
ABSTRACT
Introduction
The group of vascular tumors contains many different entities, and is considered difficult by pathologists, as they often have overlapping histological characteristics. Chromosomal translocations have been identified in ~20% of mesenchymal tumors and are considered the drivers of tumor formation. Many translocations have been discovered over the past decade through next-generation sequencing. This technological advancement has also revealed several recurrent gene fusions in vascular tumors.
Areas covered
This review will discuss the various vascular tumors for which recurrent gene fusions have been identified. The gene fusions and the presumed molecular mechanisms underlying tumorigenesis are shown, and potential implications for targeted therapies discussed. The identification of these gene fusions in vascular tumors has improved diagnostic accuracy, especially since several of these fusions can be easily detected using surrogate immunohistochemical markers.
Expert opinion
The identification of gene fusions in a subset of vascular tumors over the past decade has improved diagnostic accuracy, and has provided the pathologists with novel diagnostic tools to accurately diagnose these often difficult tumors. Moreover, the increased understanding of the underlying molecular mechanisms can guide the development of targeted therapeutic strategies.
1. Introduction
Technologies have led to the identification of many genetic alterations in sarcomas. In the late 20th century, the discovery of chromosomes led to the development of karyotyping. The first chromosomal translocation in a solid tumor, Ewing sarcoma, was identified using such classic G-banding karyotyping methods [Citation1]. Molecular technologies such as reverse transcription PCR (RT-PCR) analysis, or fluorescence in situ hybridization (FISH) have been utilized for the detection of fusion genes. However, there are limitations in each method and their sensitivity differs. Since 2005, the development of high throughput next generation sequencing (NGS) rapidly increased the number of identified recurrent translocations in tumors. NGS, also known as massive parallel sequencing, reads single DNA strands within a sample, instead of bulk of DNA. These reads can be obtained for a number of specific DNA fragments/genes within one single test (‘targeted NGS’), or even for all coding fragments within the genome (whole-exome sequencing (WES)), for the whole genome (whole-genome sequencing (WGS)) or for all actively transcribed genes (RNA or transcriptome sequencing). The chosen technique and the corresponding coverage (number of reads obtained per fragment) determine the sensitivity of a test. In routine diagnostics, in addition to the use of single gene FISH tests to detect a translocation, NGS based panels can be used interrogating multiple genes in one single test [Citation2]. Sarcomas can be genetically classified into two categories: a. sarcomas with simple genetic alterations comprising tumor-specific translocations, specific amplifications, or activating mutations or b. sarcomas with complex and unbalanced karyotypes [Citation3]. In the former, approximately 20% of the cases harbor a pathognomonic chromosomal translocation [Citation4]. In the latter, unbalanced chromosomal translocations and/or multiple genomic aberrations including copy number alterations result in a complex karyotype. In both cases, the chromosomal rearrangements might generate fusion genes through an exchange of genetic material by translocation, inversion, deletion, or tandem duplication. Such gene rearrangements invariably impact the expression and function of other genes, potentially resulting in tumorigenesis.
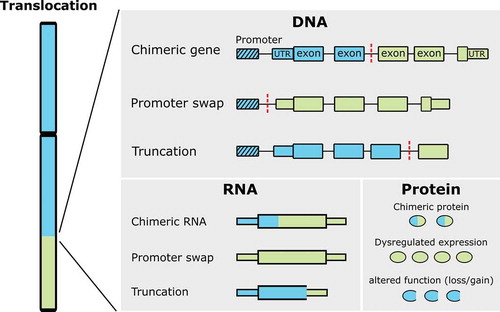
Fusion genes may alter gene function and expression in one of the following three ways: chimeric genes, promoter swap, or altered function () [Citation5]. First, chimeric genes often involve transcription factors leading to altered transcriptional regulation. A classic example is the EWSR1-FLI1 fusion in Ewing sarcoma. The N-terminal transactivation domain of EWSR1 is fused with the C-terminal ETS DNA binding domain of FLI1, resulting in a chimeric transcription factor [Citation6]. The EWSR1-FLI1 chimeric transcription factor leads to oncogenic activity by dysregulating the expression of proteins involved in crucial cellular processes such as cell cycle progression, DNA damage response, evasion of growth inhibition, escape from senescence, escape from apoptosis, angiogenesis, invasion and metastases [Citation7,Citation8]. Second, rearrangements resulting in a promoter swap lead to upregulation of expression of one single gene, as the coding sequence of that specific gene is placed under the control of a highly active promoter. This has been shown for rearrangements involving the USP6 gene with various fusion partners, as seen in nodular fasciitis and aneurysmal bone cyst, amongst others. The replacement of the USP6 promoter with a highly active promoter of another gene, such as ZNF9 and COL1A1, results in uncontrolled upregulation of USP6 gene expression [Citation9]. Third, a chromosomal translocation can disrupt the coding region of a gene, thereby altering gene function. Chromosomal translocations that result in disrupted gene function are commonly assumed to be a loss-of-function [Citation5]. However, as described later in this review for the FOS-gene, this can also result in gain-of-function.
Figure 1. Effects of translocations. A translocated chromosome is depicted on the left, while on the right resulting effects on DNA, RNA, and protein level are shown. Blue and green boxes originate from different chromosomes, dashed box represents the promoter and red dashed lines represent the breakpoint site (modified after Roukos et al., 2014)
Vascular tumors are tumors displaying endothelial differentiation occurring in bone and soft tissue and at visceral sites. They are presumed to be of vascular origin [Citation10,Citation11]. There are many different entities, with biological behavior ranging from benign to intermediate (locally aggressive/rarely metastasizing) to frankly malignant. Various molecular alterations can be found, ranging from single nucleotide variations (such as mutations in the various GNA genes in benign vascular tumors) [Citation12], recurrent reciprocal translocations (predominantly in the group of intermediate vascular tumors (hemangioendotheliomas) to a complex genome (in angiosarcoma (AS)). All the entities and their corresponding genetic alterations identified to date are summarized in . Of note, recurrent genetic alterations are not always specific for one tumor type, for instance GNA mutations can be identified not only in multiple distinct vascular tumor entities (congenital hemangioma, capillary malformation, anastomosing hemangioma, and cherry (senile) hemangioma) [Citation12] but also in melanocytic lesions (including uveal melanoma and blue nevus) [Citation13] and other pathologies (neuroendocrine tumors and fibrous dysplasia) [Citation14,Citation15]. The occurrence of recurrent chromosomal translocations is relatively frequent, especially in the group of vascular tumors displaying intermediate biologic behavior (). Recurrent chromosomal translocations mostly occur in a background with simple genomics. Understanding the oncogenic mechanisms behind these fusion genes will aid in targeted therapies.
Table 1. Genetic alterations in vascular tumors
Table 2. Recurrent chromosomal translocations and immunohistochemical markers in translocation driven vascular tumors
Currently, the detection of fusion genes is frequently used as an auxiliary diagnostic tool to diagnose these vascular tumors, which are sometimes difficult to distinguish from each other, as reviewed recently [Citation18]. Identification of the involved genes increases our understanding of the oncogenic mechanisms behind these fusion genes that will ultimately aid in targeted therapies. The current review will provide a comprehensive overview of vascular tumor entities with gene fusions, their underlying molecular mechanism, and their impact on tumorigenesis and implications for targeted therapy.
