
ABSTRACT
Introduction
Dysfunction of the immune system is a hallmark of cancer. Through increased understanding of the complex interactions between immunity and cancer, immunotherapy has emerged as a treatment modality for different types of cancer. Promising activity with immunotherapy has been reported in numerous malignancies, but challenges such as limited response rates and treatment resistance remain. Furthermore, outcomes with this therapeutic approach in hematologic malignancies are even more limited than in solid tumors. T-cell immunoglobulin domain and mucin domain 3 (TIM-3) has emerged as a potential immune checkpoint target in both solid tumors and hematologic malignancies. TIM-3 has been shown to promote immune tolerance, and overexpression of TIM-3 is associated with more aggressive or advanced disease and poor prognosis.
Areas covered
This review examines what is currently known regarding the biology of TIM-3 and clinical implications of targeting TIM-3 in cancer. Particular focus is given to myeloid malignancies.
Expert opinion
The targeting of TIM-3 is a promising therapeutic approach in cancers, including hematologic cancers such as myeloid malignancies which have not benefited much from current immunotherapeutic treatment approaches. We anticipate that with further clinical evaluation, TIM-3 blockade will emerge as an important treatment strategy in myeloid malignancies.
1. Introduction
Immune system dysfunction plays a key role in oncogenesis, and tumor immune evasion has impeded traditional treatment strategies in many cancers [Citation1]. Advances in understanding the interplay between cancer and the immune system as well as the immunosuppressive tumor microenvironment, have furthered the development of therapies targeting immune checkpoints, cell-surface receptors that modulate immune response and regulate immune homeostasis [Citation2–5]. In cancer, immune checkpoint pathways are often co-opted to subvert the immune system [Citation2,Citation6]. Targeting of these pathways initially showed clinical promise in solid malignancies, for which efforts primarily focused on the blockade of the immune checkpoints cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death 1 (PD-1), resulting in increased survival rates and durable responses in multiple malignancies, including melanoma and non-small cell lung cancer [Citation4,Citation7–9]. While immune checkpoint inhibition has doubtlessly revolutionized oncology, treatment challenges remain: primary and acquired resistance are common, and there is a paucity of reliable predictive biomarkers to identify the minority of patients who will derive the greatest benefit [Citation4,Citation5,Citation10]. As a result, the precise role of immune checkpoint inhibition varies across cancers; hematologic malignancies – and in particular, myeloid neoplasms – have thus far only seen limited clinical benefit, potentially due to increased immunosuppression and lower immunogenicity compared with solid tumors [Citation11–13].
More recently, the cell surface molecule T-cell immunoglobulin domain and mucin domain 3 (TIM-3) has emerged as a potential immune checkpoint target in the treatment of cancer, although much of the detail regarding its signaling pathway remains unknown [Citation2,Citation14]. TIM-3 is a regulator of both innate and adaptive immunity and is expressed on multiple immune cell types, including effector T cells, monocytes, natural killer cells, and dendritic cells (DCs); it is also aberrantly expressed on leukemic progenitor cells, while notably absent from the surface of healthy hematopoietic stem cells [Citation15–21]. TIM-3 has been shown to promote immune tolerance, and overexpression of TIM-3 is associated with more aggressive or advanced disease and poor prognosis [Citation22–26]; in myelodysplastic syndrome (MDS) and myeloproliferative neoplasms, TIM-3 expression is associated with leukemic transformation [Citation21]. Further, given that immunotherapy resistance may arise from tumor utilization of alternate inhibitory pathways, the simultaneous targeting of TIM-3 with other immune checkpoints may enhance clinical response [Citation14,Citation27]. This review summarizes what is currently understood regarding the underlying biology of TIM-3 and clinical implications of targeting TIM-3 in cancer in general, with a focus on myeloid malignancies.
2. TIM-3 biology
The TIM gene family comprises eight genes in mice (TIM-1 through TIM-8) and three genes in humans (TIM-1, TIM-3, and TIM-4), which are orthologs of the corresponding mouse genes [Citation28]. TIM family proteins are type-I cell-surface glycoproteins that consist of signal peptides, an extracellular N-terminal variable immunoglobulin (IgV) domain, a mucin-like domain, a single transmembrane domain, and a C-terminal intracellular cytoplasmic tail [Citation29]. The highly conserved IgV domain consists of two antiparallel β-sheets with four cysteines in the variable region and, as in other IgV domains, contains a CC′ loop and an FG loop [Citation29]. Conventionally, these loops are located at opposite ends of the IgV domain; however, in TIM-1, -3, and -4 proteins, the CC′ loop (which determines receptor–ligand interaction) is oriented more closely to the FG loop, creating a unique ligand-binding pocket [Citation30,Citation31]. The cytoplasmic tail is critical for TIM-3 signaling through the phosphorylation of tyrosine residues, but downstream signaling mechanisms in T cells are less well defined [Citation32]. A soluble form of TIM-3 also exists, which lacks the transmembrane and mucin domains and may negatively regulate T-cell–mediated immune response [Citation33].
Full details regarding TIM-3 signaling pathways remain to be elucidated. At present, it is understood that tyrosine residues 256 and 263, located on the cytoplasmic tail, facilitate interactions with HLA-B–associated transcript 3 (BAT3) and the tyrosine kinase FYN [Citation32,Citation34]. When TIM-3 is unbound by ligands, BAT3 attaches to the cytoplasmic tail of TIM-3 and recruits LCK, which blocks FYN and maintains T-cell activation [Citation32,Citation34–36]. The binding of the ligands galectin-9 (GAL-9) or carcinoembryonic antigen–related cell adhesion molecule 1 (CEACAM1) to TIM-3 has been shown to phosphorylate Tyr256 and Tyr263 via the interleukin (IL)-inducible T-cell kinase, which releases BAT3 from TIM-3, resulting in FYN signaling and subsequent immune inhibition () [Citation37,Citation38].
