
ABSTRACT
Rapid and sensitive diagnostic strategies are necessary for patient care and public health. Most of the current conventional microbiological assays detect only a restricted panel of pathogens at a time or require a microbe to be successfully cultured from a sample. Clinical metagenomics next-generation sequencing (mNGS) has the potential to unbiasedly detect all pathogens in a sample, increasing the sensitivity for detection and enabling the discovery of unknown infectious agents. High expectations have been built around mNGS; however, this technique is far from widely available. This review highlights the advances and currently available options in terms of costs, turnaround time, sensitivity, specificity, validation, and reproducibility of mNGS as a diagnostic tool in clinical microbiology laboratories. The need for a novel diagnostic tool to increase the sensitivity of microbial diagnostics is clear. mNGS has the potential to revolutionize clinical microbiology. However, its role as a diagnostic tool has yet to be widely established, which is crucial for successfully implementing the technique. A clear definition of diagnostic algorithms that include mNGS is vital to show clinical utility. Similarly to real-time PCR, mNGS will one day become a vital tool in any testing algorithm.
1. Introduction
Rapid identification and characterization of microbial pathogens are the main goals of any new microbiological diagnostic technique. Rapid diagnostics of an infectious agent will ensure the most appropriate treatment option and patient management decision. In the last 50 years, several diagnostic approaches have been introduced in medical microbiology, namely amplification-based (PCR), MALDI-TOF, DNA-microarray-based hybridization technology, T2 magnetic resonance and next-generation sequencing [Citation1]. However, none of these methods could fully replace standard techniques (microscopy, culture, and serology) [Citation2,Citation3]. Molecular biology revolutionized the diagnosis of infectious diseases [Citation4], especially for detecting viruses and identifying bacteria involved in sexually transmitted infections, gastrointestinal infections, and tuberculosis. Still, today’s clinical microbiology laboratories have not changed dramatically since the early 2000s. This is mainly due to the advantages of traditional standard techniques. Firstly, cost-effectiveness and extensive clinical validation [Citation3] and secondly, limitations of newer methods such as limited spectrum, sensitivity and specificity, the lack of differentiation between living and dead cells [Citation5] and the importance of phenotypic antimicrobial susceptibility testing [Citation1]. Moreover, one existing challenge in diagnostics remains; a priori knowledge of what to expect from a particular clinical sample or patient. In most cases, a priori knowledge is enough to request the most appropriate test, such as multiplexed panels or specific culture media, but this is not always the case.
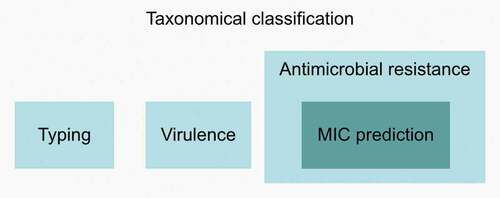
mNGS has the potential to surpass many limitations of current routine diagnostics methods. It can reveal information at different levels, including detecting and characterizing all microorganisms and viruses (DNA and RNA) without a priori knowledge from a single test (). Identification of pathogens is the first level of information that may be sufficient for some diagnostic purposes. Rapid identification is also crucial to inform the attending clinician to stop/prevent unnecessary antimicrobial prescription, particularly if a virus has been determined to be the infectious causative agent. However, for most patients who would benefit from an mNGS-based diagnostic approach, additional information to guide proper treatment is critical. This includes the detection of virulence factors, such as Shiga-toxin genes in Escherichia coli to avoid antimicrobial treatment [Citation6,Citation7], while on the other hand, identification of antimicrobial resistance markers and the prediction of minimum inhibitory concentrations [Citation8,Citation9] may help in the future to guide the appropriate antimicrobial therapy and prevent treatment failure. Finally, typing and phylogenetic inference may be essential in outbreak scenarios with unknown and/or untypeable pathogens or rare clinical presentations. The 2019 cluster of patients with pneumonia of unknown cause linked to a seafood wholesale market in Wuhan, China and later identified as SARS-CoV-2, is an excellent example of such a scenario [Citation10].
Figure 1. Different levels of information obtainable from mNGS data. The first level includes taxonomic classification, i.e. identification of pathogens, and may be sufficient for some diagnostic purposes. The second level includes detection of virulence factors, identification of antimicrobial resistance markers and typing. MIC prediction is included in the third level, although this is still in its early stages. Abbreviations: MIC, minimum inhibitory concentration
This review starts by describing the expectations of mNGS and how these can be achieved, and concludes with how mNGS could be implemented in clinical laboratories.
2. What to expect from metagenomics and how can we achieve the expectations?
2.1. Why do we need metagenomics in the clinical setting?
Over the years, new diagnostic techniques have been developed owing to constraints from current in-use techniques. Although there is strong evidence that an infectious agent is present (e.g. elevated synovial fluid leukocyte cell count [> 3000 cells/µl] in the diagnosis of a prosthetic joint infection), the reality is that up to 50% of the samples can be culture-negative. The proportion of culture-negative samples can differ between sample types ().
Table 1. Proportion of culture-negative infections in different sample types
Several reasons can account for culture-negative results: (i) true negative (ii) preemptive antimicrobial therapy (iii) unculturable (e.g. Treponema pallidum), anaerobic or fastidious microorganisms (e.g. Mycobacterium tuberculosis), (iv) poor sampling, transport and storage conditions and finally (v) time between sampling and culturing [Citation16]. Molecular tests (such as PCR) and serology are techniques used to replace or complement traditional culture techniques. This is particularly important in cases where a viral infection is suspected. These techniques rely on predetermined targets, which are usually limited to common infectious agents and require further testing if found negative. On the other hand, mNGS has the potential to detect all pathogens present in a sample. Therefore, it can be more suited when an etiological agent is suspected, but no pathogen is detected through conventional diagnostic approaches. In clinical cases that require detecting a broader spectrum of pathogens, most commonly for immunocompromised patients, mNGS could be applied [Citation17]. Nevertheless, mNGS remains a challenging option compared to multiplexed molecular assays such as real-time PCR or point of care (POC) syndromic panels in terms of cost, turnaround time, reproducibility and sensitivity/specificity () [Citation18].
