
ABSTRACT
Introduction: Advances in the biology of non-small-cell lung cancer, especially adenocarcinoma, reveal multiple molecular subtypes driving oncogenesis. Accordingly, individualized targeted therapeutics are based on mutational diagnostics.
Areas covered: Advances in strategies and techniques for individualized treatment, particularly of adenocarcinoma, are described through literature review. Approved therapies are established for some molecular subsets, with new driver mutations emerging that represent increasing proportions of patients. Actionable mutations are de novo oncogenic drivers or acquired resistance mediators, and mutational profiling is important for directing therapy. Patients should be monitored for emerging actionable resistance mutations. Liquid biopsy and associated multiplex diagnostics will be important means to monitor patients during treatment.
Expert commentary: Outcomes with targeted agents may be improved by integrating mutation screens during treatment to optimize subsequent therapy. In order for this to be translated into impactful patient benefit, appropriate platforms and strategies need to be optimized and then implemented universally.
1. Introduction
Lung cancer is the most frequently diagnosed cancer and a leading cause of cancer death worldwide [Citation1]. Non-small-cell lung cancer (NSCLC) is the most commonly diagnosed form of the disease (>85% of cases) and includes a heterogeneous group of histologies, the most common being adenocarcinoma, then squamous cell carcinoma, and less so large-cell carcinoma [Citation2]. These histologies possess different clinical characteristics, and there are potential differences in response to cytotoxic chemotherapies. Approximately 40–50% of patients with NSCLC will be diagnosed with advanced or metastatic disease and are not candidates for curative therapy. Systemic chemotherapy, once the treatment of choice for all patients, is no longer universally used following the advent of targeted therapy. For example, randomized Phase III trials showed a significant benefit (response rate and progression-free survival [PFS]) in patients with epidermal growth factor receptor (EGFR)-mutant disease treated with tyrosine kinase inhibitors (TKIs) versus those treated with standard chemotherapy [Citation3]. Further advances in the underlying biology of NSCLC have revealed multiple distinct molecular subtypes, increasingly supporting a model in which NSCLCs depend on oncogenic ‘driver mutations’ for the malignant phenotype [Citation4]. Along with mutations in EGFR, gene fusions involving rearrangements of the anaplastic lymphoma kinase (ALK) gene are prominent genetic markers. Personalized therapy aims at matching these genotyped lung adenocarcinomas with effective targeted therapies such as specific TKIs and is currently utilized; EGFR TKIs are US FDA approved for the first-line treatment of EGFR-mutant NSCLC, and ALK-rearranged NSCLC may be treated first-line with the multi-targeted ALK/MET/ROS1 (ROS proto-oncogene 1, receptor tyrosine kinase [RTK]) TKI crizotinib [Citation5,Citation6] and second-line with ceritinib [Citation7].
Molecular testing for EGFR and ALK is now considered standard of care [Citation2,Citation4], with other driver mutations in oncogenes such as ROS1, BRAF (v-Raf murine sarcoma viral oncogene homolog B), RET (rearranged during transfection), MEK1 (mitogen-activated protein kinase kinase 1), NTRK (neurotrophic tyrosine kinase receptor), MET, and KRAS (Kirsten Rat Sarcoma viral oncogene homolog), also increasingly being incorporated into the diagnostic workup of adenocarcinoma patients to determine eligibility for enrollment in diverse clinical studies on appropriate targeted agents. Reflecting these developments, current guidelines for advanced NSCLC treatment from the American Society for Clinical Oncology (ASCO) [Citation8], College of American Pathologists (CAP)/International Association for the Study of Lung Cancer (IASLC)/Association for Molecular Pathology [Citation3], and the US National Comprehensive Cancer Network [Citation2] support testing on tumor tissue to determine any genetic alterations and choose an appropriate therapy. Molecular testing for EGFR mutations and ALK rearrangement are recommended in the treatment guidelines, and further molecular testing may be appropriate depending on tissue availability and clinical criteria. As the number of molecular subgroups of NSCLC continues to grow and the methods for their detection improve, there is a need to review recent developments. This review gathers together recent data on driver mutations, discusses their characterization in the clinical diagnostic setting, and their impact on potential first- and second-line monotherapy and combination therapy decisions for patients with NSCLC.
This article will summarize some of the mutations that are ‘actionable’ in NSCLC. Certainly, there is a large momentum for immunotherapy in NSCLC; however, the reader is referred elsewhere for further understanding of this.
2. Initial testing for mutations and expression patterns
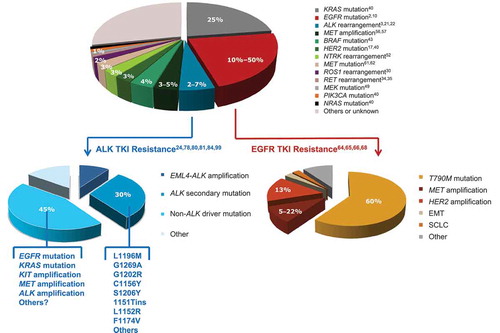
In addition to EGFR and ALK, important oncogenic driver mutations/rearrangements that may be considered for diagnostic screening include KRAS, MET, PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha), HER2 (human epidermal growth factor receptor 2), BRAF, NTRK, ROS1, RET, and MEK. Most driver oncogenes tend to occur in ≤25% of individual tumors (see for a chart showing some of the more frequent actionable mutations) and singly in tumor samples; for example, clinical data show that overlapping EGFR and KRAS mutations occur in <1% of patients with lung cancer [Citation2] and ALK rearrangements are usually mutually exclusive with mutations in EGFR or KRAS [Citation9].
Figure 1. Frequency of mutations/genomic alterations in NSCLC (adenocarcinoma) in Caucasian populations, and known mutation profiles in ALK and EGFR TKI-resistant disease. ALK: anaplastic lymphoma kinase; BRAF: v-Raf murine sarcoma viral oncogene homolog B; EGFR: epidermal growth factor receptor; EML4: echinoderm microtubule-associated protein-like 4; EMT: epithelial-mesenchymal transition; HER2: human epidermal growth factor receptor 2; KRAS: Kirsten Rat Sarcoma viral oncogene homolog; MEK: mitogen-activated protein kinase kinase; NTRK: neurotrophic tyrosine kinase receptor; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; RET: rearranged during transfection; ROS1: ROS proto-oncogene 1, receptor tyrosine kinase; SCLC: small-cell lung carcinoma; TKI: tyrosine kinase inhibitor.
2.1. ERBB family RTKs
Sensitizing EGFR mutations are found in around 10% of Caucasian patients and up to 50% of Asian patients with NSCLC [Citation2,Citation10]. The most frequent EGFR mutations result in substitution at amino acid 858 in exon 21 (Leu858Arg [L858R]) and in-frame deletions at exon 19, which alter the configuration of the kinase to preserve an activated state. Patients whose tumors have exon 19 deletions or exon 18 (G719X, G719A, G719S, G719C, G719D), exon 20 (S768I), or exon 21 (L858R, L861Q, L861R) mutations are sensitive to EGFR–TKI therapy [Citation11–Citation13]. Erlotinib is approved by the US FDA (2013) for the first-line treatment for patients with metastatic NSCLC harboring EGFR exon 19 deletions or exon 21 (L858R) substitution mutations based on a response rate of 65% compared with 16% for platinum-based chemotherapy and a median PFS of 10.4 versus 5.2 months [Citation14]. Erlotinib is also approved for maintenance treatment of locally advanced or metastatic NSCLC after platinum-based chemotherapy. Afatinib and gefitinib are now also fully US FDA-approved (2013 and 2015, respectively) for the first-line treatment of patients with the same types of EGFR-mutant NSCLC [Citation15,Citation16].
