
1. Overview of hemostasis
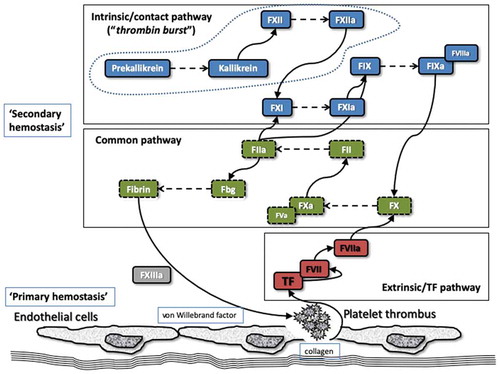
The most common bleeding disorders associated with coagulation factor deficiencies are termed hemophilia A (representing deficiency of factor VIII [FVIII]) and hemophilia B (representing deficiency of factor IX [FIX]) [Citation1]. A simple representation of various coagulation pathways is shown in . The eventual endpoint of coagulation is the formation of a fibrin clot, which is dependent on thrombin generation and conversion of fibrinogen into fibrin. This process is considered a ‘secondary’ pathway of hemostasis, the ‘primary’ pathway being principally led by the interplay of blood vessels, platelets, and von Willebrand factor (VWF). However, although convenient to separate the pathways from the perspective of the diagnosis of specific hemostasis defects, the pathways are intricately intertwined in vivo.
Figure 1. A simple representation of the melding of in vivo and in vitro hemostasis. In vitro hemostasis essentially reflects a separation of the parts of hemostasis to assist with the diagnosis of specific components, for example, FVIII or FIX deficiencies leading to hemophilia A or B. In vivo hemostasis essentially reflects an integration of the ‘different’ parts of hemostasis, which are intricately intertwined. The dashed line encompassing Prekallikrein, Kallikrein, and FXII in the ‘intrinsic/contact pathway’ indicates the portion of this pathway with the least in vivo hemostasis relevance. Thrombin is produced at various stages of the coagulation process.
a, activated; F, factor; Fbg, fibrinogen.
A deficiency of most (but not all) of the other coagulation proteins () will also lead to a bleeding diathesis. Hereditary disorders are usually associated with deficiency of individual factors (e.g., hemophilia A or B). In contrast, acquired deficiency of coagulation proteins (e.g., disseminated intravascular coagulation, liver disease, vitamin K deficiency) tends to affect multiple proteins. However, this generalization does not always hold true. Thus, there are congenital disorders that affect multiple coagulation proteins (e.g., combined FV/FVIII deficiency [Citation2]), and there are acquired conditions affecting individual coagulation proteins (e.g., acquired hemophilia A, due to development of anti-FVIII antibodies).
2. Coagulation factor testing
Testing for coagulation factors in the evaluation of bleeding disorders generally involves the performance of phenotypic assays, and only rarely are molecular tests done. Most often, testing encompasses one-stage clotting assays, using modifications of routine coagulation tests [Citation3]. For example, plasma FVIII and FIX activity (see ) is most often determined by means of a modified APTT (activated partial thromboplastin time) assay. The other main coagulation factors are also identified primarily by one-stage clotting assays, either with modified APTT (FXI, FXII) or PT (prothrombin time; FII, FV, FVII, FX) tests. In some cases, chromogenic assays (e.g., for FVIII or FIX), or other activity assays (e.g., FXIII) may be alternately employed [Citation4,Citation5]. The main reasons for these test choices are around simplicity of tests, low cost, wide availability, and because the phenotypic level of the coagulation factor will in most cases be sufficient for diagnosing a particular disorder, as well as for establishing the severity of the bleeding disorder (i.e., the lower the coagulation factor level, the more severe the bleeding phenotype [Citation1]), and also for patient management (therapy treatment/monitoring). Thus, these same simple phenotypic tests are utilized to assess coagulation factor levels during factor replacement therapy, including the new emerging factor replacement strategies, and so are used for multiple applications [Citation6].