2. Gene fusions in vascular tumors
2.1. Epithelioid hemangioma
Epithelioid hemangioma (EH) in soft tissue is a benign vascular neoplasm with well-formed blood vessels lined by epithelioid endothelial cells [Citation3]. Soft tissue EHs are predominantly located in the head and neck region and less frequently in other anatomic locations, such as distal extremities, trunk, and penis [Citation3]. In contrast, EH occurring in bone behaves more aggressively and is classified as a locally aggressive vascular neoplasm [Citation3]. The diagnosis of EH of bone might be difficult due to their multifocal tendency, making it sometimes challenging to distinguish from AS [Citation19,Citation20]. Rearrangements involving either FOS or FOSB gene were detected in one-third of EH cases, with FOS fusion twice as common as FOSB (). Histologically, three variants are now distinguished: the ‘conventional’ subtype displays mature vessels lined by epithelioid endothelial cells without a prominent inflammatory infiltrate [Citation18]. This subtype is more common in soft tissue, and in 75% of the cases, overexpression of FOSB can be seen. The ‘angiolymphoid hyperplasia with eosinophilia (ALHE)’ subtype contains inflammatory infiltrate including lymphoid follicles and eosinophils and is associated with overexpression of FOSB, exclusively, even though FOSB rearrangements are absent in ALHE [Citation21,Citation22]. Thus, FOSB expression does not necessarily coincide with FOSB rearrangement. The ‘cellular’ subtype is more common in bone and exhibits greater than 50% sheet-like growth of tumor cells. FOS rearrangements are more prevalent in EH of bone (up to 59%) (). As genetic alterations remain undetermined in two-third of EH cases, Antonescu and colleagues investigated a large cohort of fusion-negative EH cases, including patients with histological appearance of ALHE. They discovered a novel GATA6-FOXO1 fusion in 18% of them [Citation23]. None of the ALHE cases examined thus far harbor FOS, FOSB or FOXO1 fusions [Citation23,Citation24]. This evidence further supports that cutaneous ALHE, despite overexpression of FOSB, may be non-neoplastic [Citation3].
Figure 2. Epithelioid hemangioma and pseudomyogenic hemangioendothelioma. (a) Histological image of EH displaying small vessels with epithelioid endothelial cells and inflammation including eosinophils. (b) FOS expression in EH. (c) Histological image of PHE displaying spindle cells with abundant eosinophilic cytoplasm (d) overexpression of FOSB in PHE. (e) FOS rearrangements result in loss of the LALL motif, prohibiting degradation and thereby overexpression of the truncated protein. In FOSB rearrangements, most of the coding sequence of FOSB is placed under control of another promotor, causing upregulation of truncated FOSB expression. In both cases, overexpression of truncated FOS or FOSB affects the AP-1 complex and its downstream transcription activation [Citation25]. The novel chimeric GATA6-FOXO1 transcription factor results in transcriptional deregulation and affects downstream functions, presumably through PI3K–protein kinase B (PKB) signaling or Jun N-terminal kinase (JNK) signaling [Citation26]. The red points show the breakpoint. Light blue and green boxes indicate the protein regions involved in the fusion protein. Abbreviations: bZIP: basic leucine zipper domain; AA: amino acid
![Figure 2. Epithelioid hemangioma and pseudomyogenic hemangioendothelioma. (a) Histological image of EH displaying small vessels with epithelioid endothelial cells and inflammation including eosinophils. (b) FOS expression in EH. (c) Histological image of PHE displaying spindle cells with abundant eosinophilic cytoplasm (d) overexpression of FOSB in PHE. (e) FOS rearrangements result in loss of the LALL motif, prohibiting degradation and thereby overexpression of the truncated protein. In FOSB rearrangements, most of the coding sequence of FOSB is placed under control of another promotor, causing upregulation of truncated FOSB expression. In both cases, overexpression of truncated FOS or FOSB affects the AP-1 complex and its downstream transcription activation [Citation25]. The novel chimeric GATA6-FOXO1 transcription factor results in transcriptional deregulation and affects downstream functions, presumably through PI3K–protein kinase B (PKB) signaling or Jun N-terminal kinase (JNK) signaling [Citation26]. The red points show the breakpoint. Light blue and green boxes indicate the protein regions involved in the fusion protein. Abbreviations: bZIP: basic leucine zipper domain; AA: amino acid](/cms/asset/094ba4c7-ee29-47f3-b5be-0da0c74d68be/iero_a_1950533_f0002_oc.jpg)
Multiple gene fusion partners were identified for FOS including LMNA, MBNL1, VIM, or lincRNA [Citation24,Citation27,Citation28]. The FOS-VIM translocation is more frequently found than other fusion products. However, 50% of fusion partners in FOS rearrangements remained undefined as many of these rearrangements were detected by interphase-FISH tests only. FOS belongs to a basic Helix-Loop-Helix (bHLH) family of transcription factors with three other members: FOSB, FOSL1, and FOSL2. The FOS protein consists of a basic leucine zipper and a transactivation domain (). The FOS transcription family is part of the AP-1 complex that dimerize with each other through their basic leucine zipper (bZIP) domains to initiate transcriptional activities such as cellular growth, differentiation, and apoptosis [Citation29,Citation30]. Interestingly, all the breakpoints in the various FOS-rearrangements occur in exon 4 of FOS, and result in an early stop codon. Ijzendoorn et al. showed that, in vitro expression of the truncated FOS, mimicking the translocation, revealed the role of the highly conserved C-terminus LALL motif of FOS in degrading the FOS protein. In the absence of the aforementioned LALL motif, the FOS protein escapes from ubiquitin independent degradation and increases the expression of the stabilized FOS protein. The stabilization of FOS, in turn, stimulates endothelial sprouting through upregulation of matrix metalloproteinase (MMP) and components of the Notch signaling pathway [Citation31]. Taken together, this suggests that FOS rearrangements result in a gain-of-function to enhance vascular tumorigenesis by promoting endothelial sprouting by losing the C terminal part of the protein via fusion to various DNA elements. In vitro experiments showed that in the presence of MMP (Batimmastat) or Notch signaling pathway inhibitors (DAPT), the sprouting capability of truncated FOS is attenuated [Citation31]. However, further testing is required for the potential use of these drugs to treat patients with multifocal locally aggressive EH carrying FOS rearrangements.
The fusion partners of FOSB include ZFP36 and rarely WWTR1 or SETD1B [Citation32,Citation33]. The EH with SETD1B-FOSB fusion was located in the deep soft tissue of the chest wall and showed an unusual intravascular growth pattern [Citation33]. EH with FOSB rearrangements may exhibit atypical histologic features such as increased cellularity, nuclear pleomorphism, and focal necrosis [Citation32]. Like FOS, FOSB plays a role in the AP-1 transcription factor complex by dimerizing JUN proteins through their bZIP domain [Citation34]. FOSB rearrangements in EH have been reported to occur in intron 1, exon 1 or 2, with the bZIP and proline-rich transactivation domain retained, resulting in a promoter swap () [Citation32,Citation33]. Transcriptome sequencing analysis showed that FOSB chimeric transcripts uniquely upregulate the expression of FOSB and its downstream targets (IL8 and PENK) in EH [Citation32,Citation35]. The upregulation of IL-8, a chemotactic factor, may enhance angiogenic activity through modulating matrix metalloproteinases [Citation36]. Taken together, the promoter swap within the FOSB fusion propagates FOSB expression and dysregulates its transcriptional activity which may, in turn, enhance vascular tumorigenesis.
EH with FOXO1 rearrangement was found in superficial regions, either in the skin or subcutis, and showed variegated morphology, with alternating vasoformative and solid components [Citation23]. GATA6 is a transcription factor with a zinc finger-type DNA binding domain that plays a role in cellular differentiation and vertebrate development. The GATA family members have been shown to play a role in the development of breast-, gastrointestinal-, lung-, pancreatic-, ovarian-, and adrenocortical carcinomas, germ cell tumors as well as in megakaryoblastic leukemia in Down syndrome patients [Citation37–41]. Likewise, FOXO1 is also a transcription factor, which targets genes involved in apoptosis, metabolism, cell proliferation, and differentiation. FOXO1 is also involved in translocations in alveolar rhabdomyosarcoma (PAX3/PAX7-FOXO1) [Citation42–44]. Interestingly, an in vitro study that involved silencing of FOXO1 in endothelial cells revealed the role of FOXO1 in angiogenesis by regulating genes involved in vessel formation and maturation [Citation45]. In line with in vivo studies, FOXO1-deficient mice are embryonic lethal and showed impaired vascular development [Citation46]. In the GATA6-FOXO1 fusion identified in EH, all of the GATA6 functional domains were retained, although there was a partial loss of the N-terminal part of FOXO1, which disrupted the transcriptional activity of FOXO1 [Citation23]. As such, GATA6-FOXO1 translocation could disrupt angiogenic regulation of FOXO1 and increase the likelihood of tumor formation. FOXO1 gene targets, which modulate angiogenesis and vascular remodelings, such as angiopoietin-1, angiopoietin-2, and MMP10, could be drug targets in treating patients with such rearrangement [Citation45,Citation47]. However, further studies are required to verify this.