De novo mutations in HER2 typically occur in 3‒5% of NSCLC (predominantly exon 20 insertions) and are usually mutually exclusive with EGFR and KRAS mutations [Citation17,Citation18]. Clinical trials have not yet demonstrated a clear benefit, but HER2-targeted therapies such as afatinib have demonstrated signs of clinical activity in heavily pretreated patients with HER2-mutated adenocarcinoma [Citation19], including activity in patients with HER2-mutated lung cancers with exon 20 YVMA insertions, the most common variant [Citation20].
2.2. ALK
ALK gene rearrangements are present in approximately 2–7% of patients with NSCLC, typically fusions with other genes (most commonly, EML4 [echinoderm microtubule-associated protein-like 4]) [Citation3,Citation21,Citation22]. Patients diagnosed with lung tumors harboring ALK fusions can be effectively treated with ALK inhibitors, such as crizotinib [Citation3,Citation21,Citation22]. Phase I and II studies of crizotinib in ALK-rearranged NSCLC demonstrated impressive activity and clinical benefit, leading to FDA approval in 2011 [Citation23,Citation24]. In addition, a subsequent Phase III trial showed that crizotinib was superior to standard first-line pemetrexed-plus-platinum chemotherapy in patients with previously untreated advanced ALK+ NSCLC [Citation6]. However, despite high response rates (65–74%), most patients develop resistance to crizotinib within 2 years, and second-generation agents are now FDA approved (ceritinib, alectinib) or in advanced development as FDA Breakthrough Designated Therapy (brigatinib) [Citation24–Citation26]. It should be noted that as ALK+ patients are surviving longer, relapses within the central nervous system (CNS) are increasingly being diagnosed. The penetration of the blood–brain barrier by these therapeutic agents will therefore be important in controlling CNS metastases [Citation27,Citation28]. It is also interesting to note that there are unique patterns of metastases in ALK+ tumors, especially in women with metastases to the adnexa [Citation29].
2.3. ROS1
Chromosomal rearrangements of the gene encoding the ALK-related tyrosine kinase ROS1 have been identified in 1–2% of NSCLC cases that generate several distinct gene fusion partners, including SLC34A2 (solute carrier family 34 [type II sodium/phosphate cotransporter], member 2), TPM3 (tropomyosin 3), and others [Citation30–Citation32]. ROS1 rearrangements are relatively more prevalent in patients with adenocarcinoma and advanced stage disease [Citation33]. For patients with tumors with ROS1 rearrangements, crizotinib is effective and is therefore a potential first-line therapy [Citation8,Citation32].
2.4. RET
Translocations of the RTK RET occur in approximately 1% of NSCLC patients, with relatively high frequencies in young, light/never smokers with adenocarcinoma and poorly differentiated tumors [Citation34–Citation36]. A number of fusion variants are now known, the most common of which is KIF5B-RET [Citation34,Citation37,Citation38]. A Phase II trial of the RET inhibitor cabozantinib in patients with RET-positive NSCLC has shown preliminary efficacy [Citation39]. Multiple clinical trials in NSCLC with KIF5B-RET rearrangements using existing RET inhibitors (including cabozantinib, lenvatinib, vandetanib, sunitinib, ponatinib, and AUY922) are underway [Citation36].
2.5. KRAS
KRAS mutations are detected in approximately 25% of lung adenocarcinomas and 4% of lung squamous cell carcinomas, most often in codons 12 or 13 [Citation40]. KRAS mutations are most common in non-Asians and smokers [Citation41,Citation42] and are associated with intrinsic EGFR TKI resistance. KRAS mutation testing may thus identify patients who may not benefit from further molecular diagnostic testing. No direct targeted therapy is available for KRAS-mutant NSCLC, and therefore, investigations have focused on targeting downstream signaling proteins of the RAS/RAF/MEK/ERK (extracellular signal-regulated kinase) pathway, such as BRAF and MEK. The incidence of synthetic lethality associated with KRAS mutation means that these pathways need to be explored further.
2.6. BRAF
BRAF is a serine–threonine kinase belonging to the RAF kinase family lying downstream of KRAS and directly interacts with the MEK–ERK signaling cascade. BRAF mutations are found in up to 4% of lung adenocarcinomas [Citation43], half of them harboring the V600E mutation; other mutations occur within exons 11 and 15 [Citation44–Citation46]. Treatment of BRAF V600E-mutated NSCLC with BRAF inhibitor monotherapy, exemplified by dabrafenib, has demonstrated encouraging antitumor activity in early clinical trials [Citation43,Citation47]. Dabrafenib was granted FDA Breakthrough Therapy Designation (2014) for chemotherapy-pretreated BRAF V600E mutation-positive NSCLC [Citation48].
2.7. MEK
MEK1 encodes a serine–threonine kinase and is mutated in about 1% of NSCLC, largely adenocarcinoma [Citation49]. Several MEK inhibitors are in clinical development for NSCLC, including selumetinib (AZD6244) and trametinib (GSK1120212), with a focus on combination regimens that may prevent or combat resistance due to secondary MEK mutation and/or KRAS/BRAF amplification in BRAF- or KRAS-mutant NSCLC [Citation50,Citation51].
2.8. NTRK
NTRK1 fusions occur in around 3% of lung adenocarcinomas [Citation52]. Amplifications of NTRK1 and NTRK2 (among other genes) encode Src kinases that can complement loss of EGFR activity across multiple EGFR-dependent models, via EGFR-independent activation of the MEK–ERK and phosphoinositide 3-kinase (PI3K)-AKT pathways, suggesting a range of kinases capable of overcoming dependence on EGFR [Citation53]. Significant antitumor activity has been reported for the kinase inhibitor entrectinib (RXDX-101) in a patient with NSCLC harboring an SQSTM1 (sequestosome 1)-NTRK1 gene rearrangement, validating NTRK gene rearrangements as a potential clinical target in NSCLC [Citation54].
2.9. MET
MET is an RTK that binds to hepatocyte growth factor. Activation of MET promotes signaling pathway activation, including the RAS-RAF-mitogen-activated protein kinase (MAPK) and PI3K-AKT-mTOR (mammalian target of rapamycin) pathways. MET amplifications are found in 3–5% of newly diagnosed NSCLC, predominantly in adenocarcinoma [Citation55–Citation58]. Exon 14 skipping MET mutations have also been identified as oncogenic drivers, as initially discovered by the author’s laboratory in lung cancer [Citation59,Citation60], occurring in around 3% of lung adenocarcinomas and in 1–2% of other NSCLC subsets [Citation61,Citation62], and respond to MET inhibitors such as crizotinib, cabozantinib, and capmatinib (INC280) [Citation61–Citation63]. MET amplification and overexpression may confer resistance to EGFR inhibitors in EGFR-mutant lung adenocarcinoma [Citation64,Citation65].
3. Mutation patterns in relapsed patients
Acquired resistance to targeted TKIs can occur by several mechanisms during treatment, and mutations that enable escape of dependence on the initial oncogenic driver present important second-line diagnostic targets [Citation66]. Identification of resistance mutations or pathway activation upon progression is increasingly important as new and specific treatment options emerge.
