
ABSTRACT
Introduction: Biomarkers are objective indications of a medical state that can be measured accurately and reproducibly. Traditional biomarkers enable diagnosis of disease through detection of disease-specific molecules, disease-mediated molecular changes, or distinct physiological or anatomical signatures.
Areas covered: This work provides a framework for selecting biomarkers that are most likely to provide useful information about a patient’s disease state. Though the authors emphasize markers related to disease, this work is also applicable to biomarkers for monitoring physiological changes such as ovulation or pregnancy. Additionally, the scope was restricted to biomarkers that are amenable to analytical detection across a range of health care levels, including low resource settings. The authors describe trade-offs between biomarkers’ sensitivity/specificity for a disease-causing agent, the complexity of detection, and how this knowledge can be applied to the development of diagnostic tests. This report also details additional assessment criteria for successful tests.
Expert commentary: Biomarker selection should primarily be driven by an attempt to answer an explicit clinical question (preferably causative relationship of the biomarker to disease-state), and only then by test development expediency (ease of detection). This framework is useful for stakeholders from test developers to clinicians to identify the trade-offs for diagnostic biomarkers for any use case.
1. Introduction
The Biomarker Definitions Working Group defines a biomarker as ‘a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention’ [Citation1]. For this work, we have focused on biomarkers related to disease states, though the framework is applicable to biomarkers related to changes in physiological states such as pregnancy [Citation2]. The term ‘biomarker’ encompasses multiple categories of disease indicators including disease-specific molecules such as pathogen-specific proteins, host-response molecules such as immunoglobulins, physiological measurements such as blood pressure, and results from imaging technologies [Citation3]. Additionally, biomarkers must be both disease-specific and universal in a diseased population for reliable and accurate measurement across a range of patients [Citation4]. Biomarkers are used for diagnosis of both infectious and noncommunicable diseases (NCD).
There are hundreds of cataloged biomarkers described in the literature and used in diagnostic products. With this range of available information and the complexity of developing and evaluating prototypes, biomarker selection still remains one of the most important and challenging aspects of designing diagnostic systems. When designing a new assay or device, selection of an appropriate type of biomarker (nucleic acid v. protein v. other) for a given diseased state can reduce development time and cost. Often, there is more than one biomarker for a disease and selecting the right biomarker(s) usually affects aspects of a final diagnostic test such as sensitivity, specificity, cost, usability, required logistics, and, ultimately, health outcomes. All of these considerations depend on the setting in which the test will be used. These constraints and user needs assessments for diagnostic devices have been detailed elsewhere [Citation5–Citation7]. In this work, we discuss how to select appropriate analytical biomarkers for developing diagnostic applications aimed at addressing specific clinical questions and give a brief overview of general categories of biomarkers (Supplemental Information and Table S1).
Based on their biological and physiological function with respect to disease, we developed a framework for selecting biomarkers that compares the trade-offs of biomarker selectivity for identifying a disease-causing agent – namely, is a marker a primary, secondary, or tertiary disease indicator – and the relative difficulty of detecting that class of marker in a given setting. Selecting a biomarker that most directly relates to a disease can also encourage directly treating the disease rather than the symptoms. Here, biomarker selection is viewed through a clinical lens to address an explicit clinical question.
2. Assessing biomarker effectiveness
Biomarker characteristics can determine the effectiveness for answering clinical questions; some are specific to the disease (e.g. concentration) and others depend on the measurement method. The Limit of Detection (LoD) of an analytical method determines the minimum concentration of a biomarker required to produce a true positive signal, but the concentration of a biomarker in a sample is not a measure of its diagnostic efficacy. To compare the effectiveness of multiple potential biomarkers, their inherent clinical sensitivity and specificity, LoD, and required sample type and sample volume must be considered. These values should be compared across all potential biomarkers for a given test, and used as selection criteria to determine which biomarker is a more appropriate target for a desired use case.
