
1. Introduction
A major downstream effector of RAS, c-RAF (RAF1) is considered the preferential RAF isoform responsible for supporting KRAS tumor initiation [Citation1–3]. Strengthening this narrative are two convincing pre-clinical studies, where-by systemic ablation of c-RAF induced robust tumor suppression in mouse (PDX and GEM) models of KRAS mutant (KRASMT) pancreatic and lung adenocarcinomas; with limited observable toxicity [Citation4,Citation5]. These studies, and others since [Citation6,Citation7], highlighted a non-essential role for c-RAF kinase activity in KRASMT tumor progression. Instead, c-RAF kinase-independent mechanisms were believed to be primarily responsible for the observed pro-oncogenic activity. Consequently, KRASMT cancer continues to be viewed as c-RAF ‘addicted’ and novel strategies to selectively targeting the inhibition of c-RAF (beyond its catalytic activity) are becoming increasingly attractive.
2. Targeting c-RAF catalytic activity
To date, no specific inhibitor of c-RAF has been developed. Existing small molecule kinase inhibitors capable of targeting c-RAF activity represent sub-optimal therapeutic approaches to treating RASMT cancer, and have been reviewed extensively () [Citation8–12].
Table 1. Mechanism of pharmacological inhibitors capable of exploiting c-RAF activity. Compounds in blue are FDA approved for the treatment of cancer. SM, small molecule; PD, peptide disruptor; TAT, cell-penetrating cationic peptide; st, stearic acid (short chain fatty acid).
Type I and I½ ATP-competitive RAF inhibitors (RAFi) are differentiated by their ability to bind RAF in a closed conformation presenting as either DFG-IN:αC-helix-IN or DFG-IN:αC-helix-OUT, respectively. These therapies are largely limited in their therapeutic utility to B-RAFV600E/K mutant cancer (i.e. constitutively active B-RAF monomers), where acquired resistance occurs within 12 months (even when combined with a downstream MEK inhibitor). In the presence of mutant RAS, type I and I½ RAFi have been shown to drive paradoxical MAPK activation through promoting dimerization of drug-bound (inhibited) B-RAF with unbound RAF (particularly c-RAF); fostering transactivation of the RAF dimer partner and hyperactivation of the MAPK pathway.
Type II RAFi were developed as a potential strategy to mitigate against paradoxical MAPK activation. Type II RAFi can be categorized as pan-RAF, binding to RAF in a DFG-OUT:αC-helix-IN conformation and targeting the inhibition of all kinase active RAF monomers and dimers with similar potency. However, clinical responses have been variable and dose-limiting toxicities are a concern (largely due to lack of selectivity compared with type I½ RAFi and indiscriminate binding of wild-type RAF).
Paradox breaker RAFi are derived from Type I½ RAFi, binding RAF in a DFG-IN:αC-helix-OUT confirmation. What separates paradox breakers from their earlier type I½ analogues is their ability to also strongly bind B-RAF at Leucine 505, adjacent to the RTKR motif of the αC-helix. Paradox breakers disrupt the B-RAF dimer interface, preventing B-RAF homodimerisation and B-RAF – c-RAF heterodimerisation. Although these inhibitors can prevent paradoxical MAPK activation, they cannot target c-RAF homodimers or c-RAF – A-RAF heterodimers. Therefore, paradox breaker RAFi are not considered a viable approach to treating RASMT driven cancers, where c-RAF homodimerisation and heterodimerisation with A-RAF is significantly upregulated vs. B-RAF homo/heterodimers [Citation7,Citation13].
Finally, RAF-MEK complex inhibitors – capable of disrupting RAF-MEK heterodimers or locking RAF-MEK in an inactive state – prove highly effective in abolishing RAF-driven MAPK activity. However, adverse side effects induced by these inhibitors are serious – resulting in significant dose-limiting efficacy in patients.
Given that most RAFi approaches are directed at inhibiting RAF kinase activity and preventing paradoxical MAPK activation, coupled with the consistent observation that c-RAF’s kinase-independent mechanisms are capable of driving RASMT tumourigenesis, novel strategies to exploiting c-RAF activity are urgently needed [Citation14].