Table 2. Comparison of mNGS with existing molecular technologies/approaches used in routine diagnostics
2.2. Costs
The implementation of mNGS in routine diagnostics requires numerous considerations. The diagnostic laboratory will need to invest in IT infrastructure, separate sample/library preparation areas and equipment such as micropipettes, validation processes, and NGS-specialized laboratory personnel [Citation40]. The diagnostic laboratory may opt to (partially) outsource mNGS wet and dry lab processing to accredited and commercial service providers to negate infrastructure costs, such as dedicated laboratory space, high-performance e-infrastructure including networks, software stacks and large-scale storage resources [Citation40–42]. However, at the same time, routine diagnostic labs may have existing areas for pre-PCR preparation that could be incorporated into the mNGS workflow. It is important to stress that the e-infrastructure should be designed and maintained in a collaborative effort between the diagnostic laboratory and the IT department.
Processes such as nucleic acid extraction and library preparation would be ideally automated. Existing platforms such as extraction platforms, pipetting robots and thermal cyclers currently used for molecular diagnostics can also be integrated into the mNGS workflow. Several studies have evaluated the performance of available nucleic acid extraction platforms for mNGS and have found variable results [Citation43–45]. Library preparation can initially be performed manually if the sample throughput is low. The sequencing approach and platform can be selected based on sample throughput. For example, most Illumina platforms require batching to be cost-effective [Citation3]. However, these cases are most commonly used in reference laboratories or for surveillance and are less applicable in routine diagnostics where selected samples need to be immediately processed [Citation46]. Yet, cost-effective platforms such as the MinION with the Flongle adaptor from Oxford Nanopore Technologies (ONT) which offer flexible operation, and the iSeq 100 from Illumina, which provides a low-to-medium output, may overcome such limitations. mNGS requires trained laboratory technicians and bioinformaticians/computational biologists, which can increase costs. User-friendly specialized software and pipelines (such as CLC Genomics Workbench [commercial], BaseSpace [commercial], Explify [commercial], EPI2ME [free], Galaxy [free], MG-RAST [free] or SURPI+ [free]) can be utilized for automated analysis. However, results must be validated and interpreted by a multidisciplinary team of medical microbiologists (with expertise in NGS) and clinicians.
2.3. Turnaround time
The turnaround time of a diagnostic test is desired to be within a clinically actionable time frame. A conventional diagnostic workflow can take a few hours to 2–7 days from sample collection to identification and antimicrobial susceptibility determination [Citation1], compared to up to 5 days for mNGS (). Several factors affect the turnaround time of mNGS. For example, deeper sequencing by limiting the number of samples per run enables more detailed taxonomic resolution, antimicrobial drug resistance prediction, and phylogenetic analysis at the expense of extended turnaround time [Citation47,Citation48]. The number of samples per run can impact the cost efficiency of mNGS. Running individual or few samples might be necessary in the event a rapid diagnosis is required. Low-throughput platforms (such as the Flongle or the iSeq) can negate some of the extra costs, but are hampered by the inclusion of positive and negative controls in each run.
Depending on the clinical situation, the implementation of mNGS in routine diagnostics can be advantageous compared to conventional testing by circumventing some limitations, e.g. in identifying uncultivable microorganisms or slow-growing bacteria. A good example is mycobacteria which can take up to 21 days to grow in culture and another 28 days for a first-line antimicrobial susceptibility test result [Citation49], but can be recovered directly from clinical samples in 44/16 hours with Illumina MiSeq/MiniSeq or in 7.5 hours with ONT MinION sequencing [Citation50,Citation51]. Continuous technical advancements in sequencing technologies, particularly real-time ONT sequencing, could accelerate the clinically actionable results in under 6 hours following sampling, e.g. to identify pathogens based on circulating cell-free DNA from blood [Citation52]. Therefore, depending on the intended clinical use, mNGS could be a favorable choice to perform actionable results within a reasonable time.
2.4. Sensitivity/Specificity
The targeted pathogen could impact the selection of the nucleic acid extraction method, sequencing strategy (RNA and/or DNA), target enrichment/host nucleic acid depletion requirement, sequencing depth, reference database design or data analysis tools [Citation52]. Sequencing only one type of nucleic acid may decrease the overall sensitivity of the method, since some viruses may be missed (e.g. RNA viruses might be missed using DNA-sequencing and non-replicating-DNA viruses might be missed using RNA-sequencing). Furthermore, the complete recovery of bacterial/fungal/parasitic genomes will be unlikely if using RNA-sequencing. To increase sensitivity, some laboratories may opt to sequence all the nucleic acids present in a sample (DNA and cDNA), however, this may increase the overall cost per sample (if sequenced separately or if higher sequence breadth is needed).
The sensitivity of mNGS is hampered by several factors that are dependent on the specimen composition, material, volume (nucleic acid background/pathogen ratio), collection method, transport and storage. Sensitivity can also be affected by the efficiency of the nucleic acid extraction (bias toward some species), sequencing method (throughput, more reads ≈ higher sensitivity) and bioinformatics pipeline used for analysis (availability of appropriate reference sequences in databases) [Citation53]. On the other hand, specificity is influenced by contaminating nucleic acids in clinical specimens, reagents or by the accuracy of taxonomical classification algorithms [Citation53,Citation54]. NGS-related phenomena such as index hopping (also named index switching) or crosstalk (also called sample bleeding) can also introduce false-positive results, resulting in lower specificity [Citation55]. The ratio between host and microbial DNA/RNA is a major determinant of the proportion of microbial reads obtained after metagenomics sequencing [Citation53,Citation54]. The unbiased nature of mNGS, particularly shotgun metagenomics, leads to the sequencing of background (host or commensal microorganisms), as well as pathogen nucleic acids.
2.4.1. Challenges in sensitivity
Microbial identification relies on the bioinformatics pipelines and databases used for classifying sequencing reads into taxonomies. As a result, bioinformatics tools can significantly affect sensitivity. Studies have evaluated different mNGS sequence classification methods [Citation54,Citation56]. They differ not only in the algorithm for detecting infectious agents but also in the databases used. This high variability leads to inconsistent results at the taxonomical classification level and when evaluating the relative abundance of these pathogens [Citation56]. Taxonomical classification algorithms based on clade gene markers (e.g. MetaPhlAn2) may have lower sensitivity than k-mer based approaches [Citation54] since the former depends on identifying specific genes, while the latter relies on entire genomes. This has a significant impact on low biomass specimens, where genome coverage is limited. Another critical factor is the database used for taxonomic classification. Incomplete and/or unreliable taxonomic databases can lead to false-negative results or misclassifications. Hence, comprehensive, curated, and diverse reference databases are desirable [Citation53]. The databases should only include genomes which are assessed for quality (e.g. coverage, ANI, GC content, assembly size), continuity (e.g. N50, L50, number of contigs), taxonomy and metadata (e.g. species name, isolation source, submitter, orthogonal reference method) metrics and should include genomes that are representative of the circulating lineages [Citation57]. An option to increase the analytical sensitivity can be to select a platform which can offer higher outputs, such as the HiSeq (Illumina), NovaSeq (Illumina) or PromethION (ONT). However, this approach dramatically augments costs and turnaround time, precluding its use in clinical diagnostics. Finally, updated bioinformatics tools and/or databases can lead to changes in the results obtained. As a result, sequences should be kept safely and securely for long periods of time in case results need to can be reanalyzed if necessity arises (e.g. follow-up cases).