As the diagnosis of EH can be challenging, the genetic hallmarks identified in EH can help to distinguish benign EH from malignant vascular neoplasms and hence aid in clinical diagnosis and treatment. The assessment of the overexpression of FOS or FOSB in tumors using immunohistochemistry is a valuable auxiliary diagnostic tool, even though overexpression is not always correlated with an underlying gene fusion [Citation22,Citation48]. Also, FOS and FOSB rearrangements are not restricted to EH. For example, FOSB rearrangements with other gene partners (SERPINE1 and ACTB) are identified in pseudomyogenic hemangioendothelioma (PHE) () [Citation49,Citation50]. Rearrangement of FOXO1 is not only observed in EH but well-known in alveolar rhabdomyosarcoma. Also, translocations involving the FOS gene with other gene partners (ANKH, KIAA1199, MYO1B, IGR, and PPP1R100) are present in osteoid osteoma and osteoblastoma [Citation51,Citation52].
In general, patients with EH and local recurrence are cured by complete surgical excision [Citation3]. However, there is a need to develop therapeutic strategies, especially for situations where surgical excision is not possible or causes severe morbidity. Therapeutic strategies should focus on inhibiting the downstream effects caused by the upregulation of FOS/FOSB.
2.2. Pseudomyogenic hemangioendothelioma
PHE is a locally aggressive, rarely metastasizing endothelial neoplasm that occurs predominantly in young adult males. Histologically, these tumor consist of loose fascicles of plump spindle cells with abundant and brightly eosinophilic cytoplasm () [Citation3]. Immunohistochemically, there is characteristic expression of ERG, CD31, keratin AE1/AE3, while CD34 and desmin are negative and INI1 retained [Citation3,Citation18], highlighting its vascular differentiation despite the lack of vasoformation. The tumor is often multifocal, affecting different tissue planes. Similar to EH, FOSB rearrangements have been identified in PHE, causing overexpression of FOSB () and fusion partners mainly include SERPINE1 and ACTB (). Recently two additional rare FOSB fusion partners, WWTR1 and CLTC, were described [Citation53,Citation54]. Interestingly, identical FOSB fusions involving WWTR1 and ACTB were reported in EH and PHE [Citation32,Citation49,Citation53,Citation55].
SERPINE1 is an endothelial plasminogen activator inhibitor-1 (PAI1) that regulates hemostasis by inhibiting plasminogen activators. The translocation of SERPINE1-FOSB is associated with a breakpoint in exon 1 of SERPINE1 and exon 1 or 2 of FOSB [Citation56]. This genetic exchange retains the essential functional domains of FOSB, as described above, and creates a SERPINE1-FOSB chimeric fusion transcript under the control of the SERPINE1 promoter () [Citation56]. Other fusion partners of FOSB were also speculated to provide a more potent promoter for FOSB[Citation50,Citation53,Citation54]. As such, the fusion results in increased expression of FOSB. Indeed, FOSB is expressed in 96% and 54% of PHE and EH, respectively; indicating that FOSB is a useful immunohistochemical marker for PHE as well as EH [Citation48]. Interestingly, the immunoreactivity of FOSB observed in PHE was generally strong, while the reactivity varies from low to high in EH [Citation57]. Although rare, both EH and PHE may harbor identical genetic alteration (WWTR1-FOSB), however, they are clinically and morphologically distinct entities emphasizing the importance of interpreting molecular findings in the appropriate clinicopathological context. FOSB immunohistochemistry can be very helpful to distinguish PHE from epithelioid sarcoma, spindle cell carcinoma or AS [Citation18].
Most PHE patients undergo surgical excision, and about 60% of them experience local recurrences or develop additional nodules in the same anatomical region [Citation3]. Chemotherapy or radiotherapy is generally given to treat patients with multifocal disease; however, some treatments are shown to be ineffective in controlling the disease [Citation58]. Systemic therapies targeting mTOR inhibition (sirolimus, everolimus, and rapamycin) have shown clinical benefit in reported cases [Citation58–60]. Moreover, a patient treated with telatinib, a VEGFR1-4/PDGFRA multi-tyrosine kinase inhibitor, showed a complete remission [Citation61]. Due to the rarity of vascular tumors and the lack of patient-derived cell lines, functional studies are often difficult to perform. However, two in vitro models were generated to study the SERPINE1-FOSB translocation in PHE and to identify potential therapeutic targets [Citation61,Citation62]. Overexpression of truncated FOSB, mimicking the SERPINE1-FOSB fusion, in HUVECs revealed the autoregulation of the expression of the fusion. Moreover, telatinib, which was shown to induce complete remission in a single treated patient, could reduce the expression of the fusion by inhibiting FLT1, FLT4, and PDGFRA signaling [Citation61]. Subsequently, CRISPR-Cas9 was used to generate the SERPINE1-FOSB fusion in human induced pluripotent stem cells (hIPSCs) that were differentiated toward endothelial cells [Citation62]. Transcriptome analysis of these isogenic pairs revealed the PI3K-Akt-mTOR and MAPK signaling pathways as therapeutic options [Citation62]. Thus, anecdotal case reports combined with preclinical models provide a rationale for therapies involving the mTOR, VEGFR1-4/PDGFRA, PI3K-Akt, or MAPK signaling pathways to treat inoperable PHE.
2.3. Retiform hemangioendothelioma and composite hemangioendothelioma
Retiform hemangioendothelioma (RHE) is an extremely rare, locally aggressive, rarely metastasizing vascular neoplasm [Citation3]. It is histologically characterized by distinctive arborizing blood vessels lined by endothelial cells with characteristic hobnail morphology (). It is predominantly found in the skin and subcutaneous tissue of the distal extremities, particularly the lower limb [Citation3]. Classic composite hemangioendothelioma (CHE) is a very rare locally aggressive, rarely metastasizing vascular neoplasm that contains an admixture of histologically distinct components [Citation3]. At least two morphologically distinct patterns can be seen, which include EHE, RHE, spindle cell hemangioma, and angiosarcoma-like areas, as well as benign vascular lesions (lymphangioma, angiomatosis, vascular malformation, and cavernous hemangioma) making diagnosis challenging [Citation63]. Moreover, an aggressive form of CHE with neuroendocrine differentiation (expressing synaptophysin and chromogranin) has been described [Citation3]. In an attempt to elucidate the molecular genetic features of CHE, Perry et al. encountered single cases of neuroendocrine CHE bearing PTBP1-MAML2 or EPC1-PHC2 translocation [Citation64]. Lately, an additional case of neuroendocrine CHE harboring PTBP1-MAML2 was identified, again showing a highly aggressive clinical course [Citation65]. Thus, the neuroendocrine-CHE subtype harbors recurrent PTBP1-MAML2 rearrangements and behaves more aggressively, which makes it clinically relevant to separate from the classic CHE spectrum. Out of 11 classic CHE, three cases showed a YAP1-MAML2 fusion. These occurred in young girls at acral sites, while YAP1 or MAML2 fusion negative classic CHE occurred in adults [Citation65]. In the same study, a cohort of RHE cases was investigated, revealing the presence of YAP1 gene rearrangements. Among five of 13 RHE (38%) harboring YAP1 rearrangements, three have undefined fusion partners, while two remaining cases revealed a YAP1-MAML2 rearrangement. This suggests that RHE and CHE may be related entities with overlapping morphology and genetics.
