-proliferation dichotomy. Proc Natl Acad Sci U S A. 2015 Sep 1;112(35):E4874–83.
- Neumeister P, Pixley FJ, Xiong Y, et al. Cyclin D1 governs adhesion and motility of macrophages. Mol Biol Cell. 2003 May;14(5):2005–2015.
- Fuste NP, Fernandez-Hernandez R, Cemeli T, et al. Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat Commun. 2016 May 16;7:11581.
- Zhang J, Bu X, Wang H, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018 Jan 4;553(7686):91–95.
- Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017 Aug 24;548(7668):471–475.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674.
- Xu H, Wu K, Tian Y, et al. CD44 correlates with clinicopathological characteristics and is upregulated by EGFR in breast cancer. Int J Oncol. 2016 Oct;49(4):1343–1350.
- Xu HX, Wu KJ, Tian YJ, et al. Expression profile of SIX family members correlates with clinic-pathological features and prognosis of breast cancer: A systematic review and meta-analysis. Medicine (Baltimore). 2016 Jul;95(27):e4085.
- Knutson TP, Truong TH, Ma S, et al. Posttranslationally modified progesterone receptors direct ligand-specific expression of breast cancer stem cell-associated gene programs. J Hematol Oncol. 2017 Apr 17;10(1):89.
- Di Lauro L, Pizzuti L, Barba M, et al. Role of gonadotropin-releasing hormone analogues in metastatic male breast cancer: results from a pooled analysis. J Hematol Oncol. 2015 May;17(8):53.
- Albanese C, D’Amico M, Reutens AT, et al. Activation of the cyclin D1 gene by the E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. J Biol Chem. 1999 Nov 26;274(48):34186–34195.
- Motokura T, Bloom T, Kim HG, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991 Apr 11;350(6318):512–515.
- Arnold A, Motokura T, Bloom T, et al. The putative oncogene PRAD1 encodes a novel cyclin. Cold Spring Harb Symp Quant Biol. 1991;56:93–97.
- Quelle DE, Ashmun RA, Shurtleff SA, et al. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993 Aug;7(8):1559–1571.
- Choi YJ, Saez B, Anders L, et al. D-cyclins repress apoptosis in hematopoietic cells by controlling death receptor Fas and its ligand FasL. Dev Cell. 2014 Aug 11;30(3):255–267.
- Brown NE, Jeselsohn R, Bihani T, et al. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012 Dec 15;72(24):6477–6489.
- Hanai J, Dhanabal M, Karumanchi SA, et al. Endostatin causes G1 arrest of endothelial cells through inhibition of cyclin D1. J Biol Chem. 2002 May 10;277(19):16464–16469.
- Pestell TG, Jiao X, Kumar M, et al. Stromal cyclin D1 promotes heterotypic immune signaling and breast cancer growth. Oncotarget. 2017;8:81754–81775.
- Sakamaki T, Casimiro MC, Ju X, et al. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol. 2006 Jul;26(14):5449–5469.
- Bhalla K, Liu WJ, Thompson K, et al. Cyclin D1 represses gluconeogenesis via inhibition of the transcriptional coactivator PGC1alpha. Diabetes. 2014 Oct;63(10):3266–3278.
- Peiris-Pages M, Martinez-Outschoorn UE, Pestell RG, et al. Cancer stem cell metabolism. Breast Cancer Res. 2016;18(1):55.
- Yu Z, Pestell TG, Lisanti MP, et al. Cancer stem cells. Int J Biochem Cell Biol. 2012 Dec;44(12):2144–2151.
- Luo M, Wicha MS. Targeting cancer stem cell redox metabolism to enhance therapy responses. Semin Radiat Oncol. 2019 Jan;29(1):42–54.
- Jeselsohn R, Brown NE, Arendt L, et al. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010 Jan 19;17(1):65–76.
- Bizen N, Inoue T, Shimizu T, et al. A growth-promoting signaling component cyclin D1 in neural stem cells has antiastrogliogenic function to execute self-renewal. Stem Cells. 2014 Jun;32(6):1602–1615.