2.4.2. Challenges in specificity
Similarly to sensitivity, bioinformatics tools can significantly affect specificity. K-mer based approaches, for example, can incorrectly detect hundreds of species [Citation54]. This poses a significant challenge when delivering reproducible results which generates uncertainty regarding the reliability of the derived information. In addition to establishing standardized public databases with quality-controlled reference genomes [Citation57], ‘syndromic databases’ according to specimen type and clinical presentation (e.g. SIQ-db: specific database of 74 sepsis-relevant pathogens [Citation52]) can be an exciting option to achieve higher specificity. Reagent and laboratory contamination (known as the ‘kitome’) are well-known and undesirable problems impacting specificity and should be considered before applying sequence-based techniques [Citation58]. This issue should be mitigated by the sequencing of a negative control and by post-sequencing contamination removal. Contamination removal can be performed by either computational approaches, which consider the relative frequency of taxa in the samples compared to controls [Citation59,Citation60], or by manually filtering the taxa found in negative controls out from the samples [Citation42]. The latter, however, involves careful considerations: i) a biological signal can be lost because of cross-contamination from biological samples into negative controls or ii) a taxon, which is closely related to typical contaminants, could unintentionally be removed [Citation61]. Ultra-clean nucleic acid extraction kits such as the QIAamp UCP Pathogen (Qiagen) and ZymoBIOMICS DNA & RNA Miniprep have been introduced in the market and could reduce kitome contamination, according to the manufacturers.
False positives can also result from residual nucleic acid of dead microorganisms or transient bacteria (for example, in the bloodstream), leading to poor result specificity. Additionally, DNA released from pathogens following an attack from the host immune system or an efficient antimicrobial therapy can persist in the circulation for several days [Citation1], making the proper diagnosis clinically challenging. The broad nature of mNGS could invite questions into the actual cause of the infection, as detection does not necessarily indicate causation. The current limitations of data interpretation must be considered, and results must be evaluated within a clinical context [Citation62,Citation63]. This is particularly challenging in specific populations, such as immunocompromised patients, compared to generally healthy individuals, where the presence of a pathogenic microorganism usually signifies the source of the infection [Citation64]. Quantification of the abundance of pathogens is possible with mNGS and can allow the distinction between infection and colonization or contamination [Citation65]. Additionally, measuring the degree of host tissue injury from host–microorganism interactions can also be used to help differentiate between infection and colonization or contamination [Citation66,Citation67].
2.4.3. Enrichment and host depletion strategies
To increase the analytical sensitivity of mNGS, several strategies have been developed (). Enrichment is often necessary to avoid samples consisting of 100% host nucleic acids and increases confidence in a true negative result (exclude infections). Pre-lysis host depletion strategies rely on the integrity of microorganisms, as cells are separated using centrifugation [Citation44,Citation68]. Additionally, human cells can be lysed by chaotropic buffers such as saponin [Citation69] and osmotic pressure [Citation70], followed by degradation of cell-free DNA by subsequent DNase treatment. Pre-lysis methods are usually cheap and efficient, with up to 99.99% of host DNA removal, depending on the sample type [Citation69]. Several commercial kits are available that apply differential lysis, such as the QIAamp DNA microbiome kit (Qiagen) and the HostZERO Microbial DNA Kit (Zymo Research) [Citation55,Citation71]. Possible drawbacks of differential lysis need to be considered: limited suitability for viral enrichment [Citation69], significant hands-on-time, reproducibility concerns [Citation63], increased impact of reagent and laboratory contamination [Citation72]. Additionally, microorganisms without a cell wall (such as Mycoplasma species) and parasites (i.e. protozoa) might be destroyed. Furthermore, cell-free nucleic acids from dead microbes (attacked by the immune system or antimicrobials) are degraded during the procedure [Citation73]. Pelleting the intact cells prior to differential lysis could be an option to retain the supernatant containing cell-free DNA, particularly in culture-negative samples or for further viral analysis. The use of preservatives such as glycerol [Citation70] or Sputasol [Citation69] can reduce the bias of differential lysis on older or frozen samples as metagenomics is often applied retrospectively [Citation3]. However, preservatives, in general, are used sparsely in routine bacterial diagnostics.
Table 3. Enrichment and depletion strategies
For viral detection or retrospective analysis of old or frozen samples, targeted mNGS approaches can increase the sensitivity. However, targeted sequencing approaches can also limit the breadth of detectable pathogens. Targeted approaches can amplify conserved marker genes, such as 16S rRNA for bacteria and 18S/internal transcribed spacer (ITS) for fungi and are frequently applied in clinical diagnostics [Citation85,Citation86]. Whole-genome sequencing using tiled primer schemes targeting the whole viral genome [Citation87] has been proven to be highly sensitive to detect and characterize the targeted virus, as indicated during outbreaks of Ebola virus [Citation88], Zika virus [Citation89] and SARS-CoV-2 [Citation90,Citation91]. Recently, multiplexed spiked primer schemes that target several viruses (resembling conventional POC syndromic panels) have been introduced to retain the breadth of shotgun metagenomics, while increasing sensitivity for targeted organisms [Citation92]. rRNA depletion is another strategy to increase sensitivity by depleting highly abundant host or bacterial rRNA sequences that offer little diagnostic value [Citation93]. Similarly, CRISPR-Cas9 based approaches have been emerging to enrich sequences of interest [Citation94] or deplete host sequences [Citation95]. Probe capture is another targeted approach based on the hybridization of nucleic acids to targeted organisms. It is less stringent when compared to amplicon sequencing and can cover a wide breadth of targets, including DNA and RNA viruses [Citation96,Citation97], antimicrobial resistance genes [Citation98] and custom-based panels (i.e. Roche HyperDesign or Agilent SureSelect). Protocols were initially developed for Illumina sequencing, but recent approaches for other sequencing platforms are emerging [Citation99]. Although probe-based approaches increase costs and add hands-on-time, samples can be multiplexed in a run, reducing the cost per sample. New panels such as transposase-based library preparation are promising approaches that do not introduce extra hands-on-time compared to the standard library preparation procedure. For example, the recently developed CoronaHiT, which involves whole-genome sequencing of SARS-CoV-2 using ONT or Illumina technologies [Citation100], is derived from transposase-based library preparation.