Figure 1. TIM-3 signaling. APC, antigen-presenting cell; BAT3, HLA-B–associated transcript 3; CEACAM1, carcinoembryonic antigen–related cell adhesion molecule 1; PtdSer, phosphatidylserine; TCR, T-cell receptor; TIM-3, T-cell immunoglobulin domain and mucin domain 3
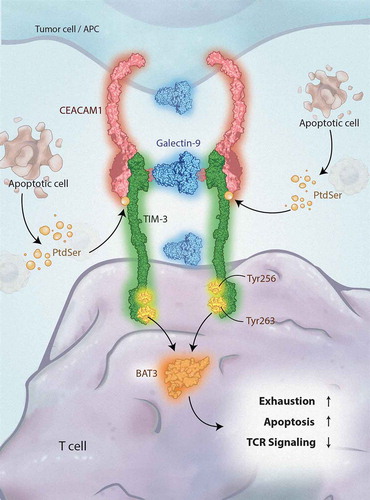
Ligands for TIM-3 include GAL-9, CEACAM1, phosphatidylserine (PtdSer), and high-mobility group box 1 (HMGB1) [Citation17,Citation38–41]. These ligands each bind to different regions of the extracellular IgV domain. GAL-9 is expressed or secreted by tumor cells, antigen-presenting cells, or leukemic stem cells (LSCs) [Citation42–44]; GAL-9 secretion may be an autocrine signal for LSC self-renewal [Citation39]. GAL-9 binds to the carbohydrate motifs on the IgV domain of TIM-3, although the exact location is not well defined [Citation30,Citation41]. The TIM-3-GAL-9 pathway has been implicated in immune evasion in cancer [Citation45]. Indeed, the binding of GAL-9 to TIM-3 has been reported to induce apoptosis in T-helper 1 effector cells as well as TIM-3+CD8+ tumor-infiltrating lymphocytes [Citation41,Citation46].
CEACAM1, the most recently discovered ligand, is expressed on the cell surface of T cells, DCs, monocytes, and macrophages, with high expression also observed on some tumor cells [Citation38,Citation47–51]. CEACAM1 is also thought to bind TIM-3 in the pocket created from the FG and CC′ loops [Citation38]. The TIM-3–CEACAM1 interaction is critical in the regulation of TIM-3–mediated antitumor immunity and autoimmunity [Citation38]. CD8+ T cells expressing both CEACAM1 and TIM-3 are profoundly dysfunctional, with limited production of interferon γ [Citation52]. Notably, anti–TIM-3 antibodies have been shown to interfere with the binding of TIM-3 to both PtdSer and CEACAM1 [Citation53].
PtdSer, the “eat me„ signal expressed on apoptotic cells, binds within the IgV domain like GAL-9 but in the pocket created from the FG and CC′ loops, opposite the predicted site for GAL-9 binding [Citation40,Citation54]. The binding of TIM-3 with PtdSer may contribute to TIM-3-mediated phagocytosis of apoptotic cells [Citation55]. Indeed, TIM-3 plays a crucial role in phagocytosis [Citation56]. Expression of TIM-3 in cancer-eliminating M1 macrophages is low. However, macrophages with the immune-tolerant M2 phenotype have increased TIM-3 expression and do not release cytokines. Macrophages that express high levels of TIM-3 do not release cytokines and thus promote immune tolerance. Anti–TIM-3 antibodies may thus lead to activation of macrophages, and if able to activate Fc receptors, may lead to increased elimination of leukemic stem cells via antibody-dependent cellular phagocytosis [Citation57].
HMGB1, a damage-associated molecular pattern molecule, is secreted from cells after stress, after which it associates with free RNA and DNA [Citation58,Citation59]. HMGB1 is implicated in immune responses toward damaged cells, and data suggest that it binds near the FG loop of TIM-3 [Citation17,Citation58,Citation60]. Binding of HMGB1 to TIM-3 impedes the innate system’s sensing of DNA via hindrance of the recruitment of nucleic acids into endosomes; this prevents DC activation in the tumor microenvironment [Citation58].
3. TIM-3 as a cancer immunotherapy target
Dysregulation of TIM-3 has been implicated in both autoimmune diseases and cancer [Citation22,Citation61,Citation62]. Increased TIM-3 expression is associated with advanced disease and poor prognosis in a variety of solid tumors as well as hematologic malignancies [Citation23–26,Citation61,Citation63–65]. Importantly, TIM-3 expression is primarily observed intratumorally, with minimal expression noted in peripheral T cells and regulatory T cells (Tregs); TIM-3 expression is also observed on T cells following relapse from stem cell transplant in myeloid malignancies [Citation22,Citation23,Citation61,Citation62]. Activation of the TIM-3 signaling pathway plays a critical role in T-cell exhaustion and death in both cancer and viral infections, impeding the immune system’s capacity to mount a response by limiting T-cell proliferation and production of inflammatory cytokines, among other effects [Citation22,Citation66]. Accordingly, TIM-3 blockade has been shown to enhance tumor antigen-specific T-cell proliferation and activity [Citation23,Citation67,Citation68]. Moreover, TIM-3 is often co-expressed with PD-1 in T cells, and this co-expression represents a severe phenotype of T-cell exhaustion [Citation22,Citation23,Citation61,Citation68,Citation69]. Notably, in solid tumors, PD-1+TIM-3+ tumor-infiltrating lymphocytes represent the largest subset of tumor-infiltrating T cells, and further, PD-1 blockade upregulates TIM-3 expression. Therefore, TIM-3 signaling may promote resistance to PD-1–targeted therapy, and combined blockade of these checkpoints has been proposed as a rational therapeutic approach [Citation22,Citation70,Citation71].
In addition to CD4+ and CD8+ T cells, TIM-3 is expressed on Tregs [Citation16]. TIM-3+ Tregs constitute the majority of Tregs present in tumor tissue and exhibit enhanced suppressor function compared with TIM-3− Tregs [Citation15,Citation16,Citation72], in addition to having a higher expression of other checkpoint receptors [Citation15,Citation72] and expression of known Treg effector molecules that contribute to their suppressive activity, such as IL-10, granzymes, and perforin [Citation15,Citation16]. TIM-3+ Tregs also express higher levels of forkhead box P3 (FOXP3), a marker denoting the most suppressive Treg subtype, predominantly found in tumor tissue and rarely observed in peripheral tissue [Citation16,Citation23,Citation72,Citation73]. Blockade of TIM-3 reduced the number of FOXP3+ Tregs and led to the enhancement of antitumor immune responses [Citation74]. TIM-3+ Tregs also co-express PD-1 [Citation16,Citation69], and additional downregulation of Treg effector molecules was found upon anti−TIM-3 therapy in combination with anti–PD-1 therapy [Citation16,Citation74].