3. Targeting c-RAF independent of its catalytic activity
Though our understanding of c-RAF’s kinase-independent mechanisms remain within their infancy, several studies have demonstrated c-RAF’s ability to regulate cellular apoptosis, cell cycle progression and cell migration, independent of its catalytic activity (). Said regulation is managed through specific c-RAF protein-protein interactions (PPIs) and has been shown to play a direct role in promoting RAS-driven oncogenesis [Citation8,Citation15,Citation16].
Figure 1. Diagram highlighting studied c-RAF kinase-dependent and kinase-independent signaling pathways. Created with BioRender.com.
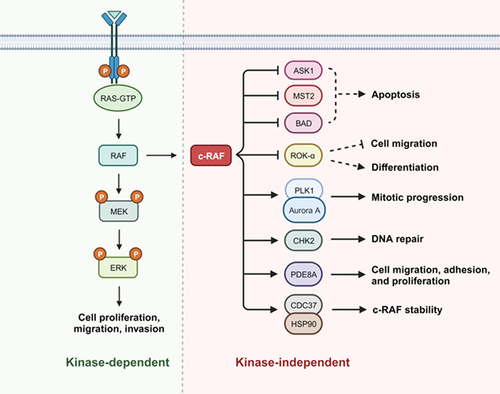
For instance, c-RAF possesses the ability to evade programmed cell death through its direct interaction with pro-apoptotic kinases ASK1 and MST2, and its indirect role as a scaffolding protein that promotes PKCθ-mediated inhibitory phosphorylation of BAD (Bcl-2 agonist of cell death).
Beyond its negative regulation of apoptosis, c-RAF can bind to and inhibit the activity of the cytoskeleton-based Rho effector kinase ROKα. c-RAF – ROKα PPI antagonizes ROKα, promoting cancer cell migration and survival. More, c-RAF – ROKα binding potentiates STAT3 and Myc expression, subsequently suppressing differentation (i.e. increased de-differentiation) and promoting RAS-induced epidermal tumourigenesis. Though no evidence of a direct interaction between c-RAF and STAT3 has been observed, increasing evidence of signaling cross-talk between c-RAF and STAT3 highlights a potential codependency in RAS – c-RAF dependent cancer [Citation17].
c-RAF can also regulate the cell cycle in a kinase-independent manner through its interaction with kinases PLK1 and Aurora A at the mitotic spindle. Phosphorylation of c-RAF at Ser338 potentiated the formation of this protein complex, consequently upregulating PLK1 kinase activity and promoting tumourigenesis through driving mitotic progression. Additionally, c-RAF Ser338 phosphorylation (but not its kinase activity) promotes the interaction with CHK2, a checkpoint kinase that regulates the cell cycle via DNA repair mechanisms and is directly linked with radiotherapeutic resistance.
Finally, recent characterization of the HSP90-CDC37-c-RAF complex highlights yet another approach to exploiting c-RAF activity [Citation18]. c-RAF binding to the HSP90-CDC37 chaperone complex is critical to stabilizing c-RAF in a folded, inactive conformation with 14-3-3 dimers. Disruption of this complex through site-directed mutagenesis promotes the degradation of c-RAF, inhibits cell proliferation, and highlights yet another vulnerability in c-RAF signaling; independent of its catalytic activity.
Selectively modulating PPIs within the oncoproteome represents a highly attractive and emerging therapeutic strategy, capable of fine-tuning the disease microenvironment and significantly reducing the potential for off-target toxicity [Citation19,Citation20]. However, given the significant size of the RAF PPI interactome, efforts aimed at characterizing c-RAF PPIs and defining their viability as drug targets are deficient [Citation21,Citation22]. This is particularly true for the above c-RAF kinase-independent targets, where PPI disruptor therapeutics are yet to be developed and evaluated in RASMT cancer. With that being said, a select number of pre-clinical compounds capable of modulating other c-RAF-associated PPIs have been developed, offering a differentiated approach to exploiting c-RAF activity vs. traditional kinase inhibitors ().