2.5. Validation
One of the biggest challenges of implementing mNGS is the validation of the test. As with other laboratory-developed tests, the requirements for validation depend on local and federal regulations. Validation is challenging due to the broad nature of the test and a large number of possible results. In addition, often, no reliable reference method with a similar scope is available. Validation is required for both the wet bench protocols, including accuracy, analytical sensitivity and specificity, reproducibility, stability, as well as bioinformatics protocols [Citation40,Citation42,Citation53]. For the latter, in silico analyses using simulated samples can be performed. During validation of the wet bench, it is crucial to define and use proper external and internal controls, which are essential to bring standardization and ensure the quality of the generated sequences in clinical settings [Citation101]. Despite the challenges to validate mNGS, examples are available for successful implementation for routine testing, such as pathogen detection in cerebrospinal fluid [Citation102] and detection of RNA and DNA viruses in respiratory samples [Citation103].
2.6. Reproducibility
Complex workflows like those for mNGS pose challenges for reproducibility [Citation53], particularly if different laboratories implement entirely different workflows. Studies on the reproducibility and validation of mNGS assays are challenging and are limited to a few reports [Citation53,Citation102–104]. As such, reference standards and external/internal controls are required to warrant quality, reproducibility and consistency of mNGS workflows. mNGS QC metrics have been established and integrated into clinical microbiology laboratories previously [Citation53,Citation102]. Validated microbial community standards, regardless of the material type or species, are the ideal choice. To the best of our knowledge, ATCC® Microbiome Standards [Citation105] and ZymoBIOMICS Microbial Community Standards [Citation106] are the only currently available standards for mNGS in the market (not including viruses). Standards can be used as external and internal controls. Examples are whole microorganisms or viruses to monitor nucleic acid extraction efficiency for different pathogen classes or, when spiked into clinical samples, as process control for the entire workflow. The latter also allows the quantification of pathogens in clinical samples. Additionally, spike-in nucleic acids can be used as a control to detect the limit of detection or estimate the sequencing error rate (i.e. phiX). Current standards developed for nucleic acid tests can also be used for mNGS. Bal and colleagues, for example, applied the bacteriophage M2 kit as an internal standard for viral metagenomics (MS2, IC1 RNA internal control; r-gene, BioMérieux) [Citation101]. Similarly, Miller and colleagues also applied MS2 (RNA) along with T1 (DNA) bacteriophages as internal controls to indicate microbial sensitivity [Citation102]. However, careful consideration must be applied when including a microbial standard as an internal control. Depending on the concentration spiked, the microbial standard could take precious sequencing reads from the pathogen of interest. Sequencing of defined standards can also be used to assess different bioinformatics pipelines. As the currently available standards are designed for specific tasks, no universal and well-defined standard for metagenomics is available. Additionally, it is important to include negative controls to negate possible contamination, which can be introduced at any step, from sampling to sequencing. Possible negative controls can consist of a sampling blank, nucleic acid extraction blank and/or no-template control [Citation107,Citation109].
3. How could the microbiology laboratory implement metagenomics?
3.1. Implementation
Before embarking on mNGS, diagnostic laboratories should decide whether to develop an in-house pipeline or implement a commercially provided solution. Commercial solutions include shipping the sample to an external laboratory that will either send the raw sequencing data back or perform the analyses and/or interpretation. Alternatively, laboratories could implement a commercial pipeline from sample preparation to reporting. Several commercial solutions have become available and are accredited either in Europe (IVD CE approved) [Citation110] or in America (CAP approved) [Citation111,Citation112]. However, they may be restricted to specific regions/continents.
3.2. Clinical conundrum
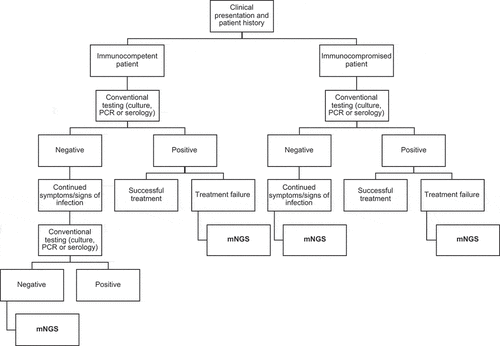
Performing mNGS on patient samples currently relies heavily on a case-by-case basis. However, mNGS has the potential to become a cost-competitive option as it could be used as a direct “rule in“ or ‘rule out’ test to confirm the presence or absence of an infectious etiology [Citation3,Citation113]. While mNGS may be more suited for immunocompetent individuals as a last resort option, with traditional methods performed first, specific patient populations could benefit from mNGS as a diagnostic complement or an alternative to conventional testing. In this respect, we propose a diagnostic algorithm that could be used to select samples for mNGS (). For example, mNGS could be used for patients with negative results from conventional testing who are still presenting with symptoms or signs consistent with infectious disease. Additionally, mNGS may be used if an infectious agent is identified, but treatment failure is observed. It can then help to detect antimicrobial resistance or co-infections, which may be the actual cause of the symptoms. For immunocompromised patients such as neonates, transplant recipients, or critically ill patients admitted to the intensive care unit, mNGS can be considered an earlier option to prevent continued sampling or to provide extra information for patients with limited care options, such as those suffering from malignancy. In this respect, mNGS can, in some cases, have an impact not only on survival but also on the quality of life gained [Citation114]. To be used in diagnostics, mNGS should have a direct impact on patient care or management. This can also involve confirming the patient no longer requires isolation, reducing the length of hospital days or medical treatment, therefore decreasing costs for both the patient and the hospital.