4. Targeting TIM-3 in myeloid malignancies
The role of immune checkpoint inhibition in MDS, acute myeloid leukemia (AML), and other myeloid malignancies is under active clinical investigation [Citation13,Citation75–77]. Although MDS and AML exhibit low neoantigen burden, these malignancies are associated with a markedly exhausted T-cell repertoire; therefore, inhibition of immune checkpoints associated with T-cell exhaustion, such as PD-1, CTLA-4, and TIM-3, may serve to reactivate immune function [Citation78–80]. Similarly, T-cell–mediated immune tolerance and increased PD-1 expression have been observed in myelofibrosis [Citation81].
In MDS and AML, immune checkpoint therapy targeting the classic pathways of CTLA-4 and PD-1/programmed cell death ligand 1 (PD-L1) and PD-L2 has so far only shown limited clinical activity as monotherapy [Citation78,Citation82–86] while early-phase trials are ongoing as combination therapy with additional checkpoint inhibitors, hypomethylating agents (HMAs), or intensive chemotherapy [Citation87,Citation88]. The only randomized clinical trial of checkpoint inhibition in myeloid malignancies combined the anti–PD-L1 antibody durvalumab with azacitidine as frontline therapy in patients with higher-risk MDS and in older patients with AML unfit for intensive chemotherapy; no improvements were reported in response rate or survival compared with the results of azacitidine monotherapy in either cohort [Citation89]. Notably, however, PD-1 blockade following stem cell transplant in AML may enhance the graft-versus-leukemia effect and T-cell function [Citation90]. Although it is clear that immuno-oncologic therapeutics have not yet revolutionized the hematologic malignancy treatment landscape as in solid tumors, the clinical success of stem cell transplant in myeloid malignancies demonstrates the potential of immunotherapeutic modalities in these diseases in particular [Citation13,Citation91–93]. Other immune checkpoint pathways involving receptors such as TIM-3, lymphocyte activation gene 3, CD47, and leukocyte immunoglobulin-like receptor 4 may be more relevant to the pathogenesis of myeloid malignancies; therefore, their targeting could prove to be more therapeutically active [Citation94].
TIM-3 is preferentially expressed on leukemic progenitor cells, often termed LSCs, but not on normal hematopoietic stem cells (HSCs) (); expression on LSCs correlates with MDS severity and progression to AML [Citation21,Citation39,Citation95–97]. Given the selective expression profile of TIM-3, targeting TIM-3 represents a unique opportunity in MDS and AML to both selectively attack LSCs and malignant blasts and enhance the immune response by targeting dysfunctional T cells [Citation39,Citation96,Citation98,Citation99]. In LSCs, activation of the protein kinase C/mechanistic target of rapamycin pathways upregulates both TIM-3 and GAL-9, resulting in activation of the TIM-3–GAL-9 autocrine loop and secretion of soluble TIM-3, leading to reduced T-cell secretion of IL-2, impaired natural killer cell killing of LSCs, and implications in leukemic progression [Citation39,Citation100]. The TIM-3 pathway is also associated with the expansion and activation of granulocytic myeloid-derived suppressor cells [Citation101].
Figure 2. TIM-3 in myeloid malignancies. TIM-3 is expressed on LSCs but not on HSCs. Treatment with hypomethylating agents increases expression of other inhibitory receptors through activation of endogenous retroviral elements, demonstrating the potential of co-targeting. CTLA-4, cytotoxic T lymphocyte antigen 4; HSC, hematopoietic stem cell; LSC, leukemic stem cell; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; TIM-3, T-cell immunoglobulin domain and mucin domain 3
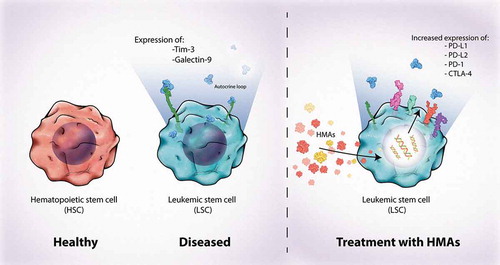
Given that TIM-3 is often co-expressed with PD-1, the simultaneous targeting of TIM-3 and PD-1 has also been proposed as a rational strategy in many tumors, including in myeloid malignancies [Citation62,Citation70]. Compared with healthy patients, those with AML have an increased frequency of PD-1+TIM-3+ T cells, and this dual expression correlates with severe T-cell exhaustion [Citation99,Citation102]. Further, HMAs upregulate the expression of PD-1 and other immune checkpoints and ligands by inducing expression of endogenous retroviral elements, and expression of TIM-3 and GAL-9 is increased in patients who experience relapse after chemotherapy [Citation98,Citation103–105]. Clinical results of such combinations are awaited, but synergistic activity in xenograft and animal models has been reported [Citation70].
Finally, it must be noted that TIM-3 has also been implicated in immune dysfunction in lymphoid malignancies. In acute B lymphoblastic leukemia, increased expression of both TIM-3 and PD-1 on CD4+ and CD8+ T cells has been observed following relapse after stem cell transplant [Citation106]. Additionally, CD200/TIM-3 signaling has been implicated in T-cell dysfunction in pediatric B-precursor acute lymphoblastic leukemia, with expression of TIM-3 on CD4+ bone marrow T cells being associated with increased risk of relapse [Citation107]. Conversely, however, immune checkpoint inhibition in early chronic lymphocytic leukemia may not reverse exhaustion of CD8+ T cells [Citation108], and TIM-3 does not appear to be widely expressed in peripheral T-cell lymphomas [Citation109]. Further in vitro and in vivo studies are needed to better identify which patient populations and disease subsets can derive immune-restoring benefit from the targeting of TIM-3.
5. Anti–TIM-3 agents/clinical studies
Anti–TIM-3 antibodies, used as either monotherapy or in combination regimens, are currently being evaluated in a variety of cancer clinical trials, primarily in solid tumors, often in combination with a PD-1 inhibitor (). While the clinical evaluation of these therapies is as of yet in early stages, early clinical data from ongoing clinical trials have been reported for three molecules: TSR-022 (Tesaro), LY3321367 (Eli Lilly and Company), and sabatolimab (Novartis Pharmaceuticals; developed as MBG453). These data are summarized in .