Figure 3. Retiform and composite hemangioendothelioma. (a) Areas of retiform HE, as evidenced by the slit-like spaces, as part of a composite HE with neuroendocrine differentiation. (b) the YAP1-MAML2 fusion has been reported in both. MAML2 plays a role in various signaling pathways. Rearrangement of MAML2 is hypothesized to change the downstream effects of these signaling pathways [Citation66]. The red points show the breakpoints. Light blue and green boxes indicate the protein regions involved in fusion protein formation. Abbreviations: AA: amino acid; GLN: glycine; TAD: Transactivation domain; TEAD: TEA domain family member 1; WW: tryptophan-tryptophan domain
![Figure 3. Retiform and composite hemangioendothelioma. (a) Areas of retiform HE, as evidenced by the slit-like spaces, as part of a composite HE with neuroendocrine differentiation. (b) the YAP1-MAML2 fusion has been reported in both. MAML2 plays a role in various signaling pathways. Rearrangement of MAML2 is hypothesized to change the downstream effects of these signaling pathways [Citation66]. The red points show the breakpoints. Light blue and green boxes indicate the protein regions involved in fusion protein formation. Abbreviations: AA: amino acid; GLN: glycine; TAD: Transactivation domain; TEAD: TEA domain family member 1; WW: tryptophan-tryptophan domain](/cms/asset/300267da-a5c5-43e2-b83a-397bb7ee4dae/iero_a_1950533_f0003_oc.jpg)
Rearrangement of MAML2 is also commonly found in mucoepidermoid carcinoma (MEC), a salivary gland neoplasm [Citation67]. In all MAML2 rearrangements identified in CHE, the breakpoint within the MAML2 gene is located in exon 2, causing the loss of its N-terminal basic domain but retaining the transactivation acidic domain in the central and C-terminus (). MAML2 is known to be involved in several signaling pathways, such as the Notch, Wnt, Shh, nuclear factor-κB (NFKB), and hippo pathway [Citation68–71]. MAML proteins function as transcriptional coactivators in multiple signal transduction pathways, including Notch, shh, p53, and Wnt/β-catenin [Citation72]. Interestingly, the N-terminal conserved basic domain of MAML2 was revealed to bind to the ankyrin repeat domain of Notch receptors contributing to the Notch signaling pathway [Citation73]. Furthermore, MAML proteins form a DNA-binding complex with regulators of the Notch signaling pathway to potentiate Notch transcriptional activity [Citation74,Citation75]. The YAP1-MAML2 fusion protein in RHE would have 150 amino acids of YAP1 preserved while losing its C-terminal TAD domain. YAP1 is a transcriptional coactivator consisting of a TEAD binding domain and is the downstream effector of the Hippo tumor suppressor pathway [Citation76]. Valouev and colleagues proposed an oncogenic model describing the TEAD1-interaction domain of YAP1-MAML2 fusion in recruiting the fusion transcript to activate TEAD1 target genes in a Notch-independent manner [Citation77]. Recently, the MAML1/2 protein function expanded to the hippo signaling pathway by localizing to the nucleus to interact with YAP/TAZ, prompting the nuclear retention of YAP/TAZ and therefore enhancing the Hippo pathway-related transcriptional activity [Citation71]. Given the multifunctionality of MAML2, its rearrangement causes a chimeric gene that could disrupt multiple pathways involved ().
In the single case of EPC1-PHC2 fusion identified in CHE, the translocation results in the fusion of the highly conserved enhancer of polycomb A (EPcA) domain of EPC1 with PHC2 [Citation64]. In yeast, a truncated Epc1 construct consists solely of the EPcA domain revealed to be essential for nuclear localization of Epc1 and interaction with Hop, a transcriptional regulator [Citation78]. EPC1 rearrangements were previously found in ossifying fibromyxoid tumors (OFMT) and endometrial stromal sarcoma, but their oncogenesis is primarily unknown [Citation79].
The standard treatment for CHE and RHE thus far is surgical excision. Future targeted therapeutic strategies may focus on the hippo pathway, as activation of this pathway is seen in multiple cancers, including other vascular tumors (e.g. EHE), which may overcome difficulties in developing therapeutic strategies caused by their extreme rarity.
2.4. Epithelioid hemangioendothelioma
EHE is a malignant vascular neoplasm composed of epithelioid endothelial cells within a distinctive myxohyaline stroma and is commonly located in either the liver, lung, or bone (2020). Two morphological subtypes, carrying either WWTR1-CAMTA1 (80%) or YAP1-TFE3 (12%), have been described for EHE [Citation80,Citation81]. In classic EH, carrying the WWTR1-CAMTA1 fusion, tumor cells are lying in nests, strands or cords, while vasoformation is always absent (). Overexpression of nuclear CAMTA1 is constantly observed and is an ideal immunohistochemical marker () to distinguish EHE from metastatic carcinoma or AS [Citation82]. In YAP1-TFE3 rearranged EHE vasoformation is common, and the tumor cells have typical abundant, eosinophilic, sometimes feathery cytoplasm (). TFE3 immunohistochemistry is less specific to diagnose YAP1-TFE3 rearranged EHE and requires additional molecular verification () [Citation83].
Figure 4. Epithelioid hemangioendothelioma. (a) Histological image of classic EHE, displaying epithelioid endothelial cells lying in strands in a myxohyaline background, (b) nuclear CAMTA1 expression in classic EHE (c) Histological image of YAP1-TFE3 rearranged EHE, displaying a vasoformative architecture, where lumina are lined by endothelial cells with abundant cytoplasm (d) nuclear TFE3 expression in the epithelioid cells in YAP1-TFE3 rearranged EHE. (e) Both the WWTR1 and the TFE3 rearrangement result in a promoter swap that dysregulates the Hippo pathway transcriptional activities and downstream effects [Citation84]. The red points show the breakpoints. Light blue and green boxes indicate the protein regions involved in fusion protein formation. Abbreviations: ANK: ankyrin repeats; AA: amino acid; bHLH: basic helix–loop–helix; CG-1 and TIG: DNA binding domains; GLN: glycine; IQ: calmodulin-binding IQ motifs; LZ: leucine zipper domain; TAD: Transactivation domain; TEAD: TEA domain family member 1; WW: tryptophan-tryptophan domain
![Figure 4. Epithelioid hemangioendothelioma. (a) Histological image of classic EHE, displaying epithelioid endothelial cells lying in strands in a myxohyaline background, (b) nuclear CAMTA1 expression in classic EHE (c) Histological image of YAP1-TFE3 rearranged EHE, displaying a vasoformative architecture, where lumina are lined by endothelial cells with abundant cytoplasm (d) nuclear TFE3 expression in the epithelioid cells in YAP1-TFE3 rearranged EHE. (e) Both the WWTR1 and the TFE3 rearrangement result in a promoter swap that dysregulates the Hippo pathway transcriptional activities and downstream effects [Citation84]. The red points show the breakpoints. Light blue and green boxes indicate the protein regions involved in fusion protein formation. Abbreviations: ANK: ankyrin repeats; AA: amino acid; bHLH: basic helix–loop–helix; CG-1 and TIG: DNA binding domains; GLN: glycine; IQ: calmodulin-binding IQ motifs; LZ: leucine zipper domain; TAD: Transactivation domain; TEAD: TEA domain family member 1; WW: tryptophan-tryptophan domain](/cms/asset/39912186-2cbb-408b-86f0-abc7a9fdd1d9/iero_a_1950533_f0004_oc.jpg)
WWTR1 is also known as TAZ. Together with its paralog YAP, these are the two main effectors of the hippo pathway. The genomic DNA breakpoints occur in WWTR1 intron 2, 3, or 4 and CAMTA1 intron 7, 8, or exon 9 [Citation20,Citation85]. In case of WWTR1-CAMTA1 fusion, the N terminal WW (tryptophan-tryptophan) domain of WWTR1 is fused in-frame to the C terminal part of CAMTA1, encoding the transcriptional activation domain. This causes a promoter swap whereby the expression of CAMTA1 is placed under the control of the WWTR1 promoter () [Citation85]. The hippo-YAP/TAZ signaling pathway has been shown to mediate angiogenesis by the stimulation of VEGF, as outlined in two reviews [Citation86,Citation87]. The hippo pathway is known to negatively regulate WWTR1/TAZ [Citation88]. The chimeric WWTR1-CAMTA1 transcription factor was speculated to be independent of the hippo signaling pathway and to be constitutively activated in the nucleus [Citation89]. In vitro overexpression of WWTR1-CAMTA1 revealed upregulation of downstream targets such as CTGF [Citation89]. CTGF expression in endothelial cells induces cell proliferation and angiogenesis [Citation90].