Figure 2. Diagnostic algorithm of potential workflow. Initially, a sample will be taken from the patient presenting with a clinical syndrome and run through conventional testing. In immunocompetent patients, usually the identified pathogen signifies the causative agent. If no pathogen is identified and the patient has continued symptoms and signs consistent with an infectious disease, another conventional test will usually be performed. If no infectious agent is found which corresponds to the clinical syndrome, mNGS could then be performed. Additionally, if antimicrobial therapy has already been initiated, mNGS could be performed to overcome the limitations of culture as fewer or no viable cells are left. Moreover, even when a positive result is achieved through conventional testing, mNGS could still be applied in the event of treatment failure (i.e. patient fails to respond to treatment), to identify co-infections and/or antimicrobial resistance genes. In immunocompromised patients, mNGS could be performed in an earlier step of the workflow since the likelihood of an infection with an unusual pathogen is higher. The patient’s clinical history should denote which workflow to follow and would most likely be on a case-by-case basis
3.3. Ethical considerations
Sequencing data is usually stored locally or in the cloud. As the data contains the personal and genetic information of the patient (either unwanted background or host response), separation, anonymization, and secure data storage are key priorities. NGS assays acquire genetic data on the patient’s current health and/or future risk factors, along with their relatives and possible future children. The presence of human data can also pose privacy issues in relation to the use of online bioinformatics tools, such as RAST [Citation115], Genome Detective [Citation116], EPI2ME (ONT) or Taxonomer [Citation117]. Removing human nucleic acid sequences by mapping usually leaves traces behind and adds additional time to downstream analysis [Citation42]. Another important ethical consideration is how to handle incidental findings, particularly HIV or other sexually transmitted diseases and should be part of the informed consent procedure. Recommendations regarding pre-test counseling, informed consent, and essential processes (ethical and clinically focused return of incidental findings) based on previous studies have been published elsewhere [Citation42,Citation118,Citation119].
4. Conclusion
This article reviewed the expectations of integrating mNGS in routine diagnostics and how this can be achieved. As many samples remain culture – or PCR-negative, clinical laboratories could benefit from mNGS. However, cost, turnaround time, variable sensitivity/specificity, validation and reproducibility remain hurdles to overcome before implementing mNGS in routine diagnostics. A commercial mNGS service provider could be applied to reduce costs before investing in infrastructures, equipment and NGS-specialized laboratory personnel. The analytical sensitivity can be increased by several host depletion and microbial enrichment strategies. Reagent and laboratory contamination should be mitigated by sequencing a negative control and post-sequencing contamination removal to increase specificity. Nevertheless, data must be interpreted and evaluated carefully within a clinical context. Furthermore, validated microbial community reference standards and external/internal controls are required to warrant quality, reproducibility, and consistency of mNGS workflows. Above all, the intended use of mNGS should be clearly defined and performed on a case-by-case basis as described in the proposed diagnostic algorithm. Additionally, careful consideration is needed to determine the most appropriate clinical approach as each have their own advantages and disadvantages (). mNGS can circumvent some of the limitations of conventional testing to obtain a clinically actionable result in a reasonable time frame. Considering the ability of some sequencing platforms to provide same-day results, mNGS can revolutionize routine diagnostics.
Table 4. Advantages and disadvantages of main diagnostic approaches and mNGS
5. Expert opinion
Looking at the reviews on real-time PCR applications in clinical microbiology from 15–20 years ago, we can find several resemblances with the current mNGS situation. The implementation of real-time PCR also required careful consideration of facility and personnel requirements, along with workflow design [Citation120]. Additionally, reports documenting the diversity of extraction methods, sample material, and protocols made a direct comparison of the methods challenging [Citation120]. Since its initial introduction, real-time PCR has been fully integrated into routine clinical diagnostics and has become a vital tool in any testing algorithm. Similarly, mNGS could become a standard microbiological method with a clearly defined role in diagnostics in the near future. However, our opinion is that large-scale prospective efforts to standardize and validate mNGS workflows should be taken by clinical laboratories that wish to implement mNGS. Such initiatives exist at the academic/reference and commercial level but most likely lack the financial capacity needed for such studies. Consequently, commercial companies that can secure large grants for development will probably be driving mNGS implementation. Additionally, economic data showing the cost-effectiveness of mNGS is needed to justify the use of such an expensive test. A clear definition of diagnostic algorithms, including mNGS, is vital to show clinical utility rather than the promise of mNGS replacing conventional techniques (at least for the time being). The need for a new diagnostic tool to increase the sensitivity of microbial diagnosis is clear. Although mNGS seems to be a promising candidate, it will still take time before it is widely applied.
Article highlights
A large proportion of samples remain culture-negative or specific-PCR-negative and could benefit from mNGS
Costs, turnaround time, sensitivity, specificity, validation and reproducibility are the main factors affecting the implementation of mNGS
Enrichment strategies have been developed to increase mNGS sensitivity and accuracy
Diagnostic laboratories should decide whether to develop an in-house mNGS workflow or employ a (partially) commercially provided solution
A diagnostic algorithm is proposed to choose samples for mNGS based on clinical presentation and patient history
Large-scale prospective cohort studies with mNGS should be performed to demonstrate clinical validity and accelerate mNGS implementation
Declaration of interest
J. W. A. Rossen is employed by IDbyDNA. This did not have any influence on the interpretation of reviewed data and conclusions drawn nor on the drafting of the manuscript, and no support was obtained from them. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
Conceptualization: N. Couto; writing—review and editing: H. Cassidy, L. Schuele, N. Peker, J.W.A. Rossen, and N. Couto. All authors have read and agreed to the published version of the manuscript.
Additional information
Funding
References
- Peker N, Couto N, Sinha B, et al. Diagnosis of bloodstream infections from positive blood cultures and directly from blood samples: recent developments in molecular approaches. Clin Microbiol Infect. 2018;24(9):944–955.
- Pallen MJ. Diagnostic metagenomics: potential applications to bacterial, viral and parasitic infections. Parasitology. 2014;141(14):1856–1862.
- Greninger AL. The challenge of diagnostic metagenomics. Expert Rev Mol Diagn. 2018;18(7):605–615.
- Isenberg HD. Clinical microbiology: past, present, and future. J Clin Microbiol. 2003;41(3):917–918.
- Maurer JJ. Rapid detection and limitations of molecular techniques. Annu Rev Food Sci Technol. 2011;2:259–279.
- Clements A, Young JC, Constantinou N, et al. Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes. 2012;3(2):71–87.
- Loman N, Constantinidou C, Christner M, et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of shiga-toxigenic escherichia coli O104:H4. JAMA. 2013;309(14):1502–1510.
- Nguyen M, Long SW, McDermott PF, et al. Using machine learning to predict antimicrobial MICs and associated genomic features for nontyphoidal Salmonella. J Clin Microbiol. 2019;57:e01260–18.