3.1. EGFR
Most patients treated with EGFR TKIs will progress after about 1 year of therapy due to acquired resistance that is generally mediated through persistence of MAPK signaling and largely due to mutations in exons 19 and 20 (most commonly, the T790M mutation; ) [Citation66–Citation68]. Secondary mutations in EGFR may be targeted by treatment with second- and third-generation TKIs [Citation68]. The FDA has recently granted accelerated approval to the EGFR TKI osimertinib (AZD9291) for patients with advanced T790M mutation-positive NSCLC based on response rates of 57–61% in two single-arm studies [Citation13,Citation69]. Other agents in clinical development include rociletinib (CO-1686) that is active against both the T790M mutation as well as the baseline activating EGFR mutations [Citation70], and in a first-in-human study of the EGFR-mutant-specific TKI EGF816, an overall response rate (ORR) of 55% and disease control rate of 86% were reported [Citation71]. Other potentially targetable EGFR TKI resistance mechanisms related to MAPK signaling that future re-biopsy diagnostics will need to detect to inform on salvage treatment strategies include amplification of HER2 or MEK1 and activating mutations in RAS or BRAF [Citation72–Citation74]. Bypass activation of other pathways also plays important roles in resistance, and includes amplification of MET [Citation64,Citation66], and acquired mutation of PIK3CA [Citation74]. The MET gene is amplified in up to 21% of NSCLC cases with EGFR inhibitor resistance [Citation65]. Acquired resistance to next-generation EGFR TKIs may emerge through increased ERK activation (via MEK1 amplification or mutation), and downstream inhibitors of this pathway such as those already described may be effective in this setting when these aberrations are detected on progression [Citation72]. RET rearrangement has also been implicated in EGFR-mutant NSCLC that has progressed on EGFR TKI therapy, and this genetic aberration should be added to the growing list of potential markers in genetic resistance screens [Citation75]. There is also evidence that with TKI resistance, there can be change of histology (to small-cell lung cancer or squamous cell carcinoma) [Citation76,Citation77].
3.2. ALK
Although crizotinib provides impressive initial responses, resistance develops within 1–2 years [Citation24]. Several bypass mechanisms have been implicated, including ALK amplification, MET activation, KIT amplification, and mutations in MAPK pathway signaling components () [Citation78–Citation81]. However, the major mechanism of resistance is through any one of a multitude of known secondary ALK mutations that either induce changes at the ATP-binding pocket and cause steric hindrance to binding of crizotinib, or destabilize the wild-type auto-inhibitory conformation of ALK to which crizotinib binds [Citation80,Citation82–Citation86]. The most commonly occurring mutations associated with crizotinib resistance are L1196M (the ‘gatekeeper’ mutation) and G1269A (see for a more comprehensive list) [Citation86,Citation87]. Mutational screens involving Ba/F3 cells expressing native EML4–ALK have identified more resistance loci [Citation88], and additional types of mutation may emerge as more patients are treated with ALK inhibitors. New-generation ALK inhibitors such as ceritinib can overcome ALK mutation-derived resistance to crizotinib [Citation78,Citation89]. In a Phase I study of ceritinib in ALK+ NSCLC, marked antitumor activity was seen in both crizotinib-relapsed and crizotinib-naïve patients (i.e. regardless of the presence of resistance mutations in ALK) [Citation7], and based on these data, ceritinib received FDA approval in 2014; confirmatory Phase II data have now been reported, including an ORR of 36% and PFS of 7.2 months in crizotinib-pretreated patients [Citation90,Citation91]. In a retrospective study of a cohort of ALK+ patients treated with crizotinib and ceritinib, data showed that ceritinib had significant antitumor activity in ALK+ NSCLC, even when crizotinib immediately preceded treatment with ceritinib; the median combined PFS for sequential treatment with crizotinib and ceritinib was 17.4 months [Citation92]. Other ALK inhibitors are in advanced clinical development with activity in crizotinib-resistant NSCLC patients; alectinib and brigatinib provide ORRs of 45–71% in patients who have progressed on crizotinib treatment [Citation93–Citation96]. However, new-generation ALK inhibitors may, in turn, induce secondary resistance mutations, for which new drugs will have to be designed [Citation78,Citation79,Citation97,Citation98]. Despite initial durable responses to ceritinib in crizotinib-resistant patients, tumors eventually develop resistance to ceritinib. Biopsies from crizotinib-resistant tumors that progressed on ceritinib showed eradication of ceritinib-sensitive mutations (S1206Y, G1269A) and the emergence of the cross-resistant G1202R mutation, which is also associated with clinical resistance to alectinib and crizotinib [Citation78,Citation87,Citation99]. In a study of 11 patients with acquired resistance, 5 patient biopsies had either G1202 or F1174 mutations and the remaining 6 biopsies had wild-type EML4–ALK with no mutation [Citation78]. The profile of ALK resistance mutations shifts depending on the ALK inhibitor, and accurate screening to match the mutational profile of tumors with the appropriate ALK inhibitor is likely to be important to maximize benefit for patients who relapse on ALK inhibitor therapy by directing subsequent sequential or combination therapy [Citation100]. Rare and complex mutational profiles have also been encountered in ALK inhibitor drug-resistant ALK+ NSCLC, including KRAS Q22K mutation and STK11 frameshift mutations; this highlights the importance of comprehensive molecular testing in progressing patients [Citation101]. The heterogeneity of ALK inhibitor resistance mechanisms means that effective monitoring of genetic changes during treatment will be important in treating or preventing the emergence of resistance.
3.3. ROS1
Crizotinib is a recommended treatment for ROS1-rearranged NSCLC [Citation102]; however, secondary mutations in ROS1 causing resistance to crizotinib have also been reported [Citation103,Citation104]. Other ALK/ROS1 inhibitors may prove effective as second-line options, although cell-based resistance profiling studies demonstrate that ROS1-selective inhibitors retain efficacy against the mutant, whereas the dual ROS1/ALK inhibitors are ineffective [Citation105]. Lorlatinib (PF-06463922), a new dual ROS1/ALK inhibitor, blocks such crizotinib-resistant mutations in preclinical studies [Citation106].
3.4. BRAF
Acquired resistance to BRAF inhibitors can occur through MAPK pathway reactivation due to a number of genetic aberrations, including BRAF V600E amplification, alternate splicing of BRAF, NRAS mutation, KRAS mutation, and MEK1 mutation [Citation107–Citation109]. Co-inhibition of BRAF and MEK may overcome resistance, and the combination of dabrafenib and trametinib has provided a response rate of 68% in BRAF-mutant NSCLC, providing the basis for FDA Breakthrough Therapy Designation (2015) for this combination regimen in this indication [Citation110]. Dual MEK–ERK inhibitors exhibit additive/synergistic effects and can delay the emergence of, and potentially overcome, acquired MEK inhibitor resistance [Citation51]. PIK3CA mutations have also been implicated in resistance to BRAF inhibitors, and diagnostic detection of this mutation during therapy may thus direct decisions on subsequent PI3K inhibitor combination therapy [Citation111].
3.5. MET
Preclinical models suggest that resistance to MET inhibitors may be mediated by KRAS amplification and overexpression that is potentially targetable with MAPK pathway inhibitors [Citation112]. Secondary MET mutations may also be targetable in the event of effective appropriate diagnostic signals [Citation113].
4. Optimal reanalysis of tumor genetic profiles
In a recent study of molecular and histologic changes in NSCLC tumors (N = 50) posttreatment, in the second tissue sampling, 54% of cases had additional genomic changes, including newly acquired alterations (81%) or losses (18%) [Citation114]. As demonstrated by the breadth and heterogeneity of potential resistance mechanisms to diverse targeted therapies, re-biopsy of growing tumors following disease progression has become increasingly important for prognosis and to direct a change in therapy [Citation66]. However, detecting mutations in resistant tumors can be challenging due to limited tissue availability; obtaining tissue from an outside institution in a timely fashion and delays in confirmation of tissue adequacy have been reported as important issues for oncologists [Citation68]. Biopsies may be required during treatment, and both ethical and bureaucratic obstacles need to be removed so that molecular testing procedures can be carried out, where required, on very sick patients. Communication between disciplines, and particularly between pathologists and treating oncologists, is paramount to ensure that appropriate tests are carried out on precious and limited biopsy material [Citation115,Citation116]. Molecular testing should be prioritized and immunostaining limited where feasible, yet sampling procedures need to be minimally invasive while still providing sufficient material for both morphologic and mutational analysis [Citation68,Citation115]. Cell blocks prepared from malignant effusions can be a useful alternative to core biopsy [Citation68]. Histologic/cytologic assessments may be required to locate tissue for macrodissection and ensure adequate tumor content above the sensitivity level of testing methods (sensitive testing methods are discussed below) [Citation117]. Aspirates may be preferable to core biopsies for obtaining tumor material from bone metastases, due to the relative ease in obtaining DNA of adequate quality for analysis; alternative tissue sampling for genetic profiling may be desirable in many cases [Citation68].