Table 1. Anti–TIM-3 agents and associated clinical trials in cancer
Table 2. Summary of available clinical trial data for anti–TIM-3 agents
In preclinical studies, TSR-022 enhanced the activity of CD4+ T cells isolated from healthy donors [Citation110]. Additionally, TSR-022 did not lead to related local irritation or adverse changes of parameters during either the dosing or recovery phase (doses ≤100 mg/kg). In the phase 1AMBER study of patients with advanced solid tumors, TSR-022 was evaluated as monotherapy and in combination with the anti–PD-1 antibody TSR-042, the anti–LAG-3 antibody TSR-033, or docetaxel (NCT02817633) [Citation111,Citation112]. Among 173 patients who received TSR-022 plus TSR-042, the rate of grade ≥3 drug-related adverse events was 7.5%. Objective responses were observed in all patients with NSCLC who had PD-L1 positive disease, with 4 of these patients achieving confirmed PR. The combination of TSR-022 and TSR-042 is also being evaluated in a phase 2 study of patients with liver cancer (NCT03680508).
LY3321367 is being evaluated alone and in combination with the anti–PD-1 antibody LY3300054 in a phase 1study of patients with advanced relapsed/refractory (R/R) solid tumors (NCT03099109) [Citation113]. In an interim analysis including 23 patients who received LY3321367 monotherapy and 18 patients who received LY3321367 plus LY3300054, treatment-emergent adverse events (TEAEs) were all mild in nature, except for 1 patient with grade 3 anemia in the combination-therapy arm. No deaths, dose-limiting toxicities, or treatment-related serious AEs were reported in either treatment arm. Tumor reduction of >20% was observed in 2 patients in the LY3321367 monotherapy arm, including 1 patient with a partial response. The recommended phase 2 dosing for LY3321367 combination therapy with LY3300054 was established as 1200 mg intravenously every 2 weeks (Q2W; cycles 1 and 2) followed by 600 mg intravenously Q2W (cycles 3+).
Presently, sabatolimab is the only anti–TIM-3 monoclonal antibody being investigated in MDS and AML with preliminary safety and efficacy data. Sabatolimab is a high-affinity IgG4 monoclonal antibody that blocks binding of TIM-3 to the ligand PtdSer [Citation114]. In vitro, sabatolimab has demonstrated increased secretion of inflammatory cytokines (both alone and synergistically when combined with the anti–PD-1 antibody spartalizumab). Additionally, sabatolimab enhanced myeloid cell and T-cell function. Following these findings, sabatolimab was investigated in a phase 1/2 study with the anti–PD-1 agent spartalizumab in patients with advanced solid tumors (NCT02608268) [Citation115]. In this study, the recommended phase 2 dose of sabatolimab was assessed as sabatolimab 800 mg every 4 weeks (600 mg every 3 weeks, 400 mg Q2W), and preliminary evidence of antitumor activity was observed. Overall, 27.6% of the patients receiving sabatolimab monotherapy and 39.5% of the patients receiving sabatolimab plus spartalizumab achieved stable disease; partial response was achieved by 4.7% of the patients in the combination therapy arm. AEs with sabatolimab monotherapy were mainly constitutional symptoms6 and were primarily grade 1–2 in severity.
Sabatolimab is presently being investigated in a phase 1b study as monotherapy and combined with spartalizumab, HMAs (either decitabine or azacitidine), or both spartalizumab and decitabine in patients with higher-risk (HR) MDS and AML (newly diagnosed or R/R) who are unfit for intensive chemotherapy [Citation116]. Interim results reported that the combination of sabatolimab plus HMA was associated with mostly grade 1 or 2 drug-related AEs and showed preliminary evidence of antileukemic activity and response durability. The most common grade 3/4 TEAE with sabatolimab plus decitabine was febrile neutropenia (47.8%; no grade 4); with sabatolimab plus azacitidine, the most common grade 3/4 TEAE was thrombocytopenia (48.6%, including 35.1% with grade 4). Overall, TEAEs with sabatolimab combined with either HMA were similar to those historically reported with the HMA alone. No treatment-related deaths were reported. Among patients with HR-MDS, median duration of exposure was 8.7 months (range, 3.4–30.3 months) with sabatolimab plus decitabine, but to date is only 5.0 months (range, 2.1–11.8 months) with the sabatolimab plus azacitidine arms. Based on preliminary follow-up, overall response rates (ORRs) in patients with HR-MDS with sabatolimab plus decitabine and sabatolimab plus azacitidine were 61.1% and 57.1%, respectively, with complete response (CR) rates of 33.3% and 7.1%. In patients with newly diagnosed AML, the ORR was 47.1% with sabatolimab plus decitabine (CR rate, 29.4%) and 28.6% with sabatolimab plus azacitidine (CR rate, 14.3%). The ORR was 23% with sabatolimab plus decitabine in patients with R/R AML (all CR with incomplete hematologic recovery). A subsequent retrospective analysis of patients who discontinued study treatment to proceed to hematopoietic stem cell transplant demonstrated limited graft-vs-host disease–related toxicities, indicating that sabatolimab does not appear to reduce the viability of progression to stem cell transplant in patients with MDS or AML or introduce new toxicities, though more experience is needed [Citation117].
Furthermore, phase 2 and phase 3 trials of sabatolimab in patients with HR-MDS and AML are ongoing as part of the ongoing STIMULUS clinical trial program [Citation118]. STIMULUS-MDS1 is a randomized phase 2 trial evaluating sabatolimab plus decitabine vs placebo plus decitabine in patients with HR-MDS (NCT03946670); the primary endpoints are rate of CR and progression-free survival. STIMULUS-MDS2 is a randomized phase 3 trial of sabatolimab plus azacitidine vs placebo plus azacitidine in patients with HR-MDS or chronic myelomonocytic leukemia-2 (NCT04266301). The primary endpoint of this study is OS. Finally, STIMULUS-AML1 is a single-arm phase 2 study investigating sabatolimab, azacitidine, and venetoclax in patients with newly diagnosed AML who are unfit for intensive chemotherapy (NCT04150029). Primary endpoints of the study are DLTs and the rate of CR.
One other anti-TIM3 compound undergoing evaluation in myeloid malignancies is SHR-1702 (Jiangsu HengRui Medicine). SHR-1702 is being investigated in a phase 1study of patients with HR-MDS and R/R AML (NCT04443751) as well as a phase 1study in combination with the anti–PD-1 antibody camrelizumab in patients with advanced R/R solid tumors (NCT03871855). Both of these studies are not yet recruiting.