For example, Plasmodium falciparum malaria can be identified from dried blood spots (DBS) using parasite nucleic acids or a malaria-specific antigen such as PfHRP2. Comparing the abundance of nucleic acids and antigens in a given sample can identify which biomarker may be more useful based on the sensitivity of NAATs or immunoassays for biomarker detection. Assuming a DBS contains 50 µl of blood, the lower end of the NAAT sensitivity range based on literature values would be 50 parasites which equates to 5.5 × 105–6.5 × 106 PfHRP2 molecules [Citation8–Citation10] or 1 × 102–1 × 108 pg/ml of PfHRP2 [Citation9,Citation10]. Therefore, to match the sensitivity of a malaria NAAT from a DBS, an immunoassay would need to detect 5.5 × 105 PfHRP2 molecules. Based on the available literature, the LoD for PfHRP2 from DBS is ~5 × 107 molecules, which is two orders of magnitude higher than the equivalent LoD for parasite nucleic acids [Citation9,Citation10]. These biomarker values would allow a test developer or clinician to determine which marker would be appropriate for a given setting and available detection method. These details are summarized in .
Table 1. Summary of NAAT/immunoassay sensitivity parity for common P. falciparum biomarkers.
Concentration is also closely linked to disease progression. If a test aims to measure past disease, or maximize sensitivity, then effective biomarkers should have a long half-life. For example, research suggests that malaria-specific IgG1 and IgG3 levels remain elevated up to 1-year postinfection, enabling the tracking of recent P. falciparum infection [Citation15,Citation17,Citation18]. Alternatively, biomarkers for measurements of disease progression should have relatively short half-lives and concentrations proportional to disease severity [Citation19]. For example, viral RNA and p24 antigen are used as HIV biomarkers. During disease onset (10–30 days), concentrations of both RNA and p24 are relatively high and therefore easy to detect. After this initial state, RNA remains abundant while the p24 concentration significantly decreases. The p24 antigen would only be an effective biomarker during early disease onset, whereas viral RNA is more stable over the course of the disease [Citation20]. This detection window can identify useful biomarkers during different disease stages.
Another important quality of effective biomarkers is robustness to variations in use, such as differing patient demographics and variations in sample acquisition and handling prior to testing [Citation21]. For example, levels of malaria-specific IgG subclasses can vary significantly among different host genetic populations [Citation22]. Ideally, an effective biomarker would also be able to identify asymptomatic and early disease states which would, in turn, decrease the timing between disease onset and treatment. Biomarkers that can effectively diagnose asymptomatic diseases would reduce adverse outcomes [Citation23] and disease transmission [Citation24].
Additional considerations related to biomarker effectiveness are sample type [Citation19], biomarker stability, and presence of potential contaminants that may inhibit detection. Stability is important because laboratory-based diagnostic systems often collect and store samples from a remote setting and then transport them to a centralized lab for testing. For these systems, effective biomarkers must store and travel well.
2.1. Biomarker selectivity for identifying a disease-causing agent
A vital characteristic for any biomarker is its specificity in determining disease state. A useful biomarker would uniquely and directly represent the disease-causing agent, be easy to detect in multiple settings, and identify whether a disease was currently active. Many common biomarkers – such as cells, nucleic acids, and proteins – are direct or primary indicators of the agent causing a disease. For example, detection of pathogen-specific nucleic acids may directly identify what pathogen has caused the disease, but could also represent a latent infection, in which case the observed symptoms might actually be caused by a different pathogen. Identifying a viable cell within a sample often indicates active infection, whereas most other molecular biomarkers may remain for a period of days or weeks after active infection.
On the other hand, some biomarkers indirectly identify a disease-causing agent such as immunoglobulins or cytokines, both of which are produced as immune responses [Citation25]. Immunoglobulins are only indirect disease indicators because they represent the body’s reaction to a disease, and not the disease itself. Immunoglobulins can identify past versus present disease and usually remain detectable in a patient longer than pathogen-specific antigens [Citation18]. Similarly, basophil or eosinophil leukocytes measured as part of a complete blood count show elevated levels during general bacterial or viral infections, but do not indicate the actual agent causing the disease [Citation26].
2.2. Biomarker detection
Some biomarkers are easy to detect in most settings. Biomarkers that fall into the ‘easy to detect’ category are often present at high concentration in a sample and show a large difference between clinically high and low values. For example, disease-specific proteins have been demonstrated as easy to detect biomarkers for both minimally invasive early cancer diagnosis [Citation27] and infectious diseases [Citation28].
‘Challenging to detect’ biomarkers often require complex equipment, trained laboratory personnel, or both for identification. For example, lipids have been used to identify disease states by measuring lipid panels through metabolic profiling [Citation29] which requires expensive, complex laboratory equipment making detection challenging in many settings. Similarly, detection of cytokines related to disease-state requires complex, multistep biochemical assays performed by laboratory personnel or a highly specialized piece of laboratory equipment. Balancing these trade-offs can help identify appropriate biomarkers for addressing specific clinical goals. summarizes the general categories of biomarkers by comparing their selectivity for identifying a disease-causing agent and their relative ease of detection.