- Pataki BÁ, Matamoros S, van der Putten BCL, et al. Understanding and predicting ciprofloxacin minimum inhibitory concentration in Escherichia coli with machine learning. Sci Rep. 2020;10:15026.
- Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733.
- Palan J, Nolan C, Sarantos K, et al. Culture-negative periprosthetic joint infections. EFORT Open Rev. 2019;4:585–594.
- Gupta S, Sakhuja A, Kumar G, et al. Culture-negative severe sepsis: nationwide trends and outcomes. Chest. 2016;150:1251–1259.
- Wilson MR, Sample HA, Zorn KC, et al. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N Engl J Med. 2019;380:2327–2340.
- Torres A, Lee N, Cilloniz C, et al. Laboratory diagnosis of pneumonia in the molecular age. Eur Respir J. 2016;48(6):1764–1778.
- Foxman B. The epidemiology of urinary tract infection. Nat Rev Urol. 2010;7(12):653–660.
- Lagier J-C, Edouard S, Pagnier I, et al. Current and past strategies for bacterial culture in clinical microbiology. Clin Microbiol Rev. 2015;28:208–236.
- Hasan MR, Rawat A, Tang P, et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J Clin Microbiol. 2016;54(4):919–927.
- Cassidy H, van Genne M, Lizarazo-Forero E, et al. QIAG: a discussion of syndromic molecular testing for clinical care. J Antimicrob Chemother. 2021;76:iii58–iii66.
- Gunson RN, Bennett S, and Carman WF, et al. Using multiplex real time PCR in order to streamline a routine diagnostic service. J Clin Virol. 2008;43(4):372–375 .
- Templeton KE, Scheltinga SA, and Beersma MF et al, et al. Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza a and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. J Clin Microbiol. 2004;42(4):1564–1569.
- Li Y, Ding S, and Wang S. A TaqMan-based multiplex real-time PCR assay for the rapid detection of tigecycline resistance genes from bacteria, faeces and environmental samples. BMC Microbiol. 2020;20(1):1–7.
- ThermoFisher Scientific [Internet]. [2021 Accessed25 May 2021]. : https://www.thermofisher.com/nl/en/home/life-science/sequencing/sequencing-learning-center/sequencing-basics/sanger-next-generation-technology.html
- Eurofins [Internet]. [2021 Accessed25 May 2021]. : https://eurofinsgenomics.eu/en/custom-dna-sequencing/gatc-services/lightrun-barcodes/
- Heflin Center for Genomics Sciences [Internet]. Accessed25 May 2021 . Available from: https://www.uab.edu/hcgs/genomics-core-lab/sanger-sequencing
- Leber AL, Everhart K, Daly JA, et al. Multicenter evaluation of biofire filmarray respiratory panel 2 for detection of viruses and bacteria in nasopharyngeal swab samples. J Clin Microbiol. 2018;56(6):e01945–17.
- Creager HM, Cabrera B, Schnaubelt A, et al. Clinical evaluation of the BioFire® Respiratory Panel 2.1 and detection of SARS-CoV-2. J Clin Virol. 2020;129:104538.
- Eckbo EJ, Locher K, Caza M, et al. Evaluation of the BioFire® COVID-19 test and Respiratory Panel 2.1 for rapid identification of SARS-CoV-2 in nasopharyngeal swab samples. Diagn Microbiol Infect Dis. 2021;99(3):115260.
- Buss SN, Leber A, Chapin K, et al. Multicenter evaluation of the BioFire FilmArray Gastrointestinal Panel for etiologic diagnosis of infectious gastroenteritis. J Clin Microbiol. 2015;53(3):915–925.
- Gingras BA, and Maggiore JA. Performance of a new molecular assay for the detection of gastrointestinal pathogens. Access Microbiol. . . 2020;2(10):acmi000160.
- Tansarli GS, Chapin KC. Diagnostic test accuracy of the BioFire® FilmArray® meningitis/encephalitis panel: a systematic review and meta-analysis. Clin Microbiol Infect. 2020;26(3):281–290.
- Visseaux B, Le Hingrat Q, Collin G, et al. Evaluation of the QIAstat-Dx Respiratory SARS-CoV-2 Panel, the first rapid multiplex PCR commercial assay for SARS-CoV-2 detection. J Clin Microbiol. 2020;58(8):e00630–20.
- Boers SA, Peters CJA, Wessels E, et al. Performance of the QIAstat-Dx Gastrointestinal Panel for diagnosing infectious gastroenteritis. J Clin Microbiol. 2020;58(3):e01737–19.
- Chen JH, Yip CC, Chan JF, et al. Clinical performance of the Luminex NxTAG CoV Extended Panel for SARS-CoV-2 detection in nasopharyngeal specimens from COVID-19 patients in Hong Kong. J Clin Microbiol. 2020;58(8):e00936–20.
- Duong VT, Phat VV, Tuyen HT, et al. Evaluation of Luminex xTAG Gastrointestinal Pathogen Panel Assay for detection of multiple diarrheal pathogens in fecal samples in Vietnam. J Clin Microbiol. 2016;54(4):1094–1100.
- Ramanan P, Bryson AL, Binnicker MJ, et al. Syndromic panel-based testing in clinical microbiology. Clin Microbiol Rev. 2017;31(1):e00024–17.
- Kosai K, Suzuki H, Tamai K, et al. Multicenter evaluation of Verigene Enteric Pathogens Nucleic Acid Test for detection of gastrointestinal pathogens. Sci Rep. 2021;11(1):3033.
- Nijhuis RHT, Guerendiain D, Claas ECJ, et al. Comparison of ePlex respiratory pathogen panel with laboratory-developed real-time PCR assays for detection of respiratory pathogens. J Clin Microbiol. 2017;55(6):1938–1945.
- Illumina [Internet]. [2021 Accessed25 May 2021]. : https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/respiratory-pathogen-id-panel.html
- Gohl DM, Garbe J, Grady P, et al. A rapid, cost-effective tailed amplicon method for sequencing SARS-CoV-2. BMC Genomics. 2020;21:863.
- López-Labrador FX, Brown JR, Fischer N, et al. Recommendations for the introduction of metagenomic high-throughput sequencing in clinical virology, part I: Wet lab procedure. J Clin Virol. 2021;134:104691.
- Spjuth O, Bongcam-Rudloff E, Dahlberg J, et al. Recommendations on e-infrastructures for next-generation sequencing. GigaScience. 2016;5(1):s13742–016–0132–7.