Liquid biopsies are assuming increasing importance as a noninvasive means of performing dynamic genetic surveillance in patients receiving targeted therapy treatment where acquired resistance may be expected and may overcome problems associated with tumor heterogeneity, as has been reported for ALK+ NSCLC small biopsy and excision samples [Citation118]. During treatment and at the time of progression, circulating tumor (ct)DNA, circulating tumor cells (CTCs), and microRNAs (miRNAs) can be used to monitor treatment response noninvasively, and track and reveal molecular mechanisms of resistance [Citation119]. This disease-related genetic information can be obtained through the analysis of ctDNA in the blood of NSCLC patients [Citation120,Citation121]. Real-world data from a large multicenter clinical study also suggest ctDNA samples are suitable for upfront EGFR mutation analysis when tumor samples are unavailable [Citation122]. Nonetheless, robust and sensitive analysis methods are recommended to minimize false negative results. CTCs themselves may also be analyzed through liquid biopsy for individualizing and monitoring treatments and resistance [Citation123]. Changes in miRNAs from liquid biopsies are predictive of response and may also help monitor resistance to treatment and detect progression early [Citation124,Citation125]. A number of methods are available for liquid biopsy analysis (there is currently no formal consensus on preferred techniques), and turnaround times need to be optimized so that treatment decisions can be made quickly when resistance to ongoing targeted therapy needs to be addressed urgently. Liquid biopsy allows detection of emerging mutations during treatment but cannot always be relied upon alone to identify all potential resistance mechanisms; for example, overexpression of potential bypass pathway driver proteins would not be identified and re-biopsy at progression would still be required in such cases where resistance mutations have not been detected in relapsed patients. Although having potential utility in the dynamic monitoring for resistance mutations in previously diagnosed patients, liquid biopsy techniques are some way from routine clinical application as a primary diagnostic tool. Future developments will need to address the sensitivity of the method for application in asymptomatic or as yet undiagnosed patients.
5. Optimal diagnostics for routine mutation screening
A wide variety of commercially available molecular assays may be used to detect mutations in lung adenocarcinomas. An ideal assay is sensitive and specific enough to comprehensively cover all clinically relevant targets using limited samples, while being cost-effective and efficient. The method of choice depends on the type of mutation to be detected, the scale of throughput expected, and the type of sample available.
5.1. Direct Sanger sequencing
DNA mutational analysis via direct (traditional Sanger) sequencing is considered the gold standard for characterizing mutations and is generally performed on polymerase chain reaction (PCR) products using sequencing primers spanning the DNA region of interest [Citation117]. Direct sequencing is the standard to detect EGFR mutations for determining whether patients are eligible for first-line EGFR–TKI therapy [Citation3,Citation126,Citation127]. This can be targeted to specific mutations or aimed at screening or scanning larger regions [Citation128]; direct sequencing of DNA corresponding to exons 18–21 of the EGFR gene is a reasonable initial approach [Citation3]. Targeted assays are available from various manufacturers, for example the non-digital Cobas EGFR Mutation Test (Roche Molecular Systems) and the Therascreen EGFR Kit (Qiagen) [Citation117,Citation129].
Additional clinically relevant mutations implicated in resistance can be added to these assays, for example a new version of the Cobas test adds the T790M mutation to those detected in the original test and has been recently approved as a companion diagnostic test for osimertinib [Citation130]. Digital PCR of plasma cell-free DNA has been successful for noninvasive detection of drug resistance mechanisms in EGFR-mutant NSCLC and detects the T790M mutation with 82% sensitivity and 86% specificity; the method was less successful in detecting MET gene copy number gain in plasma DNA [Citation131]. Multiplex droplet digital PCR has been used to detect low-frequency mutations, such as KRAS point mutations, with a detection limit that compares favorably with next-generation sequencing (NGS; also known as massively parallel sequencing [MPS]) and Sanger sequencing [Citation132]. Direct sequencing is limited by its low sensitivity, and it is estimated that a mutation should be present in ~20% of the sampled DNA to be reliably detected [Citation128]. High-resolution melt analysis may be an alternative to direct sequencing that has provided 100% sensitivity and specificity in detecting EGFR mutations in surgically resected NSCLC [Citation133].
5.2. Multiplex screening technologies
Screening technologies such as NGS and pyrosequencing have the potential to detect all EGFR mutations and allow detection of these and other mutations in tumor samples at levels as low as 5% [Citation117]. Targeted NGS/exome sequencing enriches the target of interest and allows higher coverage, read depth, or simultaneous detection of mutations across different genes, using material from small biopsies and cytological samples. The CellSearch System coupled with NGS has proved successful in EGFR mutation analysis of CTCs in the Phase II erlotinib (TRIGGER) study [Citation134]. More sensitive methods (supported by joint CAP/IASLC/ASCO guidelines) [Citation3] are able to detect a range of ‘actionable’ mutations occurring with a frequency of ≥1%. These include the Sequenom MassARRAY system and SNaPshot Multiplex System (Life Technologies/Applied Biosystems), and these methods are supported by clinical guidelines [Citation3,Citation117,Citation135]. In patients with NSCLC who experienced a treatment failure in response to EGFR–TKIs and had new biopsies, T790M, and other mutations outside EGFR have been successfully detected using NGS technology, by DNA sequencing on an Ion Torrent Personal Genome Machine (PGM) system (the Ion AmpliSeq Cancer Hotspot Panel version 2) [Citation136]. Acquired T790M resistance mutations were detected in 60% of patients; other non-EGFR mutations identified included TP53 P72R mutations (87%), KDR Q472H (33%), and KIT M541L (13%). PCR-based NGS was thus able to detect EGFR T790M mutations in cases not readily diagnosed by other conventional methods. In addition, the detection of coexisting oncogenic mutations that may play a role in acquired EGFR–TKI resistance may help direct alternative treatment strategies in relapsed patients [Citation136]. The Ion Torrent PGM system was clinically validated in a retrospective study of 39 NSCLC samples (and 51 colorectal cancer samples), interrogating 1850 hotspots in 22 genes [Citation137]. Sensitivity and accuracy for detecting variants at an allelic frequency (AF) >4% was 100% for commercial reference standards, and the concordance between NGS and the reference test (EGFR mutation was used for the NSCLC samples) was >95%. The AmpliSeq panel was thus specific and sensitive for mutation analysis of gene panels and may be suitable for incorporation into clinical daily practice [Citation137].
MPS methodologies are now being developed for the detection of gene rearrangements (e.g. ALK, ROS1, RET), as well as gene mutations in single-tube assays [Citation138,Citation139]. A novel multiplexed transcript-based assay, the Nanostring nCounter, detects overexpression of the 3′ end of transcripts versus the 5′ end, common in fusion genes [Citation117]. Whole-transcriptome sequencing has been used to detect RET fusion oncogenes [Citation38]. Microdissected tumor samples and cytology samples (including fine needle aspirates, pleural effusion) can be used to detect rare mutations if sensitive testing methods are used [Citation128]. However, small tissue samples often preclude microdissection and may be prone to false negative results; equally, with high-sensitivity assays, clinical laboratories must be extremely careful to guard against false positive results [Citation68]. NGS and parallel-sequencing technologies enable efficient, simultaneous detection of driver and drug-targeted mutations in NSCLC. GS Junior-based sequencing has been used to screen samples for multiple mutations across several driver genes [Citation140], and similarly, MiSeq™, GS Junior, and PGM Ion Torrent™ have been validated across several centers [Citation141]. NGS applied to formalin-fixed paraffin-embedded tissue has become established as a routine diagnostic tool in some centers [Citation142].