Figure 1. Evaluating biomarker usefulness for addressing clinical needs. A useful biomarker would directly and uniquely identify a disease-causing agent, be easy to detect in multiple settings, and clearly identify whether a disease is currently active. The placement of the categories of biomarkers on the graph is a generalization – individual biomarkers of a particular category may be easier or harder to detect, or be more or less specific, than their categorical placement indicates. The color of the markers indicates specificity for an active disease: green is high specificity for determining active disease, yellow is moderate specificity for an active disease (ex: residual ciruculating biomarker in a sample post-disease), and red is limited or no specifically to distinguish active disease states. *Carbohydrates are grouped separately from glucose because of the ease of measuring and high specificity of glucose for identifying type I diabetes.
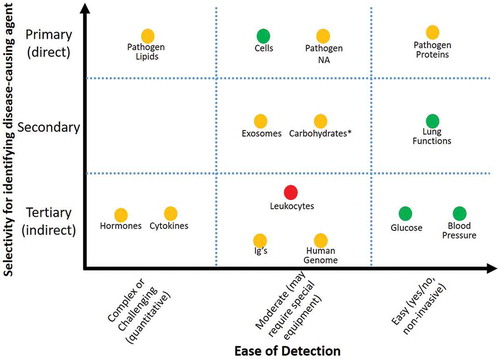
There are some exceptions within these categories; in comparison to most other carbohydrates, glucose has been a focus of research and diagnostic assay design for decades, and as a result there are tests for easily measuring it. Additionally, the ease of detection for these biomarker categories is likely to change over time as new methods and detection techniques and developed. An overview of the biomarker categories and examples from the literature are provided in the Supplemental Information and summarized in Table S1.
2.3. Comparing biomarkers using the framework
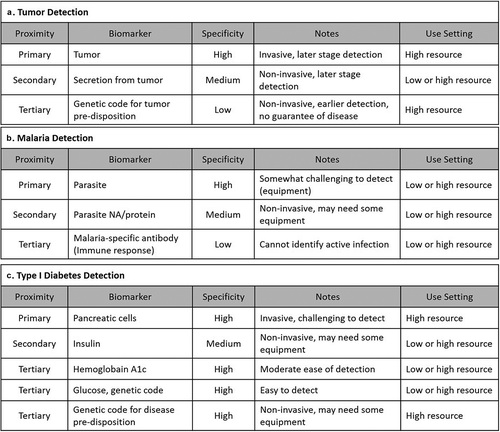
Using the framework provided above, biomarkers can be compared for a given clinical question and location of diagnosis. For example, type I diabetes can be diagnosed by identifying failing pancreatic beta cells, reduced concentration of insulin, increased concentration of blood glucose, or the genetic sequence that codes for destruction of beta cells [Citation30]. Identification of failing pancreatic beta cells is the primary cause of diabetes, but the required cell analysis can be invasive, require laboratory expertise, and specialized equipment not available in many settings. Therefore, pancreatic beta cells are not an appropriate type I diabetes biomarker for lower resource settings or for patient self-care. Collecting blood for glucose measurement, alternatively, is non-invasive and easy to detect in multiple settings using rapid test strips making glucose a more useful type I diabetes biomarker in lower resource settings even though fasting, and especially random, blood glucose levels are quite variable and thus not very specific for determining diabetes status. Hemoglobin A1c, an integrative biomarker, correlates well with average blood glucose levels over longer periods of time and represents a good compromise diagnostic marker – a bit more complex and expensive than random blood glucose, but less expensive and complex than direct identification of failing pancreatic beta cells [Citation31]. This case, and two others, have been outlined below ().
Figure 2. Comparing multiple biomarkers for clinical diagnostic applications using the framework provided in . Balancing biomarker selectivity for identifying a disease-causing agent, specificity, and invasiveness can help identify appropriate biomarkers for different settings. Three examples are provided (a) Tumor Detection, (b) Malaria Detection, and (c) Type I Diabetes Detection.
2.4. Summary questions to address biomarker effectiveness and selection
What is the clinical range of the biomarker concentration in a given sample type? Does this range change with disease progression?
What sample volume is reasonable to collect and will the collected volume contain enough biomarker to detect in a given assay?
What is the analytical LoD of a chosen detection method?
How stable is the biomarker in a given sample before it is measured?