- Ju X, Casimiro MC, Gormley M, et al. Identification of a cyclin D1 network in prostate cancer that antagonizes epithelial-mesenchymal restraint. Cancer Res. 2014 Jan 15;74(2):508–519.
- Matsushime H, Ewen ME, Strom DK, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334.
- Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14(3):2077–2086.
- Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol. 1996 Dec;16(12):6917–6925.
- Baldin V, Lukas J, Marcote MJ, et al. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821.
- Marampon F, Casimiro MC, Fu M, et al. Nerve growth factor regulation of cyclin D1 in PC12 cells through a p21RAS ERK-pathway requires cooperative interactions between Sp1 and NF-kB. Mol Biol Cell. 2008 Jun;19(6):2566–2578.
- Jiang W, Kahn SM, Zhou P, et al. Overexpression of cyclin D1 in rat fibroblasts causes abnormalities in growth control, cell cycle progression and gene expression. Oncogene. 1993;8(12):3447–3457.
- Resnitzky D, Gossen M, Bujard H, et al. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14(3):1669–1679.
- Wang TC, Cardiff RD, Zukerberg L, et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994 Jun 23;369(6482):669–671.
- Lee RJ, Albanese C, Fu M, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol. 2000 Jan;20(2):672–683.
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001 Jun 28;411(6841):1017–1021.
- Robles AI, Rodriguez-Puebla ML, Glick AB, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998 Aug 15;12(16):2469–2474.
- Hulit J, Wang C, Li Z, et al. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004 Sep;24(17):7598–7611.
- Wang C, Pattabiraman N, Zhou JN, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol Cell Biol. 2003 Sep;23(17):6159–6173.
- Nakamura T, Ushigome H. Myeloid-derived suppressor cells as a regulator of immunity in organ transplantation. Int J Mol Sci. 2018 Aug 10;19(8):2357.
- Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016 Nov 3;375(18):1767–1778.
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016 Mar 2;8(328):328rv4.
- Dorand RD, Nthale J, Myers JT, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016 Jul 22;353(6297):399–403.
- Yu Z, Wang C, Wang M, et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol. 2008 Aug 11;182(3):509–517.
- Yu Z, Willmarth NE, Zhou J, et al. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci U S A. 2010 May 4;107(18):8231–8236.
- Yu Z, Wang L, Wang C, et al. Cyclin D1 induction of dicer governs microRNA processing and expression in breast cancer. Nat Commun. 2013;4:2812.
- Hydbring P, Wang Y, Fassl A, et al. Cell-cycle-targeting MicroRNAs as therapeutic tools against refractory cancers. Cancer Cell. 2017 Apr 10;31(4):576–590 e8.
- Fu M, Wang C, Rao M, et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J Biol Chem. 2005 Aug 19;280(33):29728–29742.
- Neuman E, Ladha MH, Lin N, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997 Sep;17(9):5338–5347.
- Zwijsen RM, Wientjens E, Klompmaker R, et al. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997 Feb 7;88(3):405–415.
- Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol. 1998 Mar;18(3):1590–1600.
- Ganter B, Fu S, Lipsick JS. D-type cyclins repress transcriptional activation by the v-Myb but not the c-Myb DNA-binding domain. Embo J. 1998 Jan 2;17(1):255–268.
- Pestell RG. New roles of cyclin D1. Am J Pathol. 2013 Jul;183(1):3–9.
- Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017 Jan;3(1):39–55.
- Bienvenu F, Barre B, Giraud S, et al. Transcriptional regulation by a DNA-associated form of cyclin D1. Mol Biol Cell. 2005 Apr;16(4):1850–1858.
- Fu M, Rao M, Bouras T, et al. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem. 2005 Apr 29;280(17):16934–16941.
- Bienvenu F, Jirawatnotai S, Elias JE, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010 Jan 21;463(7279):374–378.
- Albero R, Enjuanes A, Demajo S, et al. Cyclin D1 overexpression induces global transcriptional downregulation in lymphoid neoplasms. J Clin Invest. 2018 Aug 31;128(9):4132–4147.
- Pauklin S, Madrigal P, Bertero A, et al. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes Dev. 2016 Feb 15;30(4):421–433.