6. Conclusion
Outcomes with immune checkpoint inhibition in hematologic malignancies have been largely disappointing relative to how these compounds have changed the treatment of many solid tumors. In recent years, however, improved understanding of the biology of TIM-3 – a regulator of both innate and adaptive immunity, expressed on exhausted T cells as well as LSCs and malignant blasts but not on normal, healthy HSCs – and the roles it plays in tumor immune evasion have facilitated the investigation of therapies targeting TIM-3 in both solid tumors and hematologic malignancies. Findings thus far, while early, have been encouraging, with promising early response rates and manageable toxicities – particularly compared to other immune-oncologic targets. Notably, one such molecule, sabatolimab, a high-affinity IgG4 monoclonal anti-TIM3 antibody, has been studied to date in MDS and AML, and the reported clinical activity suggests there may be a role of TIM-3 as a novel therapeutic target in these otherwise challenging malignancies. Together, these discoveries support continued clinical investigation of TIM-3 in a broader spectrum of hematologic malignancies.
7. Expert opinion
There remains great interest in further exploring the role of immune checkpoint inhibition across numerous tumor types; the success of targeting the CTLA-4 and PD-1 pathways in some solid tumors has fundamentally altered the oncology treatment landscape, but in general, this approach has shown only limited efficacy to date in hematologic malignancies, particularly for myeloid neoplasms. However, the development of newer agents that block the inhibitory receptor TIM-3 may yet change this. Dysregulation of TIM-3 has been implicated in a number of autoimmune diseases as well as across various cancers, including myeloid malignancies such as MDS and AML. TIM-3 is expressed on LSCs and leukemic blasts, but not on normal HSCs, and has been proposed as a potential marker to distinguish LSCs from HSCs. Its functions on malignant blasts remain to be well characterized, but it may play a role in autocrine self-renewal. Given the differential expression of TIM-3 between healthy and malignant progenitors, as well as T-cell exhaustion phenotypes in AML and MDS, blockade of TIM-3 represents a promising approach for the treatment of patients with myeloid malignancies. Targeting TIM-3 may have multiple antitumor mechanisms, such reactivation of immune effector cells expressing TIM-3 and interruption or other alteration of LSCs and blasts, which may potentially include increased antibody-dependent cellular phagocytosis.
Several molecules targeting TIM-3 are under active clinical investigation, including TSR-022, LY3321367, and sabatolimab; multiple clinical trials are in more preliminary stages for other molecules. Most studies to date have evaluated these compounds in solid tumors, typically in combination with therapy targeting PD-1, based on the notion that these two immune checkpoints may act in concert to facilitate immune evasion and diminish antitumor immune response. TIM-3 inhibition with sabatolimab has also been studied in myeloid neoplasms, with other studies in myeloid neoplasia pending, including a phase 1 trial of SHR-1702 monotherapy. Although only preliminary data have been reported as of yet with sabatolimab, these data suggest that it can be safely combined with HMA therapy, and the reported response rates provide support for further study. In contrast to efforts with PD-1 and CTLA-4 blockade, the overall incidence of grade 3/4 AEs, and in particular the incidence of grade 3/4 immune-related AEs, has been relatively low, with toxicities primarily comprised of arthralgias and low-grade liver function test abnormalities and a general safety profile similar to that reported with HMAs alone [Citation116]. Of note, signals for pneumonitis or more severe immuno-oncology toxicities such as colitis have not been reported to date. Given the interpatient variability in clinical presentation of immune-related AEs as well as the potential for these toxicities to become refractory to steroid treatment [Citation119], immune-related AEs should continue to be monitored closely, especially as the clinical development of anti–TIM-3 molecules expands toward larger phase 3 studies.
Responses so far have shown indications of durability [Citation116], and furthermore, given that stem cell transplant is as of yet the only known treatment option with curative potential in MDS and AML, recent data indicating that sabatolimab does not negatively impact the safety or viability of stem cell transplant [Citation117] are particularly encouraging. Other studies have started to explore TIM-3 further as a target, including, for instance, a preclinical study of chimeric antigen receptor T cells bispecific for both TIM-3 and CD13 [Citation120]. In this study, elimination of patient-derived AML with reduced toxicity to human bone marrow stem cells and peripheral myeloid cells (mouse tissues) were observed. These results are especially interesting given the ongoing clinical development of RO7121661 (Rosche), which is a bispecific antibody for both TIM-3 and PD-1 (). Clinical evaluation of these compounds will help to elucidate therapeutic potential and may enhance our understanding of TIM-3 biology.
Overall, the preclinical evidence and preliminary clinical data surrounding the use of anti–TIM-3 antibodies in cancer have been very promising. We are hopeful that, as more mature data readouts from ongoing clinical trials are published, optimization of treatment strategies utilizing TIM-3 blockade will emerge, potentially further contributing to the growing immune-oncology landscape. With regards to this treatment modality in myeloid malignancies, we eagerly await more comprehensive clinical data from the ongoing sabatolimab trials in MDS and AML, including the ongoing phase 2 and phase 3 trials from the STIMULUS program. As the MDS/AML treatment landscape is continuously evolving, we are especially excited to see this therapy evaluated in rational combinations that do not have overlapping mechanisms of action and limited overlapping toxicity profiles.