Does the biomarker directly or indirectly identify the disease-causing agent and is it a marker of present or past disease?
Is the biomarker able to be detected in the targeted setting given resource and personnel constraints?
3. Additional factors to consider when selecting biomarkers for diagnostic tests
When evaluating diagnostic test performance, one of the first factors to consider is the intended end user. Different users and settings require different test characteristics. Ease-of-use for the end user includes sample collection, number of user steps, whether steps require nonautomated timing, and how test results are interpreted.
Cost is also an important consideration, especially for lower resource settings. Some reports have described that the cost-effectiveness of a diagnostic test is the most important factor for utilization [Citation32] [Citation33]. Therefore, cost can serve as an important constraint for biomarker selection. For example, many nucleic acid amplification assays require specialized enzymes, which can be expensive. The use of these enzymes can increase the overall cost of a test which may result in nucleic acids as a biomarker being too expensive to detect in some resource limited settings.
Other vital performance characteristics include test sensitivity, specificity, and positive and negative predictive value – which are dependent on disease prevalence. An ideal assay would have 100% sensitivity and specificity, but in reality, trade-offs in performance exist to maximize test efficacy while minimizing cost [Citation34]. For example, a diagnostic test may require high specificity if the available treatment is challenging to administer, costly, and considerably harsh on the patient, but the condition itself has low transmissibility. On the other hand, diagnosis of highly infectious diseases may require biomarkers with high sensitivity (or large sample volumes with complex concentration procedures) to avoid false negatives and prevent further disease spread. Ebola is a highly infectious disease, a corresponding diagnostic test would want high sensitivity to ensure infected patients are quarantined and treated to avoid disease transmission.
Biomarkers for diagnostic tests can also be used to recommend drug selection or dosage levels based on individual metabolism (e.g. companion diagnostics [Citation35]) or drug susceptibility [Citation36]. Test use-cases may require biomarkers with different characteristics including monitoring levels over time or the ratio between different markers. Many diseases, especially NCD, have complex, multifaceted treatment programs and the choice of a dynamic biomarker that changes in response to intervention may be desirable over tests with binary (yes/no) results.
Balancing test cost, accuracy, and usage are also related to disease prevalence among affected populations. Different population can develop lower titers of antibodies against the same infection [Citation37]; this understanding of disease presentation should be considered when selecting a biomarker. Other characteristics to consider that have been described elsewhere are test stability over time and ambient storage temperatures, failure rate, run time, ability to scale for manufacturing, reagent supply chain, and regulatory strategies [Citation7,Citation21,Citation38].
3.1. Effects of sample type on biomarker selection
Most diseases can be diagnosed from multiple sample types and sampling conditions. For example, many sexually transmitted infections can be sampled from urine or swabs. Although, effective, swab-based sampling for sexually transmitted infections can be challenging or painful to collect from male patients [Citation39,Citation40]. Therefore, a biomarker that presents well from a swab sample may not be useful if collecting that sample is not realistic for a given population. Urine, on the other hand, is simple to collect from all patients in many different settings [Citation41].
Biomarkers with excellent clinical sensitivity and specificity that are not concentrated enough to be detectable with a practical analytical method may not be useful in all settings. For example, nucleic acid biomarkers for chlamydia infection are present in urine, but the clinical concentration of chlamydia gDNA ranges from 101–106 copies/ml of urine [Citation42]. If an assay was only able to process a 100 µl sample, the amount of chlamydia gDNA would be too low to detect in many cases; therefore, gDNA would not serve as a reliable biomarker for that assay. Even though gDNA is able to be amplified, not collecting an appropriate volume of sample can limit the amount of available biomarker, and therefore limit or prevent its detection. One approach to overcome this limitation would be to collect a larger sample volume, but there are limits to how much volume can be collected without unduly impacting the patient, increasing signal background, or significantly complicating the detection assay.
Additionally, some diseases require invasive samples, such as a tissue biopsy, which would not be an appropriate sample for non-trained users to collect. The choice of sample type can critically affect choice of biomarker.
3.2. Considerations for lower resource settings
In this work, these trade-offs, as well as the questions summarized above, have been integrated into a flowchart to help guide development of diagnostic tests using appropriate biomarkers ().
Figure 3. Proposed flowchart for assessing biomarker effectiveness for diagnostics tests. Using this flowchart requires detailed knowledge about the surrounding environment of a test including end-user, setting, and clinical concentrations of selected biomarkers. Special considerations should be made for tests used in resource limited settings to ensure the diagnostic tests and associated biomarkers are cost-effective because cost is a major driving factor of usage.