5.3. In situ hybridization techniques
For ALK, ROS1, and RET fusions, fluorescence in situ hybridization (FISH) is widely used in clinical laboratories and requires only a single paraffin section [Citation117]. It is the current gold standard for the detection of ALK rearrangements regardless of the fusion partner, although it cannot identify the fusion, and is not feasible for large-scale screening of low-frequency rearrangements [Citation117]. ALK gene rearrangements can be detected using the dual probe break-apart FISH assay, approved by the FDA as a prerequisite before treatment with crizotinib [Citation3,Citation143]. FISH has also been used to evaluate gene amplification in tissue microarray sections, including MET [Citation57]. Immunohistochemistry (IHC) or reverse transcription (RT)-PCR can also be used for detection of specific fusion transcripts [Citation22,Citation143]. IHC offers an alternative and universally available option for pathology laboratories unable to carry out FISH. Fusion specific RT-PCR kits are commercially available. Combined with Sanger sequencing or NGS of PCR products, RT-PCR allows specific identification of the fusion partners; however, novel translocations may be missed, and RNA in clinical samples may be of poor quality [Citation117].
6. Therapeutic approaches to combat resistance based on multiple mutation screening
With dozens of known targets and many agents approved or in development, there are a large number of possible combinations and sequences that can potentially be investigated or deployed in to treat or prevent drug-resistant patients in the clinic, often with support from preclinical models [Citation144]. Several Phase III studies of combined targeted agents are underway. Patient-derived models of acquired resistance have provided valuable data that help identify effective drug combinations and could help direct combination therapy strategies in individual patients that are not predicted by genetic analysis alone [Citation145]. Combinations of targeted therapies based on resistance mechanisms are currently being evaluated in a variety of lung cancer settings in Phase I and II studies (clinicaltrials.gov; January 2016). For example, cyclin-dependent kinases CDK4 and CDK6 are components of cell cycle control that switch on potential resistance-mediating RAS/RAF/MEK/ERK and PI3K/AKT/mTOR bypass signaling pathways in ALK+ NSCLC. The combination of ceritinib and the CDK4/6 inhibitor ribociclib (LEE011) is being evaluated in patients with ALK+ NSCLC who have progressed on an ALK inhibitor (including ceritinib) or who are ALK inhibitor-naïve. A study of the MEK inhibitor selumetinib (AZD6244) and gefitinib in patients with EGFR-mutated NSCLC and EGFR TKI resistance is ongoing (NCT02025114). The MET inhibitor capmatinib (INC280) is being evaluated in combination with gefitinib (NCT01610336) and erlotinib (NCT02468661) in patients with EGFR-mutated, MET-amplified NSCLC who have acquired resistance to an EGFR TKI, and with the EGFR-mutant-specific irreversible EGFR inhibitor EGF816 in EGFR-mutant NSCLC (NCT02335944). The combination of gefitinib and the PI3K inhibitor buparlisib (BKM120) is being evaluated in patients with NSCLC who have EGFR TKI resistance and molecular alterations in the PI3K pathway, such as PIK3CA mutation (NCT01570296). A study in patients with EGFR-mutated NSCLC who have EGFR TKI resistance will evaluate the TORC1/2 inhibitor INK128 in combination with the third-generation irreversible EGFR TKI AZD9291 (NCT02503722). Combination therapy is also being evaluated with the HER2-targeted therapy dacomitinib (NCT01918761). The increasing number of targetable mutations poses challenges for clinical study design to allow accurate efficacy assessment, particularly with respect to combination therapies of multiple targeted drugs (with or without conventional chemotherapy) and drug sequences [Citation142]. The resulting high complexity, requiring unconventional study designs and analyzing small patient pools, may potentially limit approval [Citation142].
For patients who have progressed without a defined mutation or one with no known targeted therapy, and/or the presence of other markers (e.g. programmed death-ligand-1), the advent of immunotherapy provides an alternative option to conventional chemotherapy. However, targeted therapies may increase sensitivity to immunotherapy and the combination of a targeted agent with immunotherapy may combat or prevent the emergence of resistance; this has been reviewed elsewhere [Citation146,Citation147]. Immunotherapy is outside the scope of this review, but a large number of clinical studies are now ongoing to evaluate how to optimize its merger with targeted therapy in lung cancer patients () [Citation148].
Table 1. Ongoing clinical trials evaluating immunotherapy combined with targeted therapy in patients with NSCLC [Citation148].
7. Expert commentary
Significant advances in molecular pathology in recent years have improved our understanding of NSCLC, with actionable oncogenic mutations in multiple signaling pathways identified, leading to the rational use of targeted agents tailored to tumor characteristics. Based on this approach, agents targeted to specific EGFR or ALK mutations have become widely used. Although reflex molecular testing for driver mutations is now commonplace, for those patients who have not been comprehensively screened or suboptimal diagnostics applied, no known driver mutation will be documented and routine treatment will be standard cytotoxic chemotherapy. Outcomes with targeted agents in patients with known actionable mutations have been impressive, but acquired resistance is our next major challenge – both in terms of treatment and effective monitoring. Outcomes with targeted agents will likely be improved by integrating increasing numbers of mutation screens at the point of treatment failure or even during treatment through repeated liquid biopsies, to allow subsequent therapy to be optimized. A recommended potential treatment and testing algorithm that combines existing and potential new diagnostic strategies is represented in . Multiple clinical trials are ongoing or planned to investigate sequencing and combinations based on tailored therapy and resistance mechanisms, and results will hopefully lead to further survival improvements for patients with advanced NSCLC. In order for the results of these studies to be translated into impactful patient benefit in the real world, appropriate biopsy and diagnostic platforms and strategies will need to be implemented universally. By introducing treatment paradigms which shift the diagnostic emphasis toward both upfront pretreatment testing and timely monitoring for potential actionable resistance mutations, the continued use of inappropriate treatments may be minimized. At the same time, the clinical effectiveness of diverse targeted treatments as potent personalized treatment options for the patients who will derive the most benefit may be maximized.
Figure 2. Flow chart recommending where mutational analysis can fit (white boxes) into current NSCLC treatment algorithms to direct therapy first line or in resistant disease; current recommended treatment flow is in shaded boxes and is based on current ASCO and NCCN Guidelines [Citation8]. CT: chemotherapy; IT: immunotherapy; NGS: next-generation sequencing; PS: performance status; SD: stable disease; TKI: tyrosine kinase inhibitor.