- Kollmann K, Heller G, Schneckenleithner C, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013 Aug 12;24(2):167–181.
- Otto T, Sicinski P. The kinase-independent, second life of CDK6 in transcription. Cancer Cell. 2013 Aug 12;24(2):141–143.
- Bellutti F, Tigan AS, Nebenfuehr S, et al. CDK6 antagonizes p53-induced responses during tumorigenesis. Cancer Discov. 2018 Jul;8(7):884–897.
- Malovannaya A, Lanz RB, Jung SY, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011 May 27;145(5):787–799.
- Ju BG, Lunyak VV, Perissi V, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006 Jun 23;312(5781):1798–1802.
- Wang C, Fan S, Li Z, et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer Res. 2005 Aug 1;65(15):6557–6567.
- Marampon F, Gravina GL, Ju X, et al. Cyclin D1 silencing suppresses tumorigenicity, impairs DNA double strand break repair and thus radiosensitizes androgenindependent prostate cancer cells to DNA damage. Oncotarget. 2016 Feb 2;7(5):5383–5400.
- Casimiro MC, Di Sante G, Ju X, et al. Cyclin D1 promotes androgen-dependent DNA damage repair in prostate cancer cells. Cancer Res. 2016 Jan 15;76(2):329–338.
- Li Z, Chen K, Jiao X, et al. Cyclin D1 integrates estrogen-mediated DNA damage repair signaling. Cancer Res. 2014 Jul 15;74(14):3959–3970.
- Li Z, Jiao X, Wang C, et al. Alternative cyclin d1 splice forms differentially regulate the DNA damage response. Cancer Res. 2010 Nov 1;70(21):8802–8811.
- Jirawatnotai S, Hu Y, Michowski W, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011 Jun 08;474(7350):230–234.
- Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol. 2014;4:106.
- Levin ER. Minireview: extranuclear steroid receptors: roles in modulation of cell functions. Mol Endocrinol. 2010 Sep 22;25(3):377–384.
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010 Jun 12;9(12).
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004 Nov 18;432(7015):332–337.
- Mercier I, Casimiro MC, Wang C, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: implications for the response to hormonal therapy. Cancer Biol Ther. 2008 Aug;7(8):1212–1225.
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999 Apr 1;398(6726):422–426.
- Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999 May 11;96(10):5522–5527.
- Albanese C, Johnson J, Watanabe G, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995 Oct 6;270(40):23589–23597.
- Inoue K, Fry EA. Aberrant expression of cyclin D1 in cancer. Sign Transduct Insights. 2015;4:1–13.
- Pontano LL, Diehl JA. DNA damage-dependent cyclin D1 proteolysis: GSK3beta holds the smoking gun. Cell Cycle. 2009 Mar 15;8(6):824–827.
- Cini G, Carpi A, Mechanick J, et al. Thyroid hormones and the cardiovascular system: pathophysiology and interventions. Biomed Pharmacother. 2009 Dec;63(10):742–753.
- Gennaro VJ, Stanek TJ, Peck AR, et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc Natl Acad Sci U S A. 2018 Oct 2;115(40):E9298–E9307.
- Ganiatsas S, Dow R, Thompson A, et al. A splice variant of Skp2 is retained in the cytoplasm and fails to direct cyclin D1 ubiquitination in the uterine cancer cell line SK-UT. Oncogene. 2001 Jun 21;20(28):3641–3650.
- Okabe H, Lee SH, Phuchareon J, et al. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS One. 2006 Dec 27;1:e128.
- Barbash O, Zamfirova P, Lin DI, et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell. 2008 Jul 8;14(1):68–78.
- Wei S, Yang HC, Chuang HC, et al. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008 Sep 26;283(39):26759–26770.
- Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000 Jul 7;102(1):55–66.
- Russell A, Thompson MA, Hendley J, et al. Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene. 1999 Mar 18;18(11):1983–1991.
- Diehl JA, Cheng M, Roussel MF, et al. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998 Nov 15;12(22):3499–3511.
- Casanovas O, Miro F, Estanyol JM, et al. Osmotic stress regulates the stability of cyclin D1 in a p38SAPK2-dependent manner. J Biol Chem. 2000 Nov 10;275(45):35091–35097.