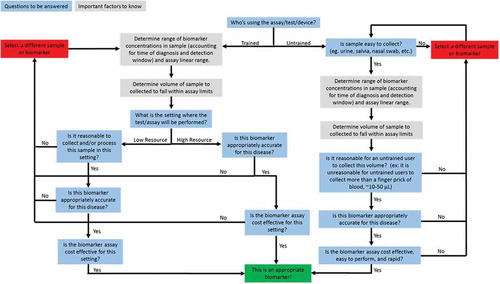
An early decision point in the flowchart focuses on sample collection which is vitally important in resource-limited settings that can have limited access to mains electricity, trained laboratory personnel, and refrigeration, which is required for many diagnostic tests. Additional specific considerations including test cost are especially important for biomarker and test selection in resource-limited settings, as noted above [Citation38,Citation43]. Understanding these types of constraints for a given use-setting will help identify appropriate biomarkers to develop successful diagnostic tests.
3.3. Using the biomarker selection flowchart
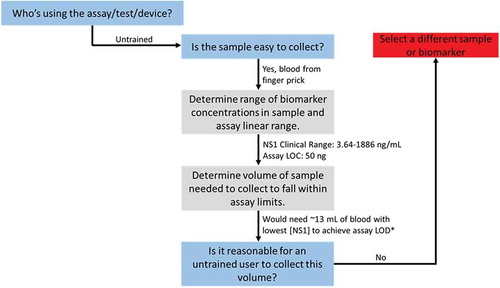
Using the chart provided above, diagnostic tests can be evaluated to assess potential usefulness for a given user and setting. For example, a researcher may want to develop a home-based serum test for dengue. Using information from , an appropriate biomarker that is easy to collect would be the dengue non-structural glycoprotein-1 (NS1) in serum. Since the hypothetical goal is a home-based test, the end user will be untrained indicating that a simple finger-stick blood sample should be used. There is a limitation, however; the literature reports the clinical concentration of NS1 in serum as 3.64–1886 ng/ml [Citation44]. Because the average finger-stick of blood only collects ~10–50 µl, the amount of NS1 in a finger-stick sample would range from 0.182 to 94.3 ng. If the researcher’s assay LOD is 50 ng, then most of the clinical range is not detectable by this assay using only 50 µl of blood. A more appropriate sample volume would come from venipuncture, but this procedure is not reasonable for an untrained, home-user to perform. The end-result of this work indicates that NS1 is not an appropriate biomarker for the desired home-based application (). The researcher should either select a different biomarker available at higher concentrations or further develop the assay to reduce the LoD to match the clinical range. Understanding these constraints before spending time and money developing a test helps better direct efforts toward projects that have a higher chance of succeeding.
Figure 4. Example assessment of a theoretical dengue diagnostic using the flowchart in above. *The lowest clinical range of 3.64 ng/mL would equal 0.182 ng in a 50 µL blood finger prick sample (0.182 ng/50 µL). If the assay LoD was 50 ng, solve for the volume of blood that would be required to achieve the same ratio (50 ng/x µL) gives ~13 mL of blood. This is far too much to collect from a finger prick, but may be possible using venipuncture. Requiring venipuncture to collect sample would not be reasonable for untrained users or in many resource limited settings. Alternatively, the research could spend more time developing the assay to improve its analytical LoD to better align with clinical biomarker levels.
4. Expert commentary
As less complex assays are starting to achieve sensitivities compared to gold-standard tests and novel device design improves capabilities in low resource settings, more people will have access to reliable diagnostics. This expanded access will only be meaningful if it is accompanied by appropriately chosen biomarkers. This expansion of access should be coupled with the continued need for basic research to identify new biomarkers [Citation45,Citation46].
Appropriate selection of biomarkers with innovative test design can help transform patient care by providing earlier diagnosis, regular treatment monitoring, and ultimately reduced burden of disease. These results will be best achieved through collaborations between researchers, device designers, and clinicians to drive diagnostic test development for addressing explicit clinical questions. Accurate biomarker selection will continue to be a critical part of this process. Additionally, it is important to consider cost-effectiveness of a diagnostic test because it drives utilization [Citation32,Citation33], especially in resource limited settings.