In the YAP1-TFE3 rearrangement, the truncated N-terminal TEAD binding domain of YAP1 is fused to TFE3, with most functional domains retained. As such, YAP1 serves as a stronger promoter to TFE3 [Citation81,Citation91]. TFE3 functional domains are also preserved in other TFE3 rearranged cancers, such as renal cell carcinoma (RCC) and alveolar soft part sarcoma (ASPS) [Citation92,Citation93]. Morphologically, these tumors share the abundance of deep eosinophilic sometimes feathery cytoplasm. TFE3 is a member of the MiT/TFE family of transcription factors that bind to DNA and activate the transcription of target genes [Citation94]. Other than TFE3, the MiT/TFE family of transcription factors consists of MITF, TFEB, and TFEC, having a bZIP domain and a transactivation domain. MiT/TFE family of transcription factors hetero- and homodimerize with each other and enhance DNA binding affinity and subsequent transcriptional activation [Citation94,Citation95]. Considerable evidence has suggested an essential role for MIT/TFE in angiogenesis [Citation96,Citation97].
The treatment of EHE includes surgery, cytotoxic chemotherapy, immunomodulatory agents (thalidomide or lenalidomide), or targeted therapies (apatinib, sorafenib, pazopanib, or sirolimus) documented in a review [Citation98]. Currently, therapeutic targeting of CTGF in EHE with the MEK inhibitor trametinib, which has been shown to reduce the expression of CTGF, is under phase 2 clinical trial (Trial ID: NCT03148275). Even though various EHE treatments have shown positive responses, the efficacy of these treatments is inconsistent.
2.5. Angiosarcoma
AS is a high-grade malignant vascular neoplasm that variably recapitulates the morphological and immunohistochemical features of endothelial cells [Citation3]. The histological characteristic of AS varies from well-formed, anastomosing vessels to solid sheets of high-grade epithelioid or spindled cells without clear vasoformation () [Citation3]. These tumors are aggressive and frequently occur in cutaneous sites, followed by deep soft tissue, breast, bone or viscera. Unlike the group of haemangioendotheliomas, they are genetically heterogeneous; most have complex karyotypes with various chromosomal alterations such as point mutations (PTPBR, PLCG1, KDR, or CIC) or amplification (MYC or FLT4). Some AS have simple genomes, for example, with amplification of SKI or KDR mutation [Citation99]. Genes involved in such alterations tend to affect the regulation of angiogenesis or vascular-specific receptor tyrosine kinases (including TIE1, TEK, KDR, and FLT4) [Citation100,Citation101]. CIC gene abnormalities were reported in nine percent of young adult patients with primary AS [Citation102]. These abnormalities include CIC-LEUTX fusion, CIC rearrangement (unknown partner), and CIC missense mutation. Interestingly, CIC rearranged AS patients lack vasoformative features. CIC rearrangements (with fusion partners such as DUX4, FOXO4, LEUTX, NUTM1, and NUTM2A) were previously described to characterize an undifferentiated round cell sarcoma that is now referred to as CIC-rearranged sarcoma [Citation3,Citation103]. CIC rearranged AS are visceral and have small cell features similar to CIC rearranged sarcoma [Citation102].
Figure 5. Angiosarcoma. (a) Histological image of AS displaying highly atypical endothelial cells (b) CIC rearrangement is found in a small subset of AS and results in a chimeric gene that dysregulates transcriptional repression of the PEA3 family of transcription factors [Citation104]. The red points show the breakpoints. Abbreviations: ATXN1: Ataxin 1 domain; C1, C1 motif; HMG: high mobility group box domain; NLS: nuclear localization signal
![Figure 5. Angiosarcoma. (a) Histological image of AS displaying highly atypical endothelial cells (b) CIC rearrangement is found in a small subset of AS and results in a chimeric gene that dysregulates transcriptional repression of the PEA3 family of transcription factors [Citation104]. The red points show the breakpoints. Abbreviations: ATXN1: Ataxin 1 domain; C1, C1 motif; HMG: high mobility group box domain; NLS: nuclear localization signal](/cms/asset/1b634060-768c-4fef-9fb0-cd77066ee617/iero_a_1950533_f0005_oc.jpg)
CIC is a tissue-specific transcriptional repressor. It is a highly conserved gene reported to repress the receptor tyrosine kinase (RTK) signaling pathway in metazoan [Citation105]. CIC can appear in two isoforms that differ in size: a short (CIC-S) and long (CIC-L) isoform. Both isoforms have an N terminal binding domain that interacts with ataxin-1 or 14-3-3, a high mobility group (HMG) box domain, and a C terminal nuclear localization signal, as well as a highly conserved motif (C1) (). Both the HMG box and C1 motif function are required for the DNA binding capability of CIC to repress target genes such as the ETV family of transcription factors [Citation106]. This family (ETV1, ETV4, and ETV5) belongs to the PEA3 subfamily of ETS transcription factors. In the CIC-LEUTX rearrangement, exon 20 of CIC is fused to exon 3 of LEUTX, thereby disrupting the C1 motif and affecting the repressor activity of CIC. Overexpression of the CIC protein and upregulation of target genes such as ETV1, ETV4, and ETV5 were observed in angiosarcoma with CIC rearrangement [Citation102]. The effect of upregulation of ETV1, ETV4, and ETV5 was studied in various contexts, all of which point to enhancing tumor progression by affecting the tumor-suppressive function of CIC [Citation107,Citation108]. For instance, the overexpression of ETV4 has been reported to promote angiogenesis by increasing VEGF activity in breast cancer cells [Citation109].
PEA3 is activated by the ERK-MAP kinase pathway [Citation110]. In turn, potential therapeutic therapies targeting the ERK MAP kinase pathway might suppress the upregulation of PEA3 transcription factors in CIC rearranged. Various activation and deactivation mechanisms controlled by ERK and DUSP6 might give therapeutic possibilities depending on the mechanism of CIC inactivation, but due to the complex regulatory mechanism, no clear therapeutic option can be offered [Citation111]. Further research is needed in this field. In general, treatment options for AS include surgery, radiotherapy, and chemotherapy. AS is heterogeneous at the molecular level, which provides fuel to drug resistance, making targeted drug selection difficult [Citation112]. Emerging data have shown the possible use of targeted and immunotherapy. For example, targeted therapies using tyrosine kinase inhibitors such as pazopanib and sorafenib targeting the VEGF signaling pathway showed promising results [Citation113]. Currently, multiple therapies are under clinical trial, and an optimal treatment strategy for the various molecular subtypes of AS has yet to be achieved.
3. Conclusion
Although various chromosomal translocations were identified in different vascular tumor entities, in a substantial number of vascular tumors no genetic alterations have (yet) been identified. The identification of fusion genes causing overexpression of one of the fusion partners (FOS, FOSB, CAMTA1, and TFE3) has led to the routine use of surrogate immunohistochemical markers by specialized pathologists in the differential diagnosis of vascular tumors. However, not all fusions and immunohistochemical markers are entirely specific (for example, identical FOSB gene alterations occurring in both PHE and EH) and the diagnosis should always be made in the appropriate clinical, radiological and histological context. In addition, gene fusions in vascular tumors are not always exclusive to vascular tumors, as they can be found in other tumor entities as well (for instance FOS and FOSB fusions in osteoid osteoma and osteoblastoma) [Citation13]. Gene fusions additionally provide us with more insight into understanding the possible underlying mechanisms leading to vascular tumor formation. With this knowledge, model systems can be developed to test potential targeted therapies.