- Hitomi M, Yang K, Stacey AW, et al. Phosphorylation of cyclin D1 regulated by ATM or ATR controls cell cycle progression. Mol Cell Biol. 2008 Sep;28(17):5478–5493.
- Vaites LP, Lian Z, Lee EK, et al. ATM deficiency augments constitutively nuclear cyclin D1-driven genomic instability and lymphomagenesis. Oncogene. 2014 Jan 2;33(1):129–133.
- Aggarwal P, Vaites LP, Kim JK, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010 Oct 19;18(4):329–340.
- Li Y, Chitnis N, Nakagawa H, et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015 Mar;5(3):288–303.
- Hamada T, Keum N, Nishihara R, et al. Molecular pathological epidemiology: new developing frontiers of big data science to study etiologies and pathogenesis. J Gastroenterol. 2017 Mar;52(3):265–275.
- Ogino S, Nowak JA, Hamada T, et al. Insights into pathogenic interactions among environment, host, and tumor at the crossroads of molecular pathology and epidemiology. Annu Rev Pathol. 2019 Jan;24(14):83–103.
- Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013 Nov;13(11):800–812.
- Fernandez MF, Reina-Perez I, Astorga JM, et al. Breast cancer and its relationship with the microbiota. Int J Environ Res Public Health. 2018 Aug 14;15(8):1747–1767.
- Lu J. Palbociclib: a first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J Hematol Oncol. 2015 Aug 13;8:98.
- Zhang P, Tong Z, Tian F, et al. Phase II trial of utidelone as monotherapy or in combination with capecitabine in heavily pretreated metastatic breast cancer patients. J Hematol Oncol. 2016 Aug 11;9(1):68.
- Song DG, Ye Q, Poussin M, et al. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol. 2016 Jul 20;9(1):56.
- Yu S, Li A, Liu Q, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017 Mar 29;10(1):78.
- Malumbres M, Pevarello P, Barbacid M, et al. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci. 2008 Jan;29(1):16–21.
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016 Apr 6;(4):353–367.
- O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016 Jul;13(7):417–430.
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009 Jul;8(7):547–566.
- Sledge GW Jr., Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017 Sep 1;35(25):2875–2884.
- Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017 Nov 10;35(32):3638–3646.
- Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016 Nov 3;375(18):1738–1748.
- Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016 Apr;17(4):425–439.
- Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016 Nov 17;375(20):1925–1936.
- Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013 Nov 15;19(22):6173–6182.
- Puyol M, Martín A, Dubus P, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010 Jul 13;18(1):63–73.
- Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77.
- Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427–1438.
- Whiteway SL, Harris PS, Venkataraman S, et al. Inhibition of cyclin-dependent kinase 6 suppresses cell proliferation and enhances radiation sensitivity in medulloblastoma cells. J Neurooncol. 2013 Jan;111(2):113–121.
- Rascon K, Flajc G, De Angelis C, et al. Ribociclib in HR+/HER2- advanced or metastatic breast cancer patients. Ann Pharmacother. 2018;7:1060028018817904.
- Jansen VM, Bhola NE, Bauer JA, et al. Kinome-wide RNA interference screen reveals a role for PDK1 in acquired resistance to CDK4/6 inhibition in ER-positive breast cancer. Cancer Res. 2017 May 1;77(9):2488–2499.
- Wu T, Chen Z, To KKW, et al. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem Pharmacol. 2017 Jan 15;124:29–42.
- Flaherty KT, Lorusso PM, Demichele A, et al. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568–576.
- Infante JR, Cassier PA, Gerecitano JF, et al. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2016 Dec 1;22(23):5696–5705.
- Patnaik A, Rosen LS, Tolaney SM, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016 Jul;6(7):740–753.
- Lee KWC, Lord S, Finn RS, et al. The impact of ethnicity on efficacy and toxicity of cyclin D kinase 4/6 inhibitors in advanced breast cancer: a meta-analysis. Breast Cancer Res Treat. 2018 Nov 21;174(1):271–278.
- O’Leary B, Hrebien S, Morden JP, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018 Mar 1;9(1):896.