Our framework aims to assist stakeholders in selecting appropriate markers for any use case. There are few, if any, ideal biomarkers for diagnostic testing due to trade-offs based on performance, cost, and usability. It is important to consider how and where a test will be used in order to select an appropriate biomarker. The framework provided is intended to help assess possible trade-offs to design new diagnostic systems and enhance those already available.
5. Five-year view
Looking forward, diagnostic test designs are beginning to strive for ultrasensitivity and quantification that can identify single target molecules; these types of tests are often referred to as digital assays because they rely on digitization of a signal where a positive signal indicates the presence of one target molecule and a negative indicates no target molecule. There are examples of digital NAATs in both the academic [Citation47] and commercial sectors [Citation48]. Recently, commercially available systems have been pushing toward digital immunoassays for quantification of proteins and larger-scale biomarker panels [Citation49–Citation53]. Currently, these types of tests are limited to higher resource facilities, but as they become more ubiquitous across a variety of settings they can lead to earlier disease detection. Additionally, future multiplexed or panel-based tests will help identify biomarkers indicative of specific treatment pathways [Citation54,Citation55].
In this work, we have focused on a biomarker described as a specific molecule; but in the future, biomarker may refer to a set of molecules that together describe the molecular signature of a disease state. As techniques such as highly parallel sequencing become more readily available, multiple molecules can simultaneously be probed to more completely understand a disease state. The principles and framework we have described in this work would still hold true for these groups of biomarkers. This is especially important for NCDs as their diagnosis and treatment begins to become more prevalent and significant around the world [Citation56].
Key issues
Biomarker selection should primarily be driven by an attempt to answer an explicit clinical question (e.g., direct correlation of the biomarker with disease-state), and only then be guided by test development expediency (e.g., which biomarker is easier to detect).
Balancing biomarker selectivity for identifying a disease-causing agent, specificity, and invasiveness can help identify appropriate biomarkers for different settings. To compare the effectiveness of different classes of biomarkers – such as proteins v. nucleic acids – their inherent clinical sensitivity and specificity, LoD, and needed sample type and volume must be considered.
Selecting an appropriate marker for a given use-case requires detailed knowledge about the surrounding environment of a test including the end-user, setting, and clinical concentrations of a biomarker. These trade-offs can be evaluated using the framework provided in this manuscript.
Trade-offs may lead to different biomarkers or tests for different settings.
Declaration of interest
During manuscript preparation, SA Byrnes began working at Intellectual Ventures Laboratory, Bellevue, WA, USA. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
SByrnes_ExpertReview_ESI_2017.pdf
Download PDF (667.5 KB)Acknowledgments
The authors gratefully acknowledge the Bill and Melinda Gates Foundation Trust for their sponsorship through Intellectual Ventures’ Global Good Fund.
Supplemental data
Supplemental data can be accessed here.
Additional information
Funding
Related Research Data
References
- Atkinson A.J., Colburn, W.A., DeGruttola, V.G., et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95.
- EPT do-it-yourself early-pregnancy test. Med Lett Drugs Ther. 1978;20(8):30–40.
- Zhao X, Modur V, Carayannopoulos LN, et al. Biomarkers in pharmaceutical research. Clin Chem. 2015;61(11):1343–1353.
- Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS. 2011;5(6):463–466.
- Derda R, Gitaka J, Klapperich CM, et al. Enabling the development and deployment of next generation point-of-care diagnostics. PLoS Negl Trop Dis. 2015;9(5):1–16.
- Vashist SK, Luppa PB, Yeo LY, et al. Emerging technologies for next-generation point-of-care testing. Trends Biotechnol. 2015; 33(11): 692–705 .
- Kumar AA, Hennek JW, Smith BS, et al. From the bench to the field in low-cost diagnostics: two case studies. Angew Chemie Int Ed. 2015;54(20):5836–5853.
- Dondorp AM, Desakorn V, Pongtavornpinyo W, et al. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2005;2(8):0788–0797.
- Rogier E, Plucinski M, Lucchi N, et al. Bead-based immunoassay allows sub-picogram detection of histidine-rich protein 2 from Plasmodium falciparum and estimates reliability of malaria rapid diagnostic tests. PLoS One. 2017;12(2):e0172139.
- Ramutton T, Hendrisksen ICE, Mwanga-Amumpaire J, et al. Sequence variation does not confound the measurement of plasma PfHRP2 concentration in African children presenting with severe malaria. Malar J. 2012;11(1):276.