![Figure 2. Flow chart recommending where mutational analysis can fit (white boxes) into current NSCLC treatment algorithms to direct therapy first line or in resistant disease; current recommended treatment flow is in shaded boxes and is based on current ASCO and NCCN Guidelines [Citation8]. CT: chemotherapy; IT: immunotherapy; NGS: next-generation sequencing; PS: performance status; SD: stable disease; TKI: tyrosine kinase inhibitor.](/cms/asset/6d9cf498-f370-4ceb-bc2c-dc9fdb4efe1c/iero_a_1181545_f0002_c.jpg)
8. Five-year view
The spectrum of currently available approved targeted therapies is still limited, and therefore, the justification for comprehensive evaluation of mutation status of vast numbers of oncogenes not just in clinical trials, but in routine clinical practice is still controversial. However, as the pace of research into new lung cancer driver and/or resistance mutations rapidly accelerates, this is unlikely to be the case in the near future. The key challenge will be in the implementation of highly complex techniques such as NGS in a uniform and preferentially centralized way that is accessible to all patients. This uniformity will need to include the software used for evaluation of results and recommended guidelines on cutoffs and minimum read numbers required per mutation tested, as well as the basic platforms used. NGS techniques as routine diagnostic tools in the real world are currently limited by specialist sample preparation requirements and costs associated with both these techniques and the widespread application of commercial platforms in routine community clinical practice. The optimization of the above factors will need to be carried out without compromising turnaround times. Nonetheless, the potential benefit to a large proportion of patients is exemplified by , which shows that up to 30% of patients with adenocarcinoma are likely to have tumors bearing a known actionable genetic mutation, and even higher ratios in some resistance settings, with others more than likely to be discovered over the next 5 years. A further key future challenge will be in moving these diagnostic and treatment algorithms forward in the management timeline of patients with NSCLC, so that patients with earlier stage disease may also benefit from targeted personalized therapies. This may lead to improved cure rates in NSCLC, in addition to already improved survival for patients with advanced disease. Ultimately, with all of the technological developments, we will have a strong impact on the cure for lung cancer.
Key issues
In addition to EGFR and ALK, important oncogenic driver mutations/rearrangements that may be considered for diagnostic screening include KRAS, MET, PIK3CA, HER2, BRAF, NTRK, ROS1, RET, and MEK
Acquired resistance to targeted agents occur by several mechanisms during treatment, and mutations that enable escape of dependence on the initial oncogenic driver present important second-line diagnostic targets
Identification of resistance mutations or pathway activation upon progression is increasingly important as new and specific treatment options emerge
In post treatment second tissue samples over half of NSCLC cases have additional genomic changes, including newly acquired alterations or losses; re-biopsy is thus important for prognosis, and to direct a change in therapy, but can be challenging due to limited tissue availability and quality
Liquid biopsies are important as a non invasive means of performing dynamic genetic surveillance in patients receiving targeted therapy treatment where acquired resistance may be expected, and may overcome problems associated with tumor heterogeneity
Circulating tumor DNA, circulating tumor cells, and microRNAs can be used to monitor treatment response non invasively, and track and reveal molecular mechanisms of resistance
DNA mutational analysis via direct sequencing is considered the gold standard for characterizing mutations, and can be targeted to specific mutations including those associated with resistance, or aimed at screening or scanning larger regions
Next-generation sequencing and pyrosequencing have the potential for simultaneous detection of mutations across different genes, including gene rearrangements, using material from small biopsies and cytological samples
Patient-derived models of acquired resistance have provided valuable data that help identify effective drug combinations, and could help direct combination therapy strategies in individual patients that are not predicted by genetic analysis alone
Combinations of targeted therapies based on resistance mechanisms are currently being evaluated in a variety of lung cancer settings
Declaration of interests
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Editorial writing assistance was provided by Matthew Naylor, funded by Novartis Pharmaceuticals Corporation.
References
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
- Ettinger DS, Wood DE, Akerley W, et al. Non-small cell lung cancer, version 6.2015. J Natl Compr Canc Netw. 2015;13(5):515–524.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn. 2013;15(4):415–453.
- Kris MG, Johnson BE, Kwatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with adenocarcinoma: The NCI’s Cancer Mutation Consortium (LCMC). J Clin Oncol. 2011;29(suppl. 18). Abstract CRA7506.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394.
- Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177.
- Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197.
- Masters GA, Temin S, Azzoli CG, et al. Systemic therapy for stage IV non-small-cell lung cancer: American society of clinical oncology clinical practice guideline update. J Clin Oncol. 2015;33(30):3488–3515.
- Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(15):4273–4281.
- Sequist LV, Bell DW, Lynch TJ, et al. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587–595.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246.
- Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141–151.
- Khozin S, Blumenthal GM, Jiang X, et al. U.S. Food and Drug Administration approval summary: erlotinib for the first-line treatment of metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations. Oncologist. 2014;19(7):774–779.
- Boehringer-Ingelheim press release: FDA approves GILOTRIFTM (afatinib) as first-line treatment for metastatic non-small cell lung cancer with common EGFR mutations. [cited 2016 Jan 21]. Avaible from: http://us.boehringer-ingelheim.com/news_events/press_releases/press_release_archive/2013/07-12-13-fda-approves-gilotrif-afatinib-first-line-treatment-metastatic-non-small-cell-lung-cancer-common-egfr-mutations.html.
- FDA news release: FDA approves targeted therapy for first-line treatment of patients with a type of metastatic lung cancer. [cited 2016 Jan 21]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm454678.htm.
- Arcila ME, Chaft JE, Nafa K, et al. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 2012;18(18):4910–4918.
- Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65(5):1642–1646.
- De Greve J, Moran T, Graas MP, et al. Phase II study of afatinib, an irreversible ERBB family blocker, in demographically and genotypically defined lung adenocarcinoma. Lung Cancer. 2015;88(1):63–69.
- Li BT, Lee A, O’Toole S, et al. HER2 insertion YVMA mutant lung cancer: long natural history and response to afatinib. Lung Cancer. 2015;90(3):617–619.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703.
- Ali G, Proietti A, Pelliccioni S, et al. ALK rearrangement in a large series of consecutive non-small cell lung cancers: comparison between a new immunohistochemical approach and fluorescence in situ hybridization for the screening of patients eligible for crizotinib treatment. Arch Pathol Lab Med. 2014;138(11):1449–1458.
- Shaw AT, Solomon B, Kenudson MM. Crizotinib and testing for ALK. J Natl Compr Canc Netw. 2011;9(12):1335–1341.
- Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31(8):1105–1111.
- Santarpia M, Altavilla G, Rosell R. Alectinib: a selective, next-generation ALK inhibitor for treatment of ALK-rearranged non-small-cell lung cancer. Expert Rev Respir Med. 2015;9(3):255–268.
- Ariad research and development: about brigatinib (AP26113). [cited 2016 Jan 21]. Available from: http://www.ariad.com/AP26113.
- Toyokawa G, Seto T, Takenoyama M, et al. Insights into brain metastasis in patients with ALK+ lung cancer: is the brain truly a sanctuary? Cancer Metastasis Rev. 2015;34(4):797–805.
- Zhang I, Zaorsky NG, Palmer JD, et al. Targeting brain metastases in ALK-rearranged non-small-cell lung cancer. Lancet Oncol. 2015;16(13):e510–521.
- West AH, Yamada SD, MacMahon H, et al. Unique metastases of ALK mutated lung cancer activated to the adnexa of the uterus. Case Rep Clin Pathol. 2014;1(2):151–154.
- Heist RS, Engelman JA. SnapShot: non-small cell lung cancer. Cancer Cell. 2012;21(3):448.e2.
- Ou SH, Tan J, Yen Y, et al. ROS1 as a ‘druggable’ receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther. 2012;12(4):447–456.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–1971.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res. 2015;4(3):300–309.
- Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381.
- Wang R, Hu H, Pan Y, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30(35):4352–4359.
- Song M, Kim SH, Yoon SK. Cabozantinib for the treatment of non-small cell lung cancer with KIF5B-RET fusion. An example of swift repositioning. Arch Pharm Res. 2015;38(12):2120–2123.
- Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18(3):382–384.
- Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375–377.
- Drilon A, Wang L, Hasanovic A, et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3(6):630–635.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998–2006.
- Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28(30):4616–4620.
- Riely GJ, Kris MG, Rosenbaum D, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008;14(18):5731–5734.
