
ABSTRACT
Objectives
Evidentiary requirements for relative effectiveness assessment vary among European health technology assessment (HTA) bodies, affecting the time to HTA decision-making and potentially delaying time to patient access. Improved alignment may reduce this time; therefore, we aim to analyze the differences in evidentiary requirements for oncology drug assessments among European HTA bodies and provide recommendations toward an increased alignment.
Methods
Interviews were conducted with stakeholders in drug assessments of Italy, the Netherlands, Poland, Portugal, England and Wales, and Sweden about evidentiary requirements for several subdomains to identify differences and obtain recommendations for addressing differences. The interview results were analyzed on degrees of evidence acceptability per HTA body and alignment on evidentiary requirements among HTA bodies.
Results
Subdomains demonstrating noteworthy differences concerned the acceptability of extrapolation to other populations, class effects, progression-free survival and (other) surrogate endpoints as outcomes, the absence of quality-of-life data, single-arm trials, cross-over trial designs, short trial duration, and the clinical relevance of effect size.
Conclusion
Alignment can be enhanced to reduce time to decision-making and to improve equity in patient access. Proposed recommendations to achieve this included joint early dialogues, intensified collaboration and exchange between countries, joint relative effectiveness assessments, and the use of access agreements.
1. Introduction
Of all new drugs receiving market authorization in Europe between 2017 and 2020, approximately a quarter were oncology products [Citation1].
Following marketing authorization by the European Medicines Agency (EMA), the national health technology assessment (HTA) bodies make decisions regarding reimbursement and pricing. Criteria evaluated for reimbursement decisions generally include unmet medical need, relative effectiveness and safety, drug price, as well as budget impact and/or cost-effectiveness [Citation2]. In general, all HTA bodies use the same pivotal clinical trial data for the assessment of the relative effectiveness and safety, alongside other potential nationally oriented information. However, the importance of various elements including the use of overall survival (OS), progression-free survival (PFS), quality of life (QoL), and safety on the recommendation of oncology drugs may vary among European countries [Citation3].
Of the 41 oncology medicines that received marketing authorization in Europe between 2017 and 2020, on average, around half are covered in the countries of the European Union. Differences are observed between the proportion of oncology medicines with have full public coverage, limited coverage, only private coverage, and no coverage. This demonstrated how the used criteria, evidence thresholds, and approaches vary among countries [Citation1]. Differences in evidentiary requirements may result in variations in the degree of evidence acceptability and therefore in the reimbursement decision [Citation1,Citation4]. This could significantly influence (time to) patient access to new drugs after marketing authorization, because additional data or analyses may be needed for specific countries [Citation2,Citation5]. Differences in time-to-patient access between countries may be conceived as undesirable. Therefore, improving the alignment of evidentiary requirements is warranted, as it may improve equity in patient access to novel products. After HTA decision-making, also other factors may influence the launch of a product, for example, insufficient budget to implement decisions, challenges with infrastructure, and sub-national approval processes [Citation6–8].
Although national differences in oncology assessments have been recognized, an analysis has describing and comparing the views of European HTA bodies on the requirements for the relative effectiveness assessments of oncology drugs is yet to be performed [Citation3,Citation6,Citation8–10]. This study, in collaboration with the European Federation of Pharmaceutical Industries and Associations (EFPIA), aims to analyze the differences in evidentiary requirements for the relative effectiveness assessment of oncology drugs among six national HTA bodies from different European locations and provide recommendations toward an improved alignment among HTA bodies in order to enhance equity in access in Europe. In addition, recommendations to optimally manage the remaining differences were collected.
2. Methods
This study is a follow-up on a previous study that focused on the differences in evidentiary requirements for oncology drugs between the EMA and six European HTA bodies. In that study, interviews were performed to identify areas where alignment can be enhanced and provide recommendations to improve alignment [Citation11]. The current article builds on that research by using the interviews from the HTA bodies to analyze the differences between the individual HTA bodies’ evidentiary requirements for oncology drugs and provide recommendations to increase alignment among HTA bodies. Improving alignment among HTA bodies is important for the success of the joint clinical assessment and to increase equity in patient access. The findings of the current study will be positioned in context of the upcoming EU HTA regulation, a new law which will be implemented of 2025 with the aim for EU member states to cooperate more in the assessment of medicines and devices. In addition, this article is part of a broader project called ‘time to patient access’ requested by EFPIA [Citation12], which aimed to unite stakeholders across Europe by establishing the different causes of delays in patient access to new oncology treatments as well as identifying solutions with the potential to reduce time to patient access. The overall aim of the initiative was to accelerate accessibility to new oncology drugs without compromising careful deliberations and evidence-based decision-making [Citation12]. For example, recently, the evaluations of the COVID-19 vaccines have shown the opportunities for a fast evaluation process. This current paper elucidates on the part of the project focusing on the differences in evidentiary requirements between individual HTA bodies and providing recommendations on aligning and reducing such differences.
In this analysis, we defined an evidentiary requirement as the minimum level of evidence accepted by an HTA body as being convincing. Requirements were compared among six European countries. Interview data were gathered and subsequently used to analyze evidentiary requirements on the degrees of evidence acceptability and alignment among HTA bodies. Detailed methods regarding the criteria for identification and selection of HTA bodies, development of classifications of domains and subdomains, and the design of the interviews were described in a previous publication and are summarized below [Citation11]. A summary of the data collection and analysis methods is provided below.
2.1. Data collection
To ensure a representative heterogeneous set of different systems in place in Europe, six HTA bodies were selected based on the following explicit criteria: geographic location (Northern, Eastern, Southern and Western Europe), organization of the healthcare system (national vs. regional/local) [Citation4,Citation12,Citation13], the time between marketing authorization and patient access (long, modest and short) [Citation14], and HTA orientation (e.g. focus on cost-effectiveness or clinical effectiveness or budget impact) [Citation4,Citation12,Citation13]. Based on these criteria, Italy, the Netherlands, Poland, Portugal, England and Wales, and Sweden were selected, see Appendix 1 [Citation11,Citation14]. Subsequently, 5 domains representing the elements of discussion during a drug’