- Cristofanilli M. Inflammatory breast cancer: a new approach. Lancet Oncol. 2016 May;17(5):544–546.
- O’Leary B, Cutts RJ, Liu Y, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018 Nov;8(11):1390–1403.
- Hortobagyi GN, Stemmer SM, Burris HA, et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018 Jul 1;29(7):1541–1547.
- Turner NC, Ro J, Andre F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015 Jul 16;373(3):209–219.
- Slamon DJ, Neven P, Chia S, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018 Aug 20;36(24):2465–2472.
- Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015 Jan;16(1):25–35.
- Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018 Nov 15;379(20):1926–1936.
- Lew J, Huang QQ, Qi Z, et al. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994 Sep 29;371(6496):423–426.
- Tsai LH, Delalle I, Caviness VS Jr., et al. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994 Sep 29;371(6496):419–423.
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015 Jul;15(7):397–408.
- Dean JL, Thangavel C, McClendon AK, et al. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010 Jul 15;29(28):4018–4032.
- Ismail A, Bandla S, Reveiller M, et al. Early G(1) cyclin-dependent kinases as prognostic markers and potential therapeutic targets in esophageal adenocarcinoma. Clin Cancer Res. 2011 Jul 1;17(13):4513–4522.
- Dean JL, McClendon AK, Hickey TE, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012 Jul 15;11(14):2756–2761.
- Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014 Aug 15;5(15):6512–6525.
- Heilmann AM, Perera RM, Ecker V, et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res. 2014 Jul 15;74(14):3947–3958.
- Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005 Jun 20;23(18):4215–4224.
- D’Amico M, Wu K, Di Vizio D, et al. The role of Ink4a/Arf in ErbB2 mammary gland tumorigenesis. Cancer Res. 2003 Jun 15;63(12):3395–3402.
- Shin E, Jung WH, Koo JS. Expression of p16 and pRB in invasive breast cancer. Int J Clin Exp Pathol. 2015;8(7):8209–8217.
- Dublin EA, Patel NK, Gillett CE, et al. Retinoblastoma and p16 proteins in mammary carcinoma: their relationship to cyclin D1 and histopathological parameters. Int J Cancer. 1998 Feb 20;79(1):71–75.
- Hui R, Macmillan RD, Kenny FS, et al. INK4a gene expression and methylation in primary breast cancer: overexpression of p16INK4a messenger RNA is a marker of poor prognosis. Clin Cancer Res. 2000 Jul;6(7):2777–2787.
- Anderson JJ, Tiniakos DG, McIntosh GG, et al. Retinoblastoma protein in human breast carcinoma: immunohistochemical study using a new monoclonal antibody effective on routinely processed tissues. J Pathol. 1996 Sep;180(1):65–70.
- Herrera-Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016 Apr 15;76(8):2301–2313.
- Kovatcheva M, Liu DD, Dickson MA, et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget. 2015 Apr 10;6(10):8226–8243.
- Gul A, Leyland-Jones B, Dey N, et al. A combination of the PI3K pathway inhibitor plus cell cycle pathway inhibitor to combat endocrine resistance in hormone receptor-positive breast cancer: a genomic algorithm-based treatment approach. Am J Cancer Res. 2018;8(12):2359–2376.
- Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017 Apr 20;36(16):2255–2264.
- Raspe E, Coulonval K, Pita JM, et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med. 2017 Aug;9(8):1052–1066.
- Ji W, Shi Y, Wang X, et al. Combined Androgen receptor blockade overcomes the resistance of breast cancer cells to palbociclib. Int J Biol Sci. 2019;15(3):522–532.
- Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014 Jul 14;26(1):136–149.
- Li Z, Jiao X, Di Sante G, et al. Cyclin D1 integrates G9a-mediated histone methylation. Oncogene. 2019 Feb 4:18. DOI:10.1038/s41388-019-0723-8.
- Bisi JE, Sorrentino JA, Jordan JL, et al. Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget. 2017 Jun 27;8(26):42343–42358.
- Casimiro MC, Pestell RG. Cyclin d1 induces chromosomal instability. Oncotarget. 2012 Mar;3(3):224–225.