- De Vries JJC, Brown JR, Couto N, et al. Recommendations for the introduction of metagenomic next-generation sequencing in clinical virology, part II: bioinformatic analysis and reporting. J Clin Virol. 2021;138:104812.
- Knudsen BE, Bergmark L, Munk P, et al. Impact of sample type and DNA isolation procedure on genomic inference of microbiome composition. mSystems. 2016;1(5):e00095–16.
- Lewandowski K, Bell A, Miles R, et al. The effect of nucleic acid extraction platforms and sample storage on the integrity of viral RNA for use in whole genome sequencing. J Mol Diagn. 2017;19(2):303–312.
- Sabatier M, Bal A, Destras G, et al. Comparison of nucleic acid extraction methods for a viral metagenomics analysis of respiratory viruses. Microorganisms. 2020;8(10):1539.
- Rossen JWA, Friedrich AW, Moran-Gilad J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect. 2018;24:355–360.
- Dulanto Chiang A, Dekker JP. From the pipeline to the bedside: advances and challenges in clinical metagenomics. J Infect Dis. 2020;221:S331–S340.
- Miller S, Chiu C, and Rodino KG, et al. Point-Counterpoint: should we be performing metagenomic next-generation sequencing for infectious disease diagnosis in the clinical laboratory? J Clin Microbiol. . . 2020;58(3): e01739–19.
- Forbes BA, Hall GS, Miller MB, et al. Practice guidelines for clinical microbiology laboratories: mycobacteria. Clin Microbiol Rev. 2018;31:e00038–17.
- George S, Xu Y, Rodger G, et al. DNA thermo-protection facilitates whole-genome sequencing of mycobacteria direct from clinical samples. J Clin Microbiol. 2020;58:e00670–20.
- Votintseva AA, Bradley P, Pankhurst L, et al. Same-day diagnostic and surveillance data for tuberculosis via whole-genome sequencing of direct respiratory samples. J Clin Microbiol. 2017;55:1285–1298.
- Grumaz C, Hoffmann A, Vainshtein Y, et al. Rapid Next-generation sequencing–based diagnostics of bacteremia in septic patients. J Mol Diagn. 2020;22:405–418.
- Schlaberg R, Chiu CY, Miller S, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. 2017;141:776–786.
- Couto N, Schuele L, Raangs EC, et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci Rep. 2018;8(1):13767.
- Xu Y, Lewandowski K, Lumley S, et al. Detection of viral pathogens with multiplex Nanopore MinION sequencing: be careful with cross-talk. Front Microbiol. 2018;9:2225.
- Peabody MA, Van Rossum T, Lo R, et al. Evaluation of shotgun metagenomics sequence classification methods using in silico and in vitro simulated communities. BMC Bioinformatics. 2015;16:363.
- Sichtig H, Minogue T, Yan Y, et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat Commun. 2019;10:3313.
- Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87.
- Davis NM, Proctor DM, Holmes SP, et al. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226.
- Martí JM. Recentrifuge: robust comparative analysis and contamination removal for metagenomics. PLOS Comput Biol. 2019;15:e1006967.
- Eisenhofer R, Minich JJ, Marotz C, et al. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 2019;27:105–117.
- Graf EH, Pancholi P. Appropriate use and future directions of molecular diagnostic testing. Curr Infect Dis Rep. 2020;22:5.
- Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20:341–355.
- Dhesi Z, Enne VI, O’Grady J, et al. Rapid and point-of-care testing in respiratory tract infections: an antibiotic guardian? ACS Pharmacol Transl Sci. 2020;3(3):401–417.
- Cheng A-P, Burnham P, Lee J-R, et al. A cell-free DNA metagenomic sequencing assay that integrates the host injury response to infection. PNAS USA. 2019;116(37):18738–18744.
- Gatcliffe C, Rao A, Brigger M, et al. Metagenomic sequencing and evaluation of the host response in the pediatric aerodigestive population. Pediatr Pulmonol. 2021;56(2):516–524.
- Schlaberg R, Barrett A, Edes K, et al. Fecal host transcriptomics for non-invasive human mucosal immune profiling: proof of concept in Clostridium difficile infection. Pathog Immun. 2018;3(2):164.
- Li T, Garcia-Gutierrez E, Scadden J, et al. An optimised protocol for detection of SARS-COV-2 in stool. medRxiv. 2021. DOI:https://doi.org/10.1101/2021.01.11.20248606
- Charalampous T, Kay GL, Richardson H, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37(7):783–792.
- Marotz CA, Sanders JG, Zuniga C, et al. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42.
- Heravi FS, Zakrzewski M, Vickery K, et al. Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples. J Microbiol Methods. 2020;170:105856.
- Olomu IN, Pena-Cortes LC, Long RA, et al. Elimination of “kitome” and “splashome” contamination results in lack of detection of a unique placental microbiome. BMC Microbiol. 2020;20:157.
- Oechslin CP, Lenz N, Liechti N, et al. Limited correlation of shotgun metagenomics following host depletion and routine diagnostics for viruses and bacteria in low concentrated surrogate and clinical samples. Front Cell Infect Microbiol. 2018;8:375.
- Charalampous T, Alcolea-Medina A, Snell LB, et al. Application of respiratory metagenomics for COVID-19 patients on the intensive care unit to inform appropriate initial antimicrobial treatment and rapid detection of nosocomial transmission. medRxiv. 2020. DOI:https://doi.org/10.1101/2020.11.26.20229989
- Mu S, Hu L, Zhang Y, et al. Prospective evaluation of a rapid clinical metagenomics test for bacterial pneumonia. SSRN. 2021. DOI:https://doi.org/10.2139/ssrn.3779202
- Govender KN. Precision pandemic preparedness: improving diagnostics with metagenomics. J Clin Microbiol. 2021;59(6):e02146–20.
- Greninger AL, Waghmare A, Adler A, et al. Rule-out outbreak: 24-hour metagenomic next-generation sequencing for characterizing respiratory virus source for infection prevention. J Pediatric Infect Dis Soc. 2017;6(2):168–172.
- Xu Y, Lewandowski K, Jeffery K, et al. Nanopore metagenomic sequencing to investigate nosocomial transmission of human metapneumovirus from a unique genetic group among haematology patients in the United Kingdom. J Infect. 2020;80(5):571–577.
- Peker N, Garcia-Croes S, Dijkhuizen B, et al. A comparison of three different bioinformatics analyses of the 16S-23S rRNA encoding region for bacterial identification. Front Microbiol. 2019;10:620.