- Tran TM, Aghili A, Li S, et al. A nested real-time PCR assay for the quantification of Plasmodium falciparum DNA extracted from dried blood spots. Malar J. 2014;13:393.
- Wihokhoen B, Dondorp AM, Turner P, et al. Use of blood smears and dried blood spots for polymerase chain reaction-based detection and quantification of bacterial infection and Plasmodium falciparum in severely ill febrile African children. Am J Trop Med Hyg. 2016;94(2):322–326.
- Oyola SO, Ariani CV, Hamilton WL, et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J. 2016;15:597.
- Laban NM, Kobayashi T, Hamapumbu H, et al. Comparison of a PfHRP2-based rapid diagnostic test and PCR for malaria in a low prevalence setting in rural southern Zambia: implications for elimination. Malar J. 2015;14:25.
- Dobano C, Quelhas D, Quinto L, et al. Age-dependent igg subclass responses to Plasmodium falciparum EBA-175 are differentially associated with incidence of malaria in Mozambican children. Clin Vaccine Immunol. 2012;19(2):157–166.
- Long GW, Fries L, Wall GH, et al. Polymerase chain reaction amplification from Plasmodium falciparum on dried blood spots. Am J Trop Med Hyg. 1995;52(4):344–346.
- Ahmed Ismail H, Tijani MK, Langer C, et al. Subclass responses and their half-lives for antibodies against EBA175 and PfRh2 in naturally acquired immunity against Plasmodium falciparum malaria. Malar J. 2014;13(1):425.
- Duah NO, Miles DJC, Whittle HC, et al. Acquisition of antibody isotypes against Plasmodium falciparum blood stage antigens in a birth cohort. Parasite Immunol. 2010;32(2):125–134.
- Zhou Q, Hu HG, Hou L. Discover, develop & validate–advance and prospect of tumor biomarkers. Clin Lab. 2015;61(11):1589–1599.
- Sherin K, Klekamp BG, Beal J, et al. What is new in HIV infection? Am Fam Physician. 2014;89(4):265–272.
- Fung ET. A recipe for proteomics diagnostic test development: the OVA1 test, from biomarker discovery to FDA clearance. Clin Chem. 2010;56(2):327–329.
- Bolad A, Farouk SE, Israelsson E, et al. Distinct interethnic differences in immunoglobulin G class/subclass and immunoglobulin M antibody responses to malaria antigens but not in immunoglobulin G responses to nonmalarial antigens in sympatric tribes living in West Africa. Scand J Immunol. 2005;61(4):380–386.
- Farrar D, Fairley L, Wright J, et al. Evaluation of the impact of universal testing for gestational diabetes mellitus on maternal and neonatal health outcomes: a retrospective analysis. BMC Pregnancy Childbirth. 2014;14(1):317.
- Diclemente RJ, Wingood GM, Harrington KF, et al. Efficacy of an HIV prevention intervention. Journal of the American Medical Association. 2004;292(2):171–179.
- Van Der Meide PH, Schellekens H. Cytokines and the immune response. Biotherapy. 1996;8(3–4):243–249.
- The Mayo Clinic C. Eosinophilia. Mayo Foundation for Medical Education and Research. 2017. p. 1–3.
- Luna Coronell JA, Syed P, Sergelen K, et al. The current status of cancer biomarker research using tumour-associated antigens for minimal invasive and early cancer diagnostics. J Proteomics. 2012;76:102–115.
- Ray S, Patel SK, Kumar V, et al. Differential expression of serum/plasma proteins in various infectious diseases: specific or nonspecific signatures. Proteomics Clin Appl. 2014;8(1–2):53–72.
- Wenk MR. Lipidomics in drug and biomarker development. Expert Opin Drug Discov. 2006;1(7):723–736.
- Størling J, Pociot F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes (Basel). 2017;8(2):72.
- Higgins T. HbA1c for screening and diagnosis of diabetes mellitus. Endocrine. 2013;43(2):266–273.
- Loubiere S, Moatti J-P. Economic evaluation of point-of-care diagnostic technologies for infectious diseases. Clin Microbiol Infect. 2010 Aug;16(8):1070–1076.
- Losina E, Weinstein MC, Anglaret X, et al. Cost-effectiveness of HIV treatment in resource-poor settings — the case of Côte d’Ivoire. The New England Journal of Medicine. 2006: p. 1141–1153.
- Weigl BH, Boyle DS, de los Santos T, et al. Simplicity of use: a critical feature for widespread adoption of diagnostic technologies in low-resource settings. Expert Rev Med Devices. 2009;6(5):461–464.