3. Molecular genetic testing for coagulation factors
Molecular genetic testing for coagulation factors is less often applied in diagnostic settings. The main reasons for this limited usage include the relatively higher cost and complexity, lower availability, and because existing genetic tests may only provide confirmation of the diagnosis, and not an appreciation of disease severity, nor can they be used for monitoring management. Thus, comparatively, phenotypic tests have far greater utility, and are more broadly applied. This situation may change in the future, with evolution of improved molecular methods and next-generation tools [Citation7–Citation10]. As an example of the current state of play, one FVIII gene (designated F8) database currently lists 2611 unique variants (https://databases.lovd.nl/shared/variants/F8/unique), and the associated FIX gene (F9) database currently lists 1112 unique variants (https://databases.lovd.nl/shared/variants/F9/unique). As for most situations in the field of hemostasis, it is not always clear when a variant is disease-causing (pathological) or is ‘benign’. Thus, pinpointing disease-causing genetic variants for coagulation factors is not only complex, it also requires correlation with both clinical and laboratory phenotype for diagnosis, and so is not generally applied in routine hemostasis diagnostics. Nevertheless, genetic testing is useful in select situations, especially to assist with family studies [Citation7–Citation10].
Until recently, Sanger sequencing of specific candidate genes has been the only method available for molecular diagnosis of coagulation factors [Citation7]. However, this approach is time-consuming, costly and requires some pre-identification of the proposed candidate gene(s). Thus, factor assays or family studies would have already needed to be performed, in order to identify the deficient/defective coagulation protein and therefore to identify which gene (and what primers) would be needed for specific molecular interrogation. Increasingly, high-throughput sequencing (HTS) is being performed to allow simultaneous and rapid investigation of multiple genes at a manageable cost. This newer technology includes targeted sequencing of prespecified genes, whole-exome sequencing, or whole-genome sequencing, and is revolutionizing the genetic diagnosis of human diseases, including those associated with bleeding disorders [Citation7]. Some relevant examples are provided in the references and . Nevertheless, its implementation remains limited in diagnostics, and while it is increasingly progressed into clinical practice, its main utility remains in the research field.
Table 1. Molecular diagnostic testing in the area of hemostasis, as focussed on coagulation factors*.
Additional emerging technologies, set to include the field of hemostasis, include short-read sequencing approaches, such as sequencing by synthesis, ion semiconductor sequencing, and nanoball sequencing [Citation10]. The main current applications for such methods include targeted resequencing, exome sequencing, transcriptome sequencing, small genome sequencing, along with liquid biopsy. The main advantage of the system is the low relative cost per base [Citation10]. Drawbacks include relatively low throughput and challenges to wider implementation. Third-generation long-read sequencing now promises to overcome many of the limitations of short-read sequencing, such as the ability to reliably resolve repeat sequences and large genomic rearrangements. By combining complementary methods with massively parallel DNA sequencing, a greater insight into the biological context of hematological disease mechanisms is becoming possible [Citation10]. Emerging methodologies, such as advances in nanopore technology, in situ nucleic acid sequencing, and microscopy-based sequencing, will also expectedly continue the rapid evolution of this area [Citation10]. As an example, current nanopore technology uses protein-based nanopores that are structurally precise, easily modified and able to be engineered and produced on a large scale, thus enabling accurate analysis of proteins at the single-molecule level. However, a large degree of development is required before solid-state nanodevice DNA sequencing becomes mainstream.
4. Expert Opinion
The situation we have identified for coagulation factors is not unique within the field of hemostasis and applies also (for example) to molecular testing for VWF and platelet disorders [Citation11–Citation13]. Indeed, molecular testing for disorders associated with defects/deficiency in VWF or platelets is even more complex than those for individual coagulation factors [Citation7,Citation8,Citation14]. For VWF, this is due to the size and complexity of the protein and its associated gene, the existence of a pseudo-gene that replicates some portions of the active gene, and a variety of epigenetic influences on VWF level and function [Citation11]. For platelets, this is since there are many separate defects potentially affecting different patients [Citation12,Citation13].