One such compound is MCP110, a small molecule derived from yeast two-hybrid screens that disrupts the interaction between RAS and c-RAF [Citation23,Citation24]. RAS – c-RAF disruption suppressed MEK-ERK activation inhibited the growth of KRASMT lung (A549 KRASG12S) and pancreatic (PANC1 KRASG12D) human cancer cell lines (not seen in A2058 BRAFV600E cells), and attenuated HT1080 (NRASQ61K) cell invasion.
TAT-Braftide, a cell-penetrating 10-mer peptide designed via an in silico screening strategy to block the intact dimer interface (DIF) of RAF, was developed as an allosteric approach to inhibiting B-RAF and prevent paradoxical MAPK activation [Citation25]. Unlike existing small molecule paradox breakers (e.g. PLX8394), TAT-Braftide-induced dimer disruption resulted in B-RAF and c-RAF protein degradation. RAF protein degradation was linked to the de-stabilization of the RAF-MAPK complex and was dependent upon the ubiquitin proteosome system. TAT-Braftide inhibited cell proliferation of two KRASG13D colorectal cancer human cell lines (HCT116, HCT-15).
Notably, no specific c-RAF kinase-independent mechanisms were evaluated with MCP110 or TAT-Braftide. However, given the effectiveness of these compounds in cellular models of RASMT cancer, it could be suggested that these compounds are negatively regulating c-RAF beyond its kinase-dependent activity. Thus, further investigation is justified.
Disruption of the c-RAF – PDE8A PPI represents another targeted approach to negatively regulating c-RAF activity [Citation26,Citation27]. PDE8A protects c-RAF from the surrounding cAMP microenvironment, blocking inhibitory PKA phosphorylation and enabling pro-oncogenic c-RAF activity. Targeted disruption of the c-RAF – PDE8A PPI with a cell-penetrating disruptor peptide was shown to displace PDE8A, subsequently upregulating PKA-mediated inhibitory phosphorylation of c-RAF at Ser259 (other inhibitory PKA phospho sites on c-RAF were not assessed: Ser43, Ser233, Ser621). Said disruptor was derived from the c-RAF binding epitope on PDE8A, and was later demonstrated to have a therapeutic role in overcoming BRAF inhibitor-resistant NRASQ61L malignant melanoma. Resultingly, efforts to evaluate the therapeutic relevance of c-RAF – PDE8A disruption in other RAS-RAFMT cancer models is on-going. Whether disrupting the cross-talk between c-RAF and the cAMP-PKA signaling axes influences c-RAF kinase-independent mechanisms is yet to be elucidated. However, it is worth noting that disrupting the c-RAF – PDE8A complex was shown to inhibit T cell adhesion and motility; a finding that may translate in RASMT cancer cell models [Citation28].
Key to stabilizing c-RAF in a closed-inactive conformation is its binding with the scaffolding protein 14-3-3. 14-3-3 dimers bind to phospho-serines on c-RAF, Ser233, Ser259 and Ser621 (e.g. following cAMP activated PKA phosphorylation), structurally constraining c-RAF in a closed-inactive conformation; occluding the catalytic site. Thus, Kenanova and colleagues set out to identify compounds capable of increasing 14-3-3-mediated PPI stabilization, including 14-3-3 – c-RAF stabilizers [Citation29]. Utilising a disulfide tethering – FBDD (fragment-based drug discovery) approach, they developed small molecule mimetics of a known 14-3-3 binding c-RAF phospho-peptide (Q255RSTpSTPNVH264). These mimetics act as molecular glues, selectively stabilizing the c-RAF − 14-3-3 complex. Unfortunately, the activity of these mimetics in cellular models of RASMT cancer are yet to be evaluated and warrant investigation.