Article highlights
Immunotherapy has revolutionized the treatment of solid tumors, but key challenges remain, including identifying reliable predictors of response, low response rates, and management of treatment resistance
In contrast, immune checkpoint inhibition targeting CTLA-4, PD-1, and PD-L1 in myeloid malignancies has thus far only resulted in mixed to modest clinical benefit
TIM-3—a regulator of both innate and adaptive immunity—has emerged as a potential immune checkpoint target in both solid tumors and hematologic malignancies; overexpression of TIM-3 has been associated with poor prognosis and aggressive disease
Of particular relevance is that TIM-3 is expressed on leukemic stem cells and blasts but not on normal hematopoietic stem cells, suggesting potential for therapeutic targeting beyond that of traditional immune checkpoint inhibition
A number of anti–TIM-3 agents are in active clinical development, with phase I clinical trial data available for three molecules: TSR-022 (Tesaro) and LY3321367 (Eli Lilly and Company) in advanced solid tumors, and sabatolimab (Novartis Pharmaceuticals; previously known as MBG453) in both advanced solid tumors and myeloid malignancies
Early findings for these agents are encouraging, with promising response rates and manageable toxicities, and support the continued clinical investigation of anti–TIM-3 therapies in cancer, especially in myeloid malignancies; more comprehensive clinical trial data are eagerly awaited
Declaration of interest
AM Zeidan received research funding (institutional) from Novartis, Celgene/Bristol Myers Squibb, AbbVie, Astex, Pfizer, MedImmune/AstraZeneca, Boehringer Ingelheim, Trovagene, Incyte, Takeda, Aprea, and ADC Therapeutics; participated in advisory boards and/or had a consultancy with and received honoraria from Novartis, AbbVie, Otsuka, Pfizer, Celgene/Bristol Myers Squibb, Jazz, Incyte, Agios, Boehringer Ingelheim, Acceleron, Astellas, Daiichi Sankyo, Cardinal Health, Taiho, Seattle Genetics, BeyondSpring, Trovagene, Takeda, Ionis, Amgen, Janssen, Epizyme, Syndax, and Tyme; served on steering and independent data review committees for clinical trials for Novartis, AbbVie, and Janssen; and received travel support for meetings from Novartis, Pfizer, and Trovagene. RS Komrokji participated in speaker’s bureaus with Jazz, Agios, and Bristol Myers Squibb, and participated in advisory boards with and/or received honoraria from Novartis, Jazz, Bristol Myers Squibb, Geron, Abbvie, Incyte, and Acceleron. AM Brunner received research funding (institutional) from Novartis, Celgene/Bristol Myers Squibb, Takeda, AstraZeneca, and Janssen and participated in advisory boards and/or had a consultancy with and received honoraria from Novartis, Acceleron, Celgene/Bristol Myers Squibb, Takeda, and Jazz. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(suppl):S185–S198. .
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264.
- Riva A, Chokshi S. Immune checkpoint receptors: homeostatic regulators of immunity. Hepatol Int. 2018;12(3):223–236.
- Darvin P, Toor SM, Sasidharan Nair V, et al. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50:1–11.
- Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–150.
- Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47:765–779.
- Khan M, Lin J, Liao G, et al. Comparative analysis of immune checkpoint inhibitors and chemotherapy in the treatment of advanced non-small cell lung cancer: a meta-analysis of randomized controlled trials. Medicine (Baltimore). 2018;97(33):e11936.
- Chen R, Hou X, Yang L, et al. Comparative efficacy and safety of first-line treatments for advanced non-small cell lung cancer with immune checkpoint inhibitors: a systematic review and meta-analysis. Thorac Cancer. 2019;10(4):607–623.
- Pons-Tostivint E, Latouche A, Valflard P, et al. Comparative analysis of durable responses on immune checkpoint inhibitors versus other systemic therapies: a pooled analysis of phase III trials. In: JCO Precis Oncol. Alexandria, VA, USA: American Society of Clinical Oncology; 2019. p. 3.
- Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723.
- Dong S, Ghobrial IM. Immunotherapy for hematological malignancies. J Life Sci. Westlake Village. 2019;1:46–52.
- Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol. 2017;10:103.
- JP B, Stahl M, AM Z. Immune checkpoint-based therapy in myeloid malignancies: a promise yet to be fulfilled. Expert Rev Anticancer Ther. 2019;19::393–404.
- Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 2018;52:39–52.
- Gupta S, Thornley TB, Gao W, et al. Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ Tregs. J Clin Invest. 2012;122:2395–2404.
- Sakuishi K, Ngiow SF, Sullivan JM, et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2:e23849.
- Chiba S, Baghdadi M, Akiba H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–842.
- Ndhlovu LC, Lopez-Vergès S, Barbour JD, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119:3734–3743.
- Khademi M, Illes Z, Gielen AW, et al. T Cell Ig- and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. J Immunol. 2004;172:7169–7176.
- Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97–111.
- Kikushige Y, Shima T, Takayanagi S, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708–717. .
- Sakuishi K, Apetoh L, Sullivan JM, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194.
- Gao X, Zhu Y, Li G, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676.
- Zhang Y, Cai P, Liang T, et al. TIM-3 is a potential prognostic marker for patients with solid tumors: A systematic review and meta-analysis. Oncotarget. 2017;8(19):31705–31713.
- Jiang J, Jin MS, Kong F, et al. Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PLoS One. 2013;8:e81799.
- Granier C, Dariane C, Combe P, et al. Tim-3 Expression on Tumor-Infiltrating PD-1 + CD8 + T Cells correlates with poor clinical outcome in renal cell carcinoma. Cancer Res. 2017;77(5):1075–1082.
- Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16.
- Freeman GJ, Casasnovas JM, Umetsu DT, et al. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–189.
- Monney L, Sabatos CA, Gaglia JL, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536–541.
- Cao E, Zang X, Ramagopal UA, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007;26(3):311–321.
- Kundapura SV, Ramagopal UA. The CC’ loop of IgV domains of the immune checkpoint receptors, plays a key role in receptor: ligand affinity modulation. Sci Rep. 2019;9:19191.
- Lee J, Su EW, Zhu C, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. 2011;31(19):3963–3974.
- Geng H, Zhang GM, Li D, et al. Soluble form of T cell Ig mucin 3 is an inhibitory molecule in T cell-mediated immune response. J Immunol. 2006;176:1411–1420.
- Rangachari M, Zhu C, Sakuishi K, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med. 2012;18:1394–1400.
- Clayton KL, Haaland MS, Douglas-Vail MB, et al. T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol. 2014;192:782–791.
- Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44(5):989–1004.
- van de Weyer PS, Muehlfeit M, Klose C, et al. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun. 2006;351(2):571–576.
- Huang YH, Zhu C, Kondo Y, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517:386–390.
- Kikushige Y, Miyamoto T, Yuda J, et al. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17(3):341–352.
- DeKruyff RH, Bu X, Ballesteros A, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184(4):1918–1930.
- Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–1252.
- Wang Y, Zhao E, Zhang Z, et al. Association between Tim3 and Gal9 expression and gastric cancer prognosis. Oncol Rep. 2018;40:2115–2126.
- Gonçalves Silva I, Yasinska IM, Sakhnevych SS, et al. The Tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine. 2017;22:44–57.
- Li H, Wu K, Tao K, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342–1351.
- Yasinska IM, Sakhnevych SS, Pavlova L, et al. The Tim-3-galectin-9 pathway and its regulatory mechanisms in human breast cancer. Front Immunol. 2019;10:1594.
- Kang CW, Dutta A, Chang LY, et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep. 2015;5:15659.
- Kammerer R, Stober D, Singer BB, et al. Carcinoembryonic antigen-related cell adhesion molecule 1 on murine dendritic cells is a potent regulator of T cell stimulation. J Immunol. 2001;166(11):6537–6544.