4. Expert opinion
The differential diagnosis of vascular tumors is challenging as they have overlapping clinical presentation and morphology. They tend to present as multifocal lesions, which can resemble the clinical presentation of a metastasized malignant tumor, especially since they frequently express keratin AE1/AE3. Moreover, the multifocality can make surgical treatment very difficult and sometimes impossible [Citation61].
Next generation sequencing technologies have identified a range of recurrent translocations in vascular tumors, especially those of intermediate biologic behavior (). For routine diagnostic use these can be detected in formalin fixed paraffin embedded tissue using interphase-FISH, which will reveal whether the gene of interest is involved in a translocation or not, which often is sufficient. Recently, targeted NGS panels become available, which can be applied to FFPE tissue as well, interrogating multiple genes and which may also elucidate the fusion partners, depending on the chosen NGS technology [Citation2].
The elucidation of these recurrent gene fusions has had a great impact on diagnostic accuracy in vascular tumors. Since some of these fusions can be detected using surrogate immunohistochemical markers, this has provided the expert pathologist with useful diagnostic tools to aid in the distinction between the various vascular tumors. However, the pathologist should be aware of sensitivity, specificity and other pitfalls such as the effect of decalcification on immunoreactivity of some antigens [Citation2].
As an example, for EH, it has become possible over the past decade to integrate morphology and genetics which has made it clear that EH is highly heterogeneous, both at clinical presentation, at histology [Citation18], and the molecular level. In EH of bone for instance, FOS rearrangements are most common [Citation27] and histology is usually cellular and spindled, especially at the acral sites [Citation18]. Immunohistochemistry for FOS or FOSB can be useful diagnostic tools as a surrogate for molecular testing in these cases, as sometimes it can be very difficult to distinguish the cellular variant from epithelioid angiosarcoma of bone. FOS and FOSB were so far not represented in commercially available NGS fusion panels, and no easily applicable commercial FISH probes are available. However, especially in bone, one should realize that the FOS antibody is vulnerable to decalcification and can give false-negative results, while polysomy can give false-positive results [Citation114], in which case molecular confirmation may be required. In contrast to EH of bone, the ALHE-like variant of EH expresses FOSB in almost all cases [Citation21,Citation22], while at the molecular level no FOSB fusions have been found. The underlying mechanism causing FOSB overexpression in these lesions still remains to be determined. The FOSB overexpression could be a derivative of undiscovered genetic alterations related to the AP-1 complex. Thus, the majority of EH, especially those in soft tissue, do not have FOS or FOSB rearrangement. Recently, FOXO1 rearrangements were detected in a subset of EH lacking FOS or FOSB rearrangement.
The identification of gene fusions has not only improved the classification and diagnosis of vascular tumors, it also sheds light on the underlying molecular mechanisms involved in tumorigenesis. By exploring these molecular pathways, leads can be found to develop molecularly targeted therapy. Since some of these vascular tumors, despite their intermediate biologic behavior, can be very difficult to treat there is an urgent need to develop novel therapeutic strategies [Citation61]. Functional studies are however hampered by the lack of model systems, as no cell lines or animal models are available. However, novel technologies like CRISPR-Cas9 can be used to create gene fusions in induced pluripotent stem cells which allows studying the effect of the specific gene fusion in an isogenic background and the identification of therapeutic vulnerabilities [Citation62]. For instance, the inhibition of PI3K and MAPK signaling was identified as a therapeutic option in PHE [Citation62], and indeed case reports describe the usability of mTOR and multi-tyrosine kinase inhibitor [Citation58–61].
Also, because hemangioendotheliomas are extremely rare, for the development of novel therapeutic strategies it would be good to focus on activated pathways that are shared between different vascular tumors. For instance, the hippo signaling pathway contributes to tumorigenesis in various vascular tumors, including MAML2 rearranged CHE, YAP1 rearranged RHE, and WWTR1-CAMTA1 and YAP1-TFE3 rearranged EHE. The hippo signaling pathway mediated gene upregulation observed in these tumors could be used as a therapeutic option, such as targeting CTGF in EHE which is currently explored in a clinical trial using a MEK inhibitor (trametinib) (Trial ID: NCT03148275).
Thus, the identification of gene fusions in vascular tumors has led to improved diagnosis and classification, as well as to insight in tumorigenesis and identification of potential novel leads to develop targeted therapy. Future strategies should focus on those rare entities where still a subset of the tumors is fusion negative, and on the development of model systems to enable functional studies exploring these molecular mechanisms and the potential to interfere with these to improve treatment for patients with multifocal inoperable vascular tumors.
Declaration of interest
The authors have no conflicts of interest to declare.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Article highlights
The detection of fusion genes can be used as an auxiliary diagnostic tool in vascular tumors.
FOS, FOSB, TFE3 and CAMTA1 immunohistochemistry can be helpful as surrogates for the detection of a translocation.
Expression of FOSB or TFE3 does not always correlate with an underlying gene fusion, and molecular confirmation may be required.
Gene fusions in vascular tumors are not always specific and interpretation should be done within the appropriate histopathological context by an expert pathologist.
Further understanding of the oncogenic pathways that are regulated by the various gene fusions may provide leads to develop targeted therapeutic strategies.
Common hippo pathway alteration is observed in epithelioid hemangioendothelioma (WWTR1-CAMTA1 or YAP1-TFE3) and retiform hemangioendothelioma/composite hemangioendothelioma (YAP1-MAML2).
Supplemental Material
Download Zip (7.1 MB)Acknowledgment
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Selleri L, Hermanson GG, Eubanks JH, et al. Molecular localization of the t(11;22)(q24;q12) translocation of Ewing sarcoma by chromosomal in situ suppression hybridization. Proc Natl Acad Sci U S A. 1991;88(3):887–891.
- Lam SW, IJzendoorn DGP, Cleton-Jansen AM, et al. Molecular Pathology of Bone Tumors. J Mol Diagn. 2019;21(2):171–182.
- WHO classification of tumours of soft tissue and bone. WHO classification of Tumours Editiorial Board. 5th edition Vol. 3 Lyon France: IARC; 2020.
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7(4):233–245.
- Roukos V, Misteli T. The biogenesis of chromosome translocations. Nat Cell Biol. 2014;16(4):293–300.
- Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359(6391):162–165.
- Sand LG, Szuhai K, Hogendoorn PC. Sequencing Overview of Ewing Sarcoma: a Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int J Mol Sci. 2015;16(7):16176–16215.
- Riggi N, Suva ML, Stamenkovic I. Ewing’s Sarcoma. N Engl J Med. 2021;384(2):154–164.
- Oliveira AM, Hsi BL, Weremowicz S, et al. USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res. 2004;64(6):1920–1923.
- Lamerato-Kozicki AR, Helm KM, Jubala CM, et al. Canine hemangiosarcoma originates from hematopoietic precursors with potential for endothelial differentiation. Exp Hematol. 2006;34(7):870–878.
- Kakiuchi-Kiyota S, Crabbs TA, Arnold LL, et al. Evaluation of expression profiles of hematopoietic stem cell, endothelial cell, and myeloid cell antigens in spontaneous and chemically induced hemangiosarcomas and hemangiomas in mice. Toxicol Pathol. 2013;41(5):709–721.
- Liau JY, Tsai JH, Lan J, et al. GNA11 joins GNAQ and GNA14 as a recurrently mutated gene in anastomosing hemangioma. Virchows Arch. 2020;476(3):475–481.
- Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602.
- Landis CA, Masters SB, Spada A, et al. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–696.
- Rao VV, Schnittger S, Hansmann I. G protein Gs alpha (GNAS 1), the probable candidate gene for Albright hereditary osteodystrophy, is assigned to human chromosome 20q12-q13.2. Genomics. 1991;10(1):257–261.
- Zhu G, Benayed R, Ho C, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol. 2019;32(5):609–620.
- Tsukamoto Y, Futani H, Watanabe T, et al. Two cases of WWTR1-CAMTA-1 fusion-positive epithelioid hemangioendotheliomas with extremely different outcomes. Hum Pathol. 2018;14:25–32.