- Agarwal A, Snyder G, Ressler D. The current and future state of companion diagnostics. Pharmgenomics Pers Med. 2015;8:99.
- Ely S. Personalized medicine: individualized care of cancer patients. J Lab Clin Med. 2009;154(6):303–308.
- Goulart LR, Vieira CU, Freschi APP, et al. Biomarkers for serum diagnosis of infectious diseases and their potential application in novel senor platforms. Crit Rev Immunol. 2010;30(2):201–222.
- Thompson M, Weigl B, Fitzpatrick A, et al. More than just accuracy: a novel method to incorporate multiple test attributes in evaluating diagnostic tests including point of care tests. IEEE J Transl Eng Heal Med. 2016 Apr;4.
- Sugunendran H, Birley HDL, Mallinson H, et al. Comparison of urine, first and second endourethral swabs for PCR based detection of genital Chlamydia trachomatis infection in male patients. Sex Transm Dis. 2001;77:423–426.
- Cook RL, Hutchison SL, Braithwaite RS, et al. Review systematic review: noninvasive testing for Chlamydia trachomatis. Ann Intern Med. 2005;142:914–925.
- Chernesky M, Jang D, Chong S, et al. Impact of urine collection order on the ability of assays to identify Chlamydia trachomatis infections in men. Sex Transm Dis. 2003;30(4):345–347.
- Michel C-EC, Sonnex C, Carne CA, et al. Chlamydia trachomatis load at matched anatomic sites: implications for screening strategies. J Clin Microbiol. 2007 May;45(5):1395–1402.
- Peeling RW, Smith PG, Bossuyt PMM. A guide for diagnostic evaluations. Nat Rev Microbiol. 2010 Dec;8(12 Suppl):S2–6.
- De La Cruz-Hernández SI, Flores-Aguilar H, Gonzalez-Mateos S, et al. Determination of viremia and concentration of circulating nonstructural protein 1 in patients infected with dengue virus in Mexico. Am J Trop Med Hyg. 2013;88(3):446–454.
- Leligdowicz A, Conroy AL, Hawkes M, et al. Validation of two multiplex platforms to quantify circulating markers of inflammation and endothelial injury in severe infection. PLoS One. 2017;12(4):1–14.
- Conroy AL, Hawkes M, McDonald CR, et al. Host biomarkers are associated with response to therapy and long-term mortality in pediatric severe malaria. Infect Dis Soc Am. 2016;3(3):1–10.
- Kreutz JE, Munson T, Huynh T, et al. Multiplexed quantification of nucleic acids with large dynamic range using multivolume digital RT-PCR on a rotational SlipChip tested with HIV and hepatitis C viral load. Anal Chem. 2011;83:8158–8168.
- ThermoFisher Scientific, “Digital PCR.” [Online]. Accessed October 2017. Available from: https://www.thermofisher.com/us/en/home/life-science/pcr/digital-pcr.html.
- Simon S, Ezan E. Ultrasensitive bioanalysis: current status and future trends. Bioanalysis. 2017;9:753–764.
- Yeung D, Ciotti S, Purushothama S, et al. Evaluation of highly sensitive immunoassay technologies for quantitative measurements of sub-pg/mL levels of cytokines in human serum. J Immunol Methods. 2016;437:53–63.
- Fischer SK, Joyce A, Spengler M, et al. Emerging technologies to increase ligand binding assay sensitivity. Aaps J. 2015;17(1):93–101.
- Cowan KJ, Geiger A, Hornauer H, et al. Performance evaluation of three platforms with ultrasensitive ligand-binding assay potential. Bioanalysis. 2017;9:937–946.
- Collet-Brose J, Couble P, Deehan MR, et al. Evaluation of multiple immunoassay technology platforms to select the anti-drug antibody assay exhibiting the most appropriate drug and target tolerance. J Immunol Res. 2016;2016.
- Semiglazov VF, Semiglazov VV, Dashyan GA, et al. Phase 2 randomized trial of primary endocrine therapy versus chemotherapy in postmenopausal patients with estrogen receptor-positive breast cancer. Cancer. 2007;110(2):244–254.
- Limper M, De Kruif MD, Duits AJ, et al. The diagnostic role of Procalcitonin and other biomarkers in discriminating infectious from non-infectious fever. J Infect. 2010;60(6):409–416.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012 Dec;380(9859):2095–2128.