- Horst AK, Bickert T, Brewig N, et al. CEACAM1+ myeloid cells control angiogenesis in inflammation. Blood. 2009;113(26):6726–6736.
- Wiener Z, Kohalmi B, Pocza P, et al. TIM-3 is expressed in melanoma cells and is upregulated in TGF-beta stimulated mast cells. J Invest Dermatol. 2007;127:906–914.
- Coutelier J-P, Godfraind C, Dveksler GS, et al. B lymphocyte and macrophage expression of carcinoembryonic antigen-related adhesion molecules that serve as receptors for murine coronavirus. Eur J Immunol. 1994;24(6):1383–1390.
- Gebauer F, Wicklein D, Horst J, et al. Carcinoembryonic antigen-related cell adhesion molecules (CEACAM) 1, 5 and 6 as biomarkers in pancreatic cancer. PLoS One. 2014;9(11):e113023.
- Zhang Y, Cai P, Li L, et al. Co-expression of TIM-3 and CEACAM1 promotes T cell exhaustion in colorectal cancer patients. Int Immunopharmacol. 2017;43:210–218.
- Sabatos-Peyton CA, Nevin J, Brock A, et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-TIM-3 antibodies that have functional efficacy. Oncoimmunology. 2018;7:e1385690.
- Fadok VA, Voelker DR, Campbell PA, et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216.
- Nakayama M, Akiba H, Takeda K, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113(16):3821–3830.
- Ocana-Guzman R, Torre-Bouscoulet L, Sada-Ovalle ITIM-3. regulates distinct functions in macrophages. Front Immunol. 2016;7:229.
- Jan M, Chao MP, Cha AC, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U S A. 2011;108(12):5009–5014.
- Tang D, Lotze MT. Tumor immunity times out: TIM-3 and HMGB1. Nat Immunol. 2012;13(9):808–810.
- Andersson U, Ottestad W, Tracey KJ. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol Med. 2020;26(1):42.
- Curtin JF, Liu N, Candolfi M, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6(1):e10.
- Yang ZZ, Grote DM, Ziesmer SC, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest. 2012;122:1271–1282.
- Zhou Q, Munger ME, Veenstra RG, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501–4510.
- Wang Z, Zhu J, Gu H, et al. The clinical significance of abnormal Tim-3 expression on NK cells from patients with gastric cancer. Immunol Invest. 2015;44(6):578–589.
- Yu M, Lu B, Liu Y, et al. Tim-3 is upregulated in human colorectal carcinoma and associated with tumor progression. Mol Med Rep. 2017;15:689–695.
- da Silva IP, Gallois A, Jimenez-Baranda S, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014;2:410–422.
- Sabatos CA, Chakravarti S, Cha E, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4(11):1102–1110.
- Jones RB, Ndhlovu LC, Barbour JD, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205(12):2763–2779.
- Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–2186.
- Liu J, Zhang S, Hu Y, et al. Targeting PD-1 and Tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J Immunother. 2016;39:171–180.
- Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7(1):10501.
- Lu X, Yang L, Yao D, et al. Tumor antigen-specific CD8+ T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol. 2017;313:43–51.
- Gautron AS, Dominguez-Villar M, de Marcken M, et al. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol. 2014;44:2703–2711.
- Yan J, Zhang Y, Zhang JP, et al. Tim-3 expression defines regulatory T cells in human tumors. PLoS One. 2013;8:e58006.
- Liu JF, Wu L, Yang LL, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37:44.
- Schieber M, Crispino JD, Stein B. Myelofibrosis in 2019: moving beyond JAK2 inhibition. Blood Cancer J. 2019;9:74.
- Liu Y, Bewersdorf JP, Stahl M, et al. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: the dawn of a new era? Blood Rev. 2019;34:67–83.
- Agrawal V, Gbolahan OB, Stahl M, et al. Vaccine and cell-based therapeutic approaches in acute myeloid leukemia. Curr Cancer Drug Targets. 2020;20(7):473–489.
- Boddu P, Kantarjian H, Garcia-Manero G, et al. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk Lymphoma. 2018;59(4):790–802.
- Lamble AJ, Lind EF. Targeting the immune microenvironment in acute myeloid leukemia: a focus on T cell immunity. Front Oncol. 2018;8:213.
- Lamble AJ, Kosaka Y, Laderas T, et al. Reversible suppression of T cell function in the bone marrow microenvironment of acute myeloid leukemia. Proc Natl Acad Sci U S A. 2020 Jun;23(117):14331–14341.
- Veletic I, Prijic S, Manshouri T, et al. Altered T-cell subset repertoire affects treatment outcome of patients with myelofibrosis. Haematologica. 2020. DOI:10.3324/haematol.2020.249441.
- Zeidan AM, Knaus HA, Robinson TM, et al. A multi-center phase I trial of ipilimumab in patients with myelodysplastic syndromes following hypomethylating agent failure. Clin Cancer Res. 2018;24(15):3519–3527.
- Wendelbo O, Nesthus I, Sjo M, et al. Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy-induced leukopenia. Cancer Immunol Immunother. 2004;53:740–747.
- Garcia-Manero G, Sasaki K, Montalban-Bravo G, et al. A phase II study of nivolumab or ipilimumab with or without azacitidine for patients with myelodysplastic syndrome (MDS). Blood. 2018;132. abstract 465
- Garcia-Manero G, Tallman MS, Martinelli G, et al. Pembrolizumab, a PD-1 inhibitor, in patients with myelodysplastic syndrome (MDS) after failure of hypomethylating agent treatment. Blood. 2016;128. DOI:10.1182/blood.V128.22.345.345. abstract 345
- Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375(2):143–153.
- Daver N, Boddu P, Garcia-Manero G, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia. 2018;32(5):1094–1105.
- Khaznadar Z, Henry G, Setterblad N, et al. Acute myeloid leukemia impairs natural killer cells through the formation of a deficient cytotoxic immunological synapse. Eur J Immunol. 2014;44(10):3068–3080.
- Zeidan AM, Cavenagh J, Voso MT, et al. Efficacy and safety of azacitidine (AZA) in combination with the anti-PD-L1 durvalumab (durva) for the front-line treatment of older patients (pts) with acute myeloid leukemia (AML) who are unfit for intensive chemotherapy (IC) and pts with higher-risk myelodysplastic syndromes (HR-MDS): results from a large, international, randomized phase 2 study. Blood. 2019;134. abstract 829
- Zhou M, Sacirbegovic F, Zhao K, et al. T cell exhaustion and a failure in antigen presentation drive resistance to the graft-versus-leukemia effect. Nat Commun. 2020;11(1):4227.