4. C-RAF dependency and RAS mutant heterogeneity
Activating RAS mutations are detected in over 20% of human cancers, predominantly occuring in three hotspot locations: G12, G13 or Q61 [Citation30]. The pro-oncogenic regulatory behavior(s) of mutant RAS is remarkably complex, depending on which RAS family member is mutated (K/N/HRAS), what specific RAS amino acid is substituted, and what secondary mutations exist [Citation31]. Such heterogeneity in the RASMT landscape will undoubtedly influence the degree to which a given cancer is dependent upon c-RAF. Therefore, systematic characterization of the relationship between RAS mutational status and c-RAF dependency is critical to the effective development of next-generation c-RAF inhibitors against appropriately stratified patient cohorts.
Utilising publicly available bioinformatic datasets available through the DepMap Portal database (https://depmap.org/portal/), further insights into RAS-RAF mutational status and relative c-RAF dependency can be made (). Firstly, human cancer cell lines harboring a KRASMT, NRASMT or HRASMT are significantly more dependent upon c-RAF than RAS-RAFwild-type (). This was also observed in RASG12 and RASQ61 mutant cancer cell lines, but not RASG13 or RASother (i.e. non-G12, G13 or Q61, ). Secondly, B-RAFV600 mutant cancer cell lines were expectedly less dependent upon c-RAF (). However, no significant differences between c-RAF dependency and A-RAFMT, B-RAFnon-V600 or c-RAFMT were observed (). Moreover, no correlation () was shown between RAS-RAF mutational status and c-RAF gene expression (), c-RAF protein expression () or c-RAF phospho-Ser338 protein expression (, representative of relative c-RAF kinase activity). These data not only suggest that targeted degradation of c-RAF is unlikely to prove efficacious in these RAFMT cancer cell lines, but that relative c-RAF gene expression, protein expression and kinase activity represent unsuitable biomarkers for predicting c-RAF inhibitor sensitivity (). On the other hand, RASMT cancer cell lines are significantly more c-RAF dependent than wild-type (). However, the extensive variability within all human cancer cell cohorts (including RAS-RAFwild-type) cannot be ignored (). Therefore, given the far from straight forward relationship between RAS mutational status and c-RAF dependency, it is unlikely that RASMT can be wielded as a standalone marker for future c-RAF inhibitor therapies.
Figure 2. Utilisation of a large heterogenous panel of human cancer cell lines (derived from > 25 cancer lineages), generated from the publicly available bioinformatic DepMap portal, to compare RAS-RAF mutational status and respective: (a) c-RAF dependency (RNAi, gene effect, DEMETER2, achilles + DRIVE + Marcotte), (b) c-RAF gene expression (public, 23Q2, Log2(TPM+1)), (c) c-RAF total protein expression (protein array, RPPA signal, Log2), and (d) phosphorylated serine 338 (pS338) c-RAF protein expression (protein array, RPPA signal, Log2). Data represented as MEAN ± STDEV. *p < 0.05; **p < 0.01; ****p < 0.001; ns, not significant vs. RAS-RAF WT (one-way ANOVA). # refers to number of cells per sub-group.
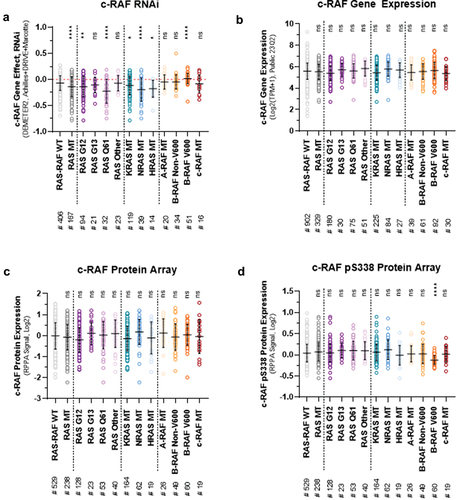
Table 2. Correlation between c-RAF RNAi (achilles + DRIVE + Marcotte, DEMETER2), c-RAF CRISPR knockout (public, 23Q2 + score, chronos), c-RAF gene expression (23Q2, Log2(TPM+1)), c-RAF total protein array (BD610151, RPPA, Log2), and c-RAF pS338 protein array (RPPA, Log2). Table represents data generated and published in DepMap Portals publicly available bioinformatic data sets. Pcoeff, Pearson’s Coefficient; Scoeff, Spearman’s Coefficient; #, No. Of data points (i.e. no. Of cell lines assessed).