- Nguyen-Ngoc T, Bouchaab H, Adjei AA, et al. BRAF alterations as therapeutic targets in non-small-cell lung cancer. J Thorac Oncol. 2015;10(10):1396–1403.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29(15):2046–2051.
- Marchetti A, Felicioni L, Malatesta S, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29(26):3574–3579.
- Naoki K, Chen TH, Richards WG, et al. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 2002;62(23):7001–7003.
- Planchard D, Kim TM, Mazieres J. Dabrafenib in patients with BRAF V600E-mutant advanced NSCLC: A multicenter, open-label, Phase II trial (BRF113928). Ann Oncol. 2014;25 (suppl. 4). 1-41-LBA38_PR
- Inman S OncLive: dabrafenib receives breakthrough designation for NSCLC. [cited 2016 Jan 21]. Available from: http://www.onclive.com/web-exclusives/Dabrafenib-Receives-Breakthrough-Designation-for-NSCLC#sthash.ggLpXltM.dpuf.
- Marks JL, Gong Y, Chitale D, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68(14):5524–5528.
- Wang H, Daouti S, Li WH, et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011;71(16):5535–5545.
- Hatzivassiliou G, Liu B, O’Brien C, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11(5):1143–1154.
- Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19(11):1469–1472.
- Sharifnia T, Rusu V, Piccioni F, et al. Genetic modifiers of EGFR dependence in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2014;111(52):18661–18666.
- Farago AF, Le LP, Zheng Z, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol. 2015;10(12):1670–1674.
- Kawakami H, Okamoto I, Okamoto W, et al. Targeting MET amplification as a new oncogenic driver. Cancers (Basel). 2014;6(3):1540–1552.
- Beau-Faller M, Ruppert AM, Voegeli AC, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol. 2008;3(4):331–339.
- Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667–1674.
- Jagadeeswaran R, Surawska H, Krishnaswamy S, et al. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res. 2008;68(1):132–142.
- Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65(4):1479–1488.
- Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63(19):6272–6281.
- Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–859.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–849.
- Jenkins RW, Oxnard GR, Elkin S, et al. Response to crizotinib in a patient with lung adenocarcinoma harboring a MET splice site mutation. Clin Lung Cancer. 2015;16(5):e101–104.
- Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043.
- Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247.
- Lovly CM, Horn L. Strategies for overcoming EGFR resistance in the treatment of advanced-stage NSCLC. Curr Treat Options Oncol. 2012;13(4):516–526.
- Oxnard GR, Arcila ME, Chmielecki J, et al. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011;17(17):5530–5537.
- Mitsudomi T, Tsai C, Shepherd FA, et al. AZD9291 in pre-treated T790M positive advanced NSCLC: AURA2 Phase II study. Presented at the 16th World Conference on Lung Cancer; 2015 Sep 6–9; Denver, CO. Abstract 1406.
- Sequist LV, Goldman JW, Wakelee HA. Efficacy of rociletinib (CO-1686) in plasma-genotyped T790M-positive non-small cell lung cancer (NSCLC) patients (pts). J Clin Oncol. 2015;33(suppl):8001.
- Tan DS, Seto T, Leighl NB. First-in-human phase I study of EGF816, a third generation, mutant-selective EGFR tyrosine kinase inhibitor, in advanced non-small cell lung cancer (NSCLC) harboring T790M. J Clin Oncol. 2015;33(suppl):8013.
- Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2(10):934–947.
- Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–133.
- Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17.
- Klempner SJ, Bazhenova LA, Braiteh FS, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer. 2015;89(3):357–359.
- Alam N, Gustafson KS, Ladanyi M, et al. Small-cell carcinoma with an epidermal growth factor receptor mutation in a never-smoker with gefitinib-responsive adenocarcinoma of the lung. Clin Lung Cancer. 2010;11(5):E1–4.
- Kuiper JL, Ronden MI, Becker A, et al. Transformation to a squamous cell carcinoma phenotype of an EGFR-mutated NSCLC patient after treatment with an EGFR-tyrosine kinase inhibitor. J Clin Pathol. 2015;68(4):320–321.
- Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673.
- Ramalingam SS, Khuri FR. Second-generation ALK inhibitors: filling the non “MET” gap. Cancer Discov. 2014;4(6):634–636.
- Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012;18(5):1472–1482.
- Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120): 120ra17.
- Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739.
- Sun HY, Ji FQ. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem Biophys Res Commun. 2012;423(2):319–324.
- Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051–6060.
- Puig De La Bellacasa R, Karachaliou N, Estrada-Tejedor R, et al. ALK and ROS1 as a joint target for the treatment of lung cancer: a review. Transl Lung Cancer Res. 2013;2(2):72–86.
- Roskoski R Jr. Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res. 2013;68(1):68–94.
- Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. 2014;12(7):429–439.
- Zhang S, Wang F, Keats J, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78(6):999–1005.
- Ni Z, Zhang TC. Computationally unraveling how ceritinib overcomes drug-resistance mutations in ALK-rearranged lung cancer. J Mol Model. 2015;21(7):175.
- Mok TS, Spigel DR, Felip E. ASCEND-2: A single-arm, open-label, multicenter phase II study of ceritinib in adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (CRZ). J Clin Oncol. 2015;33(suppl):8059.
- Felip E, Orlov S, Park K. ASCEND-3: A single-arm, open-label, multicenter phase II study of ceritinib in ALKi-naïve adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC). J Clin Oncol. 2015;33(suppl):8060.
- Gainor JF, Tan DS, De Pas T, et al. Progression-free and overall survival in ALK-positive NSCLC patients treated with sequential crizotinib and ceritinib. Clin. Cancer Res. 2015;21(12):2745–2752.
- Hotta K, Hida T, Nakagawa K, et al. Updated data from JP28927 study of alectinib in ALK+ NSCLC patients with or without history of ALK inhibitor treatment. Presented at the 16th World Conference on Lung Cancer; 2015 Sep 6–9; Denver, CO. Abstract P3.01-020.
- Barlesi F, Dingemans AMC, Ou I et al. Updated efficacy and safety results from a global phase 2, open-label, single-arm study (NP28673) of alectinib in crizotinib-refractory ALK+ non-small cell lung cancer (NSCLC). Presented at the European Society for Clinical Oncology European Cancer Congress; 2015 Sep 25–29; Vienna, Austria. Abstract 3101.
- Shaw A, West H, Socinski MA et al. Updated efficacy/safety data from the phase 2 NP28761 study of alectinib in ALK+ NSCLC. Presented at the 16th World Conference on Lung Cancer; 2015 Sep 6–9; Denver, CO. Abstract 33.03.
- Gettinger SN, Bazhenova LA, Salgia R et al. Brigatinib (AP26113) efficacy and safety in ALK+ NSCLC: phase 1/2 trial results. Presented at the 16th World Conference on Lung Cancer; 2015 Sep 6–9; Denver, CO. Abstract 33.06.
- Toyokawa G, Inamasu E, Shimamatsu S, et al. Identification of a novel ALK G1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J Thorac Oncol. 2015;10(7):e55–57.
- Ou SH, Greenbowe J, Khan ZU, et al. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer. 2015;88(2):231–234.
- Ignatius Ou SH, Azada M, Hsiang DJ, et al. Next-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014;9(4):549–553.
- Politi K, Gettinger S. Perfect ALKemy: optimizing the use of ALK-directed therapies in lung cancer. Clin. Cancer Res. 2014;20(22):5576–5578.
- Azzato EM, Deshpande C, Aikawa V, et al. Rare complex mutational profile in an ALK inhibitor-resistant non-small cell lung cancer. Anticancer Res. 2015;35(5):3007–3012.
- NCCN. Clinical practice. Guidelines in oncology. Non-small Cell Lung Cancer, Version. 2015;5: NSCL-16 and NSCL-H.