- Papke DJ Jr., Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476(1):17–28.
- Evans HL, Raymond AK, Ayala AG. Vascular tumors of bone: a study of 17 cases other than ordinary hemangioma, with an evaluation of the relationship of hemangioendothelioma of bone to epithelioid hemangioma, epithelioid hemangioendothelioma, and high-grade angiosarcoma. Hum Pathol. 2003;34(7):680–689.
- Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50(8):644–653.
- Llamas-Velasco M, Kempf W, Cota C, et al. Multiple Eruptive Epithelioid Hemangiomas: a Subset of Cutaneous Cellular Epithelioid Hemangioma With Expression of FOS-B. Am J Surg Pathol. 2019;43(1):26–34.
- Ortins-Pina A, Llamas-Velasco M, Turpin S, et al. FOSB immunoreactivity in endothelia of epithelioid hemangioma (angiolymphoid hyperplasia with eosinophilia). J Cutan Pathol. 2018;45(6):395–402.
- Antonescu CR, Huang SC, Sung YS, et al. Novel GATA6-FOXO1 fusions in a subset of epithelioid hemangioma. Mod Pathol. 2020;34:934–941.
- Huang SC, Zhang L, Sung YS, et al. Frequent FOS Gene Rearrangements in Epithelioid Hemangioma: a Molecular Study of 58 Cases With Morphologic Reappraisal. Am J Surg Pathol. 2015;39(10):1313–1321.
- Bejjani F, Evanno E, Zibara K, et al. The AP-1 transcriptional complex: local switch or remote command? Biochim Biophys Acta Rev Cancer. 2019;1872(1):11–23.
- Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97.
- IJzendoorn DGP, Jong D, Romagosa C, et al. Fusion events lead to truncation of FOS in epithelioid hemangioma of bone. Genes Chromosomes Cancer. 2015;54(9):565–574.
- Tsuda Y, Suurmeijer AJH, Sung YS, et al. Epithelioid hemangioma of bone harboring FOS and FOSB gene rearrangements: a clinicopathologic and molecular study. Genes Chromosomes Cancer. 2021;60(1):17–25.
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072(2–3):129–157.
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–2400.
- IJzendoorn DGP, Forghany Z, Liebelt F, et al., Functional analyses of a human vascular tumor FOS variant identify a novel degradation mechanism and a link to tumorigenesis. J Biol Chem. 2017;292(52):21282–21290. .
- Antonescu CR, Chen HW, Zhang L, et al. ZFP36-FOSB fusion defines a subset of epithelioid hemangioma with atypical features. Genes Chromosomes Cancer. 2014;53(11):951–959.
- Ooi LY, Goh JY, Kuick CH, et al. Intravascular epithelioid haemangioma of deep soft tissue with novel SETD1B-FOSB gene rearrangement. Pathology. 2020;23(2):270–273.
- Curran T, Franza BR Jr. Fos and Jun: the AP-1 connection. Cell. 1988;55(3):395–397.
- Yutsudo N, Kamada T, Kajitani K, et al. fosB-null mice display impaired adult hippocampal neurogenesis and spontaneous epilepsy with depressive behavior. Neuropsychopharmacology. 2013;38(5):895–906.
- Li A, Dubey S, Varney ML, et al. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170(6):3369–3376.
- Kiiveri S, Liu J, Arola J, et al. Transcription factors GATA-6, SF-1, and cell proliferation in human adrenocortical tumors. Mol Cell Endocrinol. 2005;233(1–2):47–56.
- Caslini C, Capo-chichi CD, Roland IH, et al. Histone modifications silence the GATA transcription factor genes in ovarian cancer. Oncogene. 2006;25(39):5446–5461.
- Kwei KA, Bashyam MD, Kao J, et al. Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genet. 2008;4(5):e1000081.
- Salonen J, Rajpert-De Meyts E, Mannisto S, et al. Differential developmental expression of transcription factors GATA-4 and GATA-6, their cofactor FOG-2 and downstream target genes in testicular carcinoma in situ and germ cell tumors. Eur J Endocrinol. 2010;162(3):625–631.
- Zheng R, Blobel GA. GATA Transcription Factors and Cancer. Genes Cancer. 2010;1(12):1178–1188.
- Barr FG, Galili N, Holick J, et al. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;3(2):113–117.
- Galili N, Davis RJ, Fredericks WJ, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5(3):230–235.
- Davis RJ, D’Cruz CM, Lovell MA, et al. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54(11):2869–2872.
- Potente M, Urbich C, Sasaki K, et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115(9):2382–2392.
- Furuyama T, Kitayama K, Shimoda Y, et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279(33):34741–34749.
- Daly C, Wong V, Burova E, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1. Genes Dev. 2004;18(9):1060–1071.
- Hung YP, Fletcher CDM, Hornick JL. FOSB is a Useful Diagnostic Marker for Pseudomyogenic Hemangioendothelioma. Am J Surg Pathol. 2017;41(5):596–606.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13)-a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet. 2011;204(4):211–215.
- Agaram NP, Zhang L, Cotzia P, et al. Expanding the Spectrum of Genetic Alterations in Pseudomyogenic Hemangioendothelioma With Recurrent Novel ACTB-FOSB Gene Fusions. Am J Surg Pathol. 2018;42(12):1653–1661.
- Fittall MW, Mifsud W, Pillay N, et al. Recurrent rearrangements of FOS and FOSB define osteoblastoma. Nat Commun. 2018;9(1):2150.
- Panagopoulos I, Gorunova L, Lobmaier I, et al. FOS-ANKH and FOS-RUNX2 Fusion Genes in Osteoblastoma. Cancer Genomics Proteomics. 2020;17(2):161–168.
- Panagopoulos I, Lobmaier I, Gorunova L, et al. Fusion of the Genes WWTR1 and FOSB in Pseudomyogenic Hemangioendothelioma. Cancer Genomics Proteomics. 2019;16(4):293–298.
- Bridge JA, Sumegi J, Royce T, et al. A novel CLTC-FOSB gene fusion in pseudomyogenic hemangioendothelioma of bone. Genes Chromosomes Cancer. 2021;60(1):38–42.
- Thway K, Fisher C. Mesenchymal Tumors with EWSR1 Gene Rearrangements. Surg Pathol Clin. 2019;12(1):165–190.
- Walther C, Tayebwa J, Lilljebjorn H, et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J Pathol. 2014;232(5):534–540.
- Hung YP, Fisch AS, Diaz-Perez JA, et al. Identification of EWSR1-NFATC2 Fusion in Simple Bone Cysts. Histopathology. 2020;78:849–856.
- Joseph J, Wang WL, Patnana M, et al. Cytotoxic and targeted therapy for treatment of pseudomyogenic hemangioendothelioma. Clin Sarcoma Res. 2015;5(1):22.
- Ozeki M, Nozawa A, Kanda K, et al. Everolimus for Treatment of Pseudomyogenic Hemangioendothelioma. J Pediatr Hematol Oncol. 2017;39(6):e328–e331.
- Gabor KM, Sapi Z, Tiszlavicz LG, et al. Sirolimus therapy in the treatment of pseudomyogenic hemangioendothelioma. Pediatr Blood Cancer. 2018;65(2):2.
- IJzendoorn DGP, Sleijfer S, Gelderblom H, et al. Telatinib Is an Effective Targeted Therapy for Pseudomyogenic Hemangioendothelioma. Clin Cancer Res. 2018;24(11):2678–2687.
- IJzendoorn DGP, Salvatori DCF, Cao X, et al. Vascular Tumor Recapitulated in Endothelial Cells from hiPSCs Engineered to Express the SERPINE1-FOSB Translocation. Cell Rep Med. 2020;1(9):100153.
- Requena L, Luis Diaz J, Manzarbeitia F, et al. Cutaneous composite hemangioendothelioma with satellitosis and lymph node metastases. J Cutan Pathol. 2008;35(2):225–230.