- Krejcik J. van de Donk N. Trogocytosis represents a novel mechanism of action of daratumumab in multiple myeloma. Oncotarget. 2018;9:33621–33622.
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447.
- Vandsemb EN, Kim TK, Zeidan AM. Will deeper characterization of the landscape of immune checkpoint molecules in acute myeloid leukemia bone marrow lead to improved therapeutic targeting? Cancer. 2019;125(9):1410–1413.
- Bewersdorf JP, Shallis RM, Zeidan AM. Immune checkpoint inhibition in myeloid malignancies: moving beyond the PD-1/PD-L1 and CTLA-4 pathways. In: Blood Rev. Amsterdam, the Netherland: Elsevier; 2020. p. 100709.
- Kikushige Y, Miyamoto T. Identification of TIM-3 as a leukemic stem cell surface molecule in primary acute myeloid leukemia. Oncology. 2015;89(suppl 1):28–32.
- Kikushige Y, Miyamoto T. TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J Hematol. 2013;98(6):627–633.
- Asayama T, Tamura H, Ishibashi M, et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget. 2017;8(51):88904–88917.
- Dama P, Tang M, Fulton N, et al. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J Immunother Cancer. 2019;7:175.
- Kong Y, Zhang J, Claxton DF, et al. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J. 2015;5:e330.
- Gonçalves Silva I, Rüegg L, Gibbs BF, et al. The immune receptor Tim-3 acts as a trafficker in a Tim-3/galectin-9 autocrine loop in human myeloid leukemia cells. Oncoimmunology. 2016;5(7):e1195535.
- Dardalhon V, Anderson AC, Karman J, et al. Tim-3/Galectin-9 Pathway: regulation of Th1 Immunity through Promotion of CD11b + Ly-6G + Myeloid Cells. J Immunol. 2010;185(3):1383–1392.
- Williams P, Basu S, Garcia-Manero G, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer. 2019;125:1470–1481.
- Ogata K, Kakumoto K, Matsuda A, et al. Differences in blast immunophenotypes among disease types in myelodysplastic syndromes: a multicenter validation study. Leuk Res. 2012 Oct;36(10):1229–1236.
- Zhou Q, Munger ME, Highfill SL, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116:2484–2493.
- Wolff F, Leisch M, Greil R, et al. The double-edged sword of (re)expression of genes by hypomethylating agents: from viral mimicry to exploitation as priming agents for targeted immune checkpoint modulation. Cell Commun Signal. 2017;15(1):13.
- Liu L, Chang YJ, Xu LP, et al. T cell exhaustion characterized by compromised MHC class I and II restricted cytotoxic activity associates with acute B lymphoblastic leukemia relapse after allogeneic hematopoietic stem cell transplantation. Clin Immunol. 2018;190:32–40.
- Blaeschke F, Willier S, Stenger D, et al. Leukemia-induced dysfunctional TIM-3+CD4+ bone marrow T cells increase risk of relapse in pediatric B-precursor ALL patients. Leukemia. 2020;34(10):2607–2620.
- Rezazadeh H, Astaneh M, Tehrani M, et al. Blockade of PD-1 and TIM-3 immune checkpoints fails to restore the function of exhausted CD8+ T cells in early clinical stages of chronic lymphocytic leukemia. Immunol Res. 2020;68(5):269–279.
- Murga-Zamalloa CA, Brown NA, Wilcox RA. Expression of the checkpoint receptors LAG-3, TIM-3 and VISTA in peripheral T cell lymphomas. J Clin Pathol. 2020;73(4):197–203.
- Laken H, McEachern K, Murtaza A, et al. Discovery of TSR-022, a novel, potent anti-TIM-3 therapeutic antibody. Eur J Cancer. 2016;69. DOI:10.1016/S0959-8049(16)32902-1. abstract S2102
- Weiss GJ, Luke JL, Falchook G, et al. A phase 1 study of TSR-022, an anti-TIM-3 monoclonal antibody, in patients (pts) with advanced solid tumors. J Immunother Cancer. 2017;5. abstract O13
- Davar D, Boasberg P, Eroglu Z, et al. A phase 1 study of TSR-022, an anti-TIM-3 monoclonal antibody, in combination with TSR-042 (anti-PD-1) in patients with colorectal cancer and post-PD-1 NSCLC and melanoma. J Immunother Cancer. 2018;6. DOI:10.1186/s40425-018-0393-z. abstract O42
- Harding JJ, Patnaik A, Moreno V, et al. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J Clin Oncol. abstract 12. 2019;37(suppl 8). DOI:10.1200/JCO.2019.37.8_suppl.12
- Sabatos-Peyton CA MBG453: a high affinity, ligand-blocking anti-TIM-3 monoclonal Ab. Presented at: 107th AACR Annual Meeting; April 16-20, 2016; New Orleans, LA.
- Curigliano G, Gelderblom H, Mach N, et al. Phase (Ph) I/II study of MBG453 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Cancer Res. 2019;79:13suppl; abstract CT18
- Borate U, Esteve J, Porkka K, et al. Anti-TIM-3 antibody MBG453 in combination with hypomethylating agents in patients with high-risk myelodysplastic syndrome and acute myeloid leukemia: a phase 1 study. Paper presented at: 25th EHA Congress; June 11-21, 2020 [ abstract S185].
- Brunner A, Narayan R, Esteve J, et al. Post-transplant outcomes of patients with MDS and AML after receiving hypomethylating agent therapy combined with the TIM-3 inhibitor, MBG453. Poster presented at 25th EHA Congress; June 11-21, 2020 [ abstract EP828].
- Zeidan AM, Kim HJ, Miyazaki Y, et al. The STIMULUS clinical trial program: evaluating combination therapy with sabatolimab in patients with higher-risk myelodysplastic syndrome (HR-MDS) or acute myeloid leukemia (AML). Poster presented at 8th SOHO Annual Meeting; 2020 September 9-12 [ poster AML-187].
- Martins F, Sykiotis GP, Maillard M, et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol. 2019;20(1):e54–e64.
- He X, Feng Z, Ma J, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135(10):713–723.