- Sun H, Li Y, Tian S, et al. P-loop conformation governed crizotinib resistance in G2032R-mutated ROS1 tyrosine kinase: clues from free energy landscape. PLoS Comput Biol. 2014;10(7):e1003729.
- Awad MM, Katayama R, McTigue M, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–2401.
- Davare MA, Vellore NA, Wagner JP, et al. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci U S A. 2015;112(39):E5381–390.
- Zou HY, Li Q, Engstrom LD, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. 2015;112(11):3493–3498.
- Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096.
- Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724.
- Rudin CM, Hong K, Streit M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol. 2013;8(5):e41–42.
- Planchard D, Groen HJM, Kim TM. Interim results of a phase II study of the BRAF inhibitor dabrafenib in combination with the MEK inhibitor trametinib in patients with BRAF V600E-mutated NSCLC. J Clin Oncol. 2015;33(suppl):8006.
- Falchook GS, Trent JC, Heinrich MC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4(2):310–315.
- Cepero V, Sierra JR, Corso S, et al. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res. 2010;70(19):7580–7590.
- Tiedt R, Degenkolbe E, Furet P, et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011;71(15):5255–5264.
- Vatrano S, Righi L, Vavala T, et al. Molecular and histological changes in post-treatment biopsies of non-squamous non-small cell lung cancer: a retrospective study. Target Oncol. 2016;11(2):157–166.
- Aisner DL, Marshall CB. Molecular pathology of non-small cell lung cancer: a practical guide. Am J Clin Pathol. 2012;138(3):332–346.
- Salgia R. Diagnostic challenges in non-small-cell lung cancer: an integrated medicine approach. Future Oncol. 2015;11(3):489–500.
- Khoo C, Rogers TM, Fellowes A, et al. Molecular methods for somatic mutation testing in lung adenocarcinoma: EGFR and beyond. Transl Lung Cancer Res. 2015;4(2):126–141.
- Abe H, Kawahara A, Azuma K, et al. Heterogeneity of anaplastic lymphoma kinase gene rearrangement in non-small-cell lung carcinomas: a comparative study between small biopsy and excision samples. J Thorac Oncol. 2015;10(5):800–805.
- Sun W, Yuan X, Tian Y, et al. Non-invasive approaches to monitor EGFR-TKI treatment in non-small-cell lung cancer. J Hematol Oncol. 2015;8:95. doi:10.1186/s13045-015-0193-6
- Jiang T, Ren S, Zhou C. Role of circulating-tumor DNA analysis in non-small cell lung cancer. Lung Cancer. 2015;90(2):128–134.
- Pasquale R, Fenizia F, Esposito Abate R, et al. Assessment of high-sensitive methods for the detection of EGFR mutations in circulating free tumor DNA from NSCLC patients. Pharmacogenomics. 2015;16(10):1135–1148.
- Reck M, Hagiwara K, Han B, et al. Investigating the utility of circulating-free tumour-derived DNA (ctDNA) in plasma for the detection of epiudermal growth factor receptor (EGFR) mutation status in European and Japanese patients (pts) with advanced non-small cell lung cancer (ANSCLC): ASSESS study. Ann Oncol. 2015; doi:10.1093/annonc/mdv128. 1-Abstr 350_PR
- Ilie M, Hofman V, Long E, et al. Current challenges for detection of circulating tumor cells and cell-free circulating nucleic acids, and their characterization in non-small cell lung carcinoma patients. What is the best blood substrate for personalized medicine? Ann Transl Med. 2014;2(11):107. doi:10.3978/j.issn.2305-5839.2014.08.11
- Shen Y, Tang D, Yao R, et al. MicroRNA expression profiles associated with survival, disease progression, and response to gefitinib in completely resected non-small-cell lung cancer with EGFR mutation. Med Oncol. 2013;30(4):750. doi:10.1007/s12032-013-0750-1
- Li B, Ren S, Li X, et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer. 2014;83(2):146–153.
- Sholl LM, Xiao Y, Joshi V, et al. EGFR mutation is a better predictor of response to tyrosine kinase inhibitors in non-small cell lung carcinoma than FISH, CISH, and immunohistochemistry. Am J Clin Pathol. 2010;133(6):922–934.
- Eberhard DA, Giaccone G, Johnson BE. Non-small-cell lung cancer working group. Biomarkers of response to epidermal growth factor receptor inhibitors in non-small-cell lung cancer working group: standardization for use in the clinical trial setting. J Clin Oncol. 2008;26(6):983–994.
- Ellison G, Zhu G, Moulis A, et al. EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol. 2013;66(2):79–89.
- Thress KS, Brant R, Carr TH, et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015;90(3):509–515.
- U.S. FDA News Release: FDA approves new pill to treat certain patients with non-small cell lung cancer. [cited 2016 Jan 21]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm472525.htm.
- Ishii H, Azuma K, Sakai K, et al. Digital PCR analysis of plasma cell-free DNA for non-invasive detection of drug resistance mechanisms in EGFR mutant NSCLC: Correlation with paired tumor samples. Oncotarget. 2015;6(31):30850–30858.
- Pender A, Garcia-Murillas I, Rana S, et al. Efficient genotyping of KRAS mutant non-small cell lung cancer using a multiplexed droplet digital PCR approach. PLoS One. 2015;10(9):e0139074.
- Sriram KB, Tan ME, Savarimuthu SM, et al. Screening for activating EGFR mutations in surgically resected nonsmall cell lung cancer. Eur Respir J. 2011;38(4):903–910.
- Marchetti A, Del Grammastro M, Felicioni L, et al. Assessment of EGFR mutations in circulating tumor cell preparations from NSCLC patients by next generation sequencing: toward a real-time liquid biopsy for treatment. PLoS One. 2014;9(8):e103883.
- Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158.
- Masago K, Fujita S, Muraki M, et al. Next-generation sequencing of tyrosine kinase inhibitor-resistant non-small-cell lung cancers in patients harboring epidermal growth factor-activating mutations. BMC Cancer. 2015;15(1):908.
- D’Haene N, Le Mercier M, De Neve N, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PLoS One. 2015;10(9):e0138245.
- Pfarr N, Stenzinger A, Penzel R, et al. High-throughput diagnostic profiling of clinically actionable gene fusions in lung cancer. Genes Chromosomes Cancer. 2016;55(1):30–44.
- Lira ME, Choi YL, Lim SM, et al. A single-tube multiplexed assay for detecting ALK, ROS1, and RET fusions in lung cancer. J Mol Diagn. 2014;16(2):229–243.
- Deeb KK, Hohman CM, Risch NF, et al. Routine clinical mutation profiling of non-small cell lung cancer using next-generation sequencing. Arch Pathol Lab Med. 2015;139(7):913–921.
- Fassunke J, Haller F, Hebele S, et al. Utility of different massive parallel sequencing platforms for mutation profiling in clinical samples and identification of pitfalls using FFPE tissue. Int J Mol Med. 2015;36(5):1233–1243.
- Dietel M, Johrens K, Laffert MV, et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther. 2015;22(9):417–430.
- Weickhardt AJ, Aisner DL, Franklin WA, et al. Diagnostic assays for identification of anaplastic lymphoma kinase-positive non-small cell lung cancer. Cancer. 2013;119(8):1467–1477.
- Shtivelman E, Hensing T, Simon GR, et al. Molecular pathways and therapeutic targets in lung cancer. Oncotarget. 2014;5(6):1392–1433.
- Crystal AS, Shaw AT, Sequist LV, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–1486.
- Ribas A, Wolchok JD. Combining cancer immunotherapy and targeted therapy. Curr Opin Immunol. 2013;25(2):291–296.
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251.
- ClinicalTrials.gov. [cited 2106 Jan 21]. Available from: www.clinicaltrials.gov.