5. Concluding remarks
The need for novel approaches to effectively exploiting pro-oncogenic c-RAF activity in RASMT cancer, beyond simply its catalytic activity, is clearer than ever. Selectively targeting pro-oncogenic c-RAF PPIs, and/or inducing the degradation of c-RAF completely, represent increasingly attractive therapeutic strategies in the context of RASMT – c-RAF-dependent cancer. However, succesful development of these next-generation c-RAF inhibitors are wholey dependent upon accurate characterization of the complex RASMT – c-RAF relationship. Unlocking key details in this relationship will not only allow for identification of actionable c-RAF-associated vulnerabilities in RASMT cancer, but guide future synergistic combination strategies capable of overcoming challenges associated with therapeutic resistance; including that observed with current RASMT-specific small molecule inhibitors. When considering c-RAF as a target in RASMT cancer, this time – the goal posts appear to of widened.
Declaration of interest
C Blair is the co-founder of Disruptyx Therapeutics Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contribution
C Blair wrote the article, analyzed the bioinformatic data and made the figures/tables.
S Cooke supported C Blair in writing the article and making the figures/tables.
Supplemental Material
Download Zip (242.8 KB)Acknowledgments
We aknowledge DepMap Portal, a publicly available bioinformatic database, containing in-depth polyomic characterisation of a large heterogenous library of human cancer cell lines. Data from this database was yielded to support the narrative of the manuscript submitted.
Data availability statement
All data is available within article figures, tables and supplementary files.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14737140.2024.2319035
Additional information
Funding
References
- Ehrenreiter K, Kern F, Velamoor V, et al. Raf-1 addiction in Ras induced skin carcinogenesis. Cancer Cell. 2009;16:149–160. doi: 10.1016/j.ccr.2009.06.008
- Karreth FA, Frese KK, DeNicola GM, et al. And Tuveson, D.A.C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov. 2011;1:128–136. doi: 10.1158/2159-8290.CD-10-0044
- Blasco RB, Francoz S, Santamaria D, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogenedriven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–663. doi: 10.1016/j.ccr.2011.04.002
- Sanclemente M, Francoz S, Esteban-Burgos L, et al. C-RAF ablation induces regression of advanced Kras/Trp53 mutant lung adenocarcinomas by a mechanism Independent of MAPK signaling. Cancer Cell. 2018;33(2):217–228. doi: 10.1016/j.ccell.2017.12.014
- Blasco MT, Navas C, Martin-Serrano G, et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and C-RAF. Cancer Cell. 2019;35(4):573–587. doi: 10.1016/j.ccell.2019.03.002
- Sanclemente M, Nieto P, Garcia-Alonso S, et al. RAF1 kinase activity is dispensable for KRAS/p53 mutant lung tumor progression. Cancer Cell. 2021;39(3):294–296. doi: 10.1016/j.ccell.2021.01.008
- Venkatanarayan A, Liang J, Yen I, et al. CRAF dimerization with ARAF regulates KRAS-driven tumor growth. Cell Rep. 2022;38(6):110351. doi: 10.1016/j.celrep.2022.110351
- Riaud M, Maxwell J, Soria-Bretones I, et al. The role of CRAF in cancer progression: from molecular mechanisms to precision therapies. Nat Rev Cancer. 2024;24(2):105–122. doi: 10.1038/s41568-023-00650-x
- Man RJ, Zhang YL, Jiang AQ, et al. A patent review of RAF kinase inhibitors (2010-2018). Expert Opin Ther Pat. 2019;29(9):675–688. doi:10.1080/13543776.2019.1651842
- Cook FA, Cook SJ. Inhibition of RAF dimers: it takes two to tango. Biochem Soc Trans. 2021;49(1):237–251. doi: 10.1042/BST20200485
- Degiremenci U, Yap J, Sim YRM, et al. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resist. 2021;4(3):665–683. doi: 10.20517/cdr.2021.36
- Pinzi L. On the development of B-Raf inhibitors acting through innovative mechanisms. F1000 Research. 2022;11:237. doi: 10.12688/f1000research.108761.2
- Vasta JD, Michaud A, Zimprich CA, et al. Protomer selectivity of type II RAF inhibitors within the RAS/RAF complex. Cell Chem Bio. 2023;S2451-9456(23):00247–7.