Nevertheless, this is certainly an exciting time for molecular testing in hemostasis, including coagulation factors [Citation14]. One recently proposed algorithm for molecular testing of F8, F9, and VWF had a reported overall diagnostic success rate of 99% [Citation8]. However, even such an approach currently requires much more time and effort than routine phenotypic testing for FVIII, FIX, and VWF [Citation3,Citation4,Citation15]. Accordingly, we believe that genetic testing currently can only be considered an adjunct diagnostic procedure or have potential utility in select (individual) cases, and after phenotypic tests have been performed, although of course there remains potential utility for genetic testing in prenatal diagnosis or family studies where a disease-causing variant has already been identified.
Nevertheless, molecular testing tools in the area of hemostasis are ever improving. Next-generation sequencing and adjunct tools continue to be developed for hemostasis applications [Citation7–Citation12], and these will expectedly revolutionize diagnostic strategies for bleeding disorders. However, for the time being, at least, phenotypic testing remains the current diagnostic strategy for most bleeding disorders [Citation1–Citation5,Citation15].
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. The views expressed in this editorial are those of the authors, and not necessarily those of the University of Verona or NSW Health Pathology.
Reviewers Disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Lippi G, Franchini M, Favaloro EJ. Diagnostics of inherited bleeding disorders of secondary hemostasis: an easy guide for routine clinical laboratories. Semin Thromb Hemost. 2016 Jul;42(5):471–477.
- Zheng C, Zhang B. Combined deficiency of coagulation factors V and VIII: an update. Semin Thromb Hemost. 2013 Sep;39(6):613–620.
- Duncan E, Rodgers S. One-stage factor VIII assays. Methods Mol Biol. 2017;1646:247–263.
- Rodgers S, Duncan E. Chromogenic factor VIII assays for improved diagnosis of hemophilia A. Methods Mol Biol. 2017;1646:265–276.
- Muszbek L, É K, Kerényi A. Assessment of factor XIII. Methods Mol Biol. 2017;1646:277–293.
- Kitchen S, Tiefenbacher S, Gosselin R. Factor activity assays for monitoring extended half-life FVIII and factor IX replacement therapies. Semin Thromb Hemost. 2017 Apr;43(3):331–337.
- Bastida JM, Benito R, Lozano ML, et al. Molecular diagnosis of inherited coagulation and bleeding disorders. Semin Thromb Hemost. 2019 Oct;45(7):695–707.
- Bastida JM, González-Porras JR, Jiménez C, et al. Application of a molecular diagnostic algorithm for haemophilia A and B using next-generation sequencing of entire F8, F9 and VWF genes. Thromb Haemost. 2017 Jan 5;117(1):66–74.
- Ghorbanpour E, Lillicrap D. Innovative molecular testing strategies for adjunctive investigations in hemostasis and thrombosis. Semin Thromb Hemost. 2019 Oct;45(7):751–756.
- Kumar KR, Cowley MJ, Davis RL. Next-generation sequencing and emerging technologies. Semin Thromb Hemost. 2019 Oct;45(7):661–673.
- Batlle J, Pérez-Rodríguez A, Corrales I, et al., PCM-EVW-ES Investigators Team. Update on molecular testing in von Willebrand disease. Semin Thromb Hemost. 2019 Oct;45(7):708–719.
- Mumford AD, Westbury SK. Genetic techniques used in the diagnosis of inherited platelet disorders. Semin Thromb Hemost. 2019 Oct;45(7):685–694.
- Downes K, Megy K, Duarte D, et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019 Dec 5;134(23):2082–2091.
- Bastida JM, Del Rey M, Lozano ML, et al. Design and application of a 23-gene panel by next-generation sequencing for inherited coagulation bleeding disorders. Haemophilia. 2016 Jul;22(4):590–597.
- Favaloro EJ. Diagnosis or exclusion of von Willebrand disease using laboratory testing. Methods Mol Biol. 2017;1646:391–402.