- Rivett L, Sridhar S, Sparkes D, et al. Screening of healthcare workers for SARS-CoV-2 highlights the role of asymptomatic carriage in COVID-19 transmission. Elife. 2020;9:e58728.
- Thézé J, Li T, Du Plessis L, et al. Genomic epidemiology reconstructs the introduction and spread of zika virus in Central America and Mexico. Cell Host Microbe. 2018;23(6):855–864.e7.
- Oba M, Tsuchiaka S, Omatsu T, et al. A new comprehensive method for detection of livestock-related pathogenic viruses using a target enrichment system. Biochem Biophys Res Commun. 2018;495(2):1871–1877.
- O’Flaherty BM, Li Y, Tao Y, et al. Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res. 2018;28(6):869–877.
- Nguyen AT, Tran TT, Hoang VM, et al. Development and evaluation of a non-ribosomal random PCR and next-generation sequencing based assay for detection and sequencing of hand, foot and mouth disease pathogens. Virol J. 2016;13:125.
- Gu W, Deng X, Lee M, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27(1):115–124.
- Fida M, Wolf MJ, and Hamdi A, et al. Detection of pathogenic bacteria from septic patients using 16S rRNA gene targeted metagenomic sequencing. Clin Infect Dis. 2021 73 ;1165–1172.
- Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017;12(6):1261–1276.
- Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228–232.
- Faria NR, Quick J, Claro IM, et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. 2017;546:406–410.
- Tyson JR, James P, Stoddart D, et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv. 2020. DOI:https://doi.org/10.1101/2020.09.04.283077
- Meredith LW, Hamilton WL, Warne B, et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis. 2020;20(11):1263–1271.
- Deng X, Achari A, Federman S, et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat Microbiol. 2020;5(3):443–454.
- Liu T, Chen Z, Chen W, et al. A benchmarking study of SARS-CoV-2 whole-genome sequencing protocols using COVID-19 patient samples. bioRxiv. 2020. DOI:https://doi.org/10.1101/2020.11.10.375022
- Quan J, Langelier C, Kuchta A, et al. FLASH: a next-generation CRISPR diagnostic for multiplexed detection of antimicrobial resistance sequences. Nucleic Acids Res. 2019;47(14):e83.
- Gu W, Crawford ED, O’Donovan BD, et al. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016;17:41.
- Wylie TN, Wylie KM, Herter BN, et al. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015;25:1910–1920.
- Jansen SA, Nijhuis W, Leavis HL, et al. Broad virus detection and variant discovery in fecal samples of hematopoietic transplant recipients using targeted sequence capture metagenomics. Front Microbiol. 2020;11:560179.
- Lanza VF, Baquero F, Martínez JL, et al. In-depth resistome analysis by targeted metagenomics. Microbiome. 2018;6:11.
- Schuele L, Cassidy H, Lizarazo E, et al. Assessment of viral targeted sequence capture using nanopore sequencing directly from clinical samples. Viruses. 2020;12(12):1358.
- Baker DJ, Aydin A, Le-Viet T, et al. CoronaHiT: high-throughput sequencing of SARS-CoV-2 genomes. Genome Med. 2021;13:21.
- Bal A, Pichon M, Picard C, et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC Infect Dis. 2018;18(1):537.
- Miller S, Naccache SN, Samayoa E, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29(5):831–842.
- Van Boheemen S, van Rijn AL, Pappas N, et al. Retrospective validation of a metagenomic sequencing protocol for combined detection of RNA and DNA viruses using respiratory samples from pediatric patients. J Mol Diagn. 2020;22(2):196–207.
- Junier T, Huber M, Schmutz S, et al. Viral metagenomics in the clinical realm: lessons learned from a Swiss-wide ring trial. Genes (Basel). 2019;10(9):655.
- ATCC [Internet]. [2021 Accessed25 May 2021]. : https://www.atcc.org/products/msa-2003.
- Zymo Research [Internet]. [2021 Accessed25 May 2021]. : https://www.zymoresearch.com/collections/zymobiomics-microbial-community-standards.
- Govender KN, Street TL, and Sanderson ND, et al. Metagenomic sequencing as a pathogen-agnostic clinical diagnostic tool for infectious diseases: a systematic review and meta-analysis of diagnostic test accuracy studies. J Clin Microbiol. 2021 ;59 .
- Player R, Verratti K, Staab A, et al. Comparison of the performance of an amplicon sequencing assay based on Oxford Nanopore technology to real-time PCR assays for detecting bacterial biodefense pathogens. BMC Genomics. 2020;21(1):166.
- Han D, Gao P, and Li R, et al. Multicenter assessment of microbial community profiling using 16S rRNA gene sequencing and shotgun metagenomic sequencing. J Ad Res . . 2020;26. 111–121.
- Noscendo [Internet]. [2021 Accessed25 May 2021]. : https://noscendo.com.
- Karius Test [Internet]. [2021 Accessed25 May 2021]. : https://kariusdx.com.
- Illumina IDbyDNA panel [Internet]. [2021 Accessed25 May 2021]. : https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/respiratory-pathogen-id-panel.html.
- Park ST, Kim J. Trends in next-generation sequencing and a new era for whole genome sequencing. Int Neurourol J. 2016;20(Suppl 2):S76–S83.
- Schluckebier L, Caetano R, Garay OU, et al. Cost-effectiveness analysis comparing companion diagnostic tests for EGFR, ALK, and ROS1 versus next-generation sequencing (NGS) in advanced adenocarcinoma lung cancer patients. BMC Cancer. 2020;20:875.
- Davis JJ, Wattam AR, Aziz RK, et al. The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res. 2020;48(D1):D606–D612.
- Vilsker M, Moosa Y, Nooij S, et al. Genome detective: an automated system for virus identification from high-throughput sequencing data. Bioinformatics. 2019;35(5):871–873.
- Flygare S, Simmon K, Miller C, et al. Taxonomer: an interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome Biol. 2016;17:111.
- Ayuso C, Millán JM, Mancheño M, et al. Informed consent for whole-genome sequencing studies in the clinical setting. Proposed recommendations on essential content and process. Eur J Hum Genet. 2013;21(10):1054–1059.
- Hall RJ, Draper JL, Nielsen FG, et al. Beyond research: a primer for considerations on using viral metagenomics in the field and clinic. Front Microbiol. 2015;6:224.
- Espy MJ, Uhl JR, Sloan LM, et al. Real-Time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19(1):165–256.