- McCormick F. C-Raf in Kras mutant cancers: a moving target. Cancer Cell. 2018;33(2):158–159. doi: 10.1016/j.ccell.2018.01.017
- Matallanas D, Birtwhilstle M, Romano D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2(3):22–260. doi: 10.1177/1947601911407323
- Nolan AA, Aboud NK, Kolch W, et al. Hidden targets in RAF signalling pathways to block oncogenic RAS signalling. Genes (Basel). 2021;12(4):553. doi:10.3390/genes12040553
- Dorard C, Madry C, Buhard O, et al. RAF1 contributes to cell proliferation and STAT3 activation in colorectal cancer independently of microsatellite and KRAS status. Oncogene. 2023;42(20):1649–1660. doi: 10.1038/s41388-023-02683-w
- Garcia-Alonso S, Mesa P, Ovejero LP, et al. Structure of the RAF1-HSP90-CDC37 complex reveals the basis of RAF1 regulation. Mol Cell. 2022;82(18):3438–3452. doi: 10.1016/j.molcel.2022.08.012
- Lu H, Zhou Q, He J, et al. Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct Target Ther. 2020;5(1):213. doi: 10.1038/s41392-020-00315-3
- Sorolla A, Wang E, Golden E, et al. Precision medicine by designer interference peptides: applications in oncology and molecular therapeutics. Oncogene. 2020;39(6):1167–1184. doi: 10.1038/s41388-019-1056-3
- An S, Yang Y, Ward R, et al. Raf-interactome in tuning the complexity and diversity of raf function. FEBS J. 2014;282(1):32–53. doi: 10.1111/febs.13113
- Iglesias-Martinez LF, Rauch N, Wynne K, et al. Interactome dynamics of RAF1-BRAF kinase monomers and dimers. Sci Data. 2023;10(1):203. doi: 10.1038/s41597-023-02115-0
- Kato-Stankiewicz J, Hakimi I, Zhi G, et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. PNAS. 2002;99(22):14398–14403. doi: 10.1073/pnas.222222699
- González-Pérez V, Reiner DJ, Alan JK, et al. Genetic and functional characterization of putative Ras/Raf interaction inhibitors in C. elegans and mammalian cells. J Mol Signal. 2010;5(2):2. doi: 10.1186/1750-2187-5-2
- Gunderwala AY, Nimbvikar AA, Cope NJ, et al. Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem Biol. 2019;14(7):1471–1480. doi: 10.1021/acschembio.9b00191
- Brown KM, Day JP, Huston E, et al. Phosphodiesterase-8A binds to and regulates raf-1 kinase. Proc Nat Acad Sci. 2013;110(16):E1533–E1542. doi: 10.1073/pnas.1303004110
- Blair CM, Walsh NM, Littman BH, et al. Targeting B-Raf inhibitor resistant melanoma with novel cell penetrating peptide disruptors of PDE8A - C-Raf. BMC Cancer. 2019;19(1):266. doi: 10.1186/s12885-019-5489-4
- Basole CP, Nguyen RK, Lamothe K, et al. PDE8 controls CD4+ T cell motility through the PDE8A-Raf-1 kinase signaling complex. Cell Signal. 2017;40:62–72. doi: 10.1016/j.cellsig.2017.08.007
- Kenanova DN, Visser EJ, Virta JM, et al. A systemic approach to discovery of protein-protein interaction stabilizers. ACS Cent Sci. 2023;9(5):937–946. doi: 10.1021/acscentsci.2c01449
- Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res. 2020;80(14):2969–2974. doi: 10.1158/0008-5472.CAN-19-3682
- Sealover NE, Kortum RL. Heterogeneity in RAS mutations: One size does not fit all. Sci Signal. 2022;15(746):eadc9816. doi: 10.1126/scisignal.adc9816