- Perry KD, Al-Lbraheemi A, Rubin BP, et al. Composite hemangioendothelioma with neuroendocrine marker expression: an aggressive variant. Mod Pathol. 2017;30(10):1512.
- Antonescu CR, Dickson BC, Sung YS, et al., Recurrent YAP1 and MAML2 Gene Rearrangements in Retiform and Composite Hemangioendothelioma. Am J Surg Pathol. 2020;44(12):1677–1684. .
- Zema S, Pelullo M, Nardozza F, et al. Role of Mastermind-Like 1: a Journey Through the Main (Path)ways Between Development and Cancer. Front Cell Dev Biol. 2020;8:613557.
- Chenevert J, Barnes LE, Chiosea SI. Mucoepidermoid carcinoma: a five-decade journey. Virchows Arch. 2011;458(2):133–140.
- Alves-Guerra MC, Ronchini C, Capobianco AJ. Mastermind-like 1 Is a specific coactivator of beta-catenin transcription activation and is essential for colon carcinoma cell survival. Cancer Res. 2007;67(18):8690–8698.
- Jin B, Shen H, Lin S, et al. The mastermind-like 1 (MAML1) co-activator regulates constitutive NF-kappaB signaling and cell survival. J Biol Chem. 2010;285(19):14356–14365.
- Quaranta R, Pelullo M, Zema S, et al. Maml1 acts cooperatively with Gli proteins to regulate sonic hedgehog signaling pathway. Cell Death Dis. 2017;8(7):e2942.
- Kim J, Kwon H, Shin YK, et al. MAML1/2 promote YAP/TAZ nuclear localization and tumorigenesis. Proc Natl Acad Sci U S A. 2020;117(24):13529–13540.
- McElhinny AS, Li JL, Wu L. Mastermind-like transcriptional co-activators: emerging roles in regulating cross talk among multiple signaling pathways. Oncogene. 2008;27(38):5138–5147.
- Wu L, Sun T, Kobayashi K, et al. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol. 2002;22(21):7688–7700.
- Nam Y, Sliz P, Song L, et al. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124(5):973–983.
- Wilson JJ, Kovall RA. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell. 2006;124(5):985–996.
- Li Z, Zhao B, Wang P, et al. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24(3):235–240.
- Valouev A, Weng Z, Sweeney RT, et al. Discovery of recurrent structural variants in nasopharyngeal carcinoma. Genome Res. 2014;24(2):300–309.
- Kee HJ, Kim JR, Nam KI, et al. Enhancer of polycomb1, a novel homeodomain only protein-binding partner, induces skeletal muscle differentiation. J Biol Chem. 2007;282(10):7700–7709.
- Antonescu CR, Sung YS, Chen CL, et al. Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors--molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosomes Cancer. 2014;53(2):183–193.
- Mendlick MR, Nelson M, Pickering D, et al. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am J Surg Pathol. 2001;25(5):684–687.
- Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer. 2013;52(8):775–784.
- Shibuya R, Matsuyama A, Shiba E, et al. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology. 2015;67(6):827–835.
- Sharain RF, Gown AM, Greipp PT, et al. Immunohistochemistry for TFE3 lacks specificity and sensitivity in the diagnosis of TFE3-rearranged neoplasms: a comparative, 2-laboratory study. Hum Pathol. 2019;87:65–74.
- Yang M, Liu E, Tang L, et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal. 2018;16(1):31.
- Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3(98):98ra82.
- Park JA, Kwon YG. Hippo-YAP/TAZ signaling in angiogenesis. BMB Rep. 2018;51(3):157–162.
- Taha Z, Janse van Rensburg HJ, Yang X. The Hippo Pathway: immunity and Cancer. Cancers (Basel). 2018;10(4):4.
- Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23(7):785–793.
- Tanas MR, Ma S, Jadaan FO, et al., Mechanism of action of a WWTR1(TAZ)-CAMTA1 fusion oncoprotein. Oncogene. 2016;35(7):929–938. .
- Shimo T, Nakanishi T, Nishida T, et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 1999;126(1):137–145.
- Puls F, Niblett A, Clarke J, et al. YAP1-TFE3 epithelioid hemangioendothelioma: a case without vasoformation and a new transcript variant. Virchows Arch. 2015;466(4):473–478.
- Jong B, Molenaar IM, Leeuw JA, et al. Cytogenetics of a renal adenocarcinoma in a 2-year-old child. Cancer Genet Cytogenet. 1986;21(2):165–169.
- Ladanyi M, Lui MY, Antonescu CR, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20(1):48–57.
- Pogenberg V, Ogmundsdottir MH, Bergsteinsdottir K, et al. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012;26(23):2647–2658.
- Hemesath TJ, Steingrimsson E, McGill G, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8(22):2770–2780.
- Busca R, Berra E, Gaggioli C, et al. Hypoxia-inducible factor 1{alpha} is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J Cell Biol. 2005;170(1):49–59.
- Fan Y, Lu H, Liang W, et al. Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ Res. 2018;122(7):945–957.
- Rosenberg A, Agulnik M. Epithelioid Hemangioendothelioma: update on Diagnosis and Treatment. Curr Treat Options Oncol. 2018;19(4):19.
- Verbeke SL, Jong D, Bertoni F, et al. Array CGH analysis identifies two distinct subgroups of primary angiosarcoma of bone. Genes Chromosomes Cancer. 2015;54(2):72–81.
- Itakura E, Yamamoto H, Oda Y, et al. Detection and characterization of vascular endothelial growth factors and their receptors in a series of angiosarcomas. J Surg Oncol. 2008;97(1):74–81.
- Antonescu CR, Yoshida A, Guo T, et al. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res. 2009;69(18):7175–7179.
- Huang SC, Zhang L, Sung YS, et al. Recurrent CIC Gene Abnormalities in Angiosarcomas: a Molecular Study of 120 Cases With Concurrent Investigation of PLCG1, KDR, MYC, and FLT4 Gene Alterations. Am J Surg Pathol. 2016;40(5):645–655.
- Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15(13):2125–2137.
- Qi T, Qu Q, Li G, et al. Function and regulation of the PEA3 subfamily of ETS transcription factors in cancer. Am J Cancer Res. 2020;10(10):3083–3105.
- Jimenez G, Shvartsman SY, Paroush Z. The Capicua repressor--a general sensor of RTK signaling in development and disease. J Cell Sci. 2012;125(Pt 6):1383–1391.
- Fores M, Simon-Carrasco L, Ajuria L, et al. A new mode of DNA binding distinguishes Capicua from other HMG-box factors and explains its mutation patterns in cancer. PLoS Genet. 2017;13(3):e1006622.
- Oh S, Shin S, Song H, et al. Relationship between ETS Transcription Factor ETV1 and TGF-beta-regulated SMAD Proteins in Prostate Cancer. Sci Rep. 2019;9(1):8186.
- Meng D, Li Z, Ma X, et al. ETV5 overexpression contributes to tumor growth and progression of thyroid cancer through PIK3CA. Life Sci. 2020;253:117693.
- Hua D, Chen B, Bai M, et al. PEA3 activates VEGF transcription in T47D and SKBR3 breast cancer cells. Acta Biochim Biophys Sin (Shanghai). 2009;41(1):63–68.
- Keld R, Guo B, Downey P, et al. The ERK MAP kinase-PEA3/ETV4-MMP-1 axis is operative in oesophageal adenocarcinoma. Mol Cancer. 2010;9(1):313.
- Kim JW, Ponce RK, Okimoto RA. Capicua in Human Cancer. Trends Cancer. 2021;7(1):77–86.
- Boichard A, Wagner MJ, Kurzrock R. Angiosarcoma heterogeneity and potential therapeutic vulnerability to immune checkpoint blockade: insights from genomic sequencing. Genome Med. 2020;12(1):1.
- Melincovici CS, Bosca AB, Susman S, et al. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. 2018;59(2):455–467.
- Lam SW, Cleven AHG, Kroon HM, et al. Utility of FOS as diagnostic marker for osteoid osteoma and osteoblastoma. Virchows Arch. 2020;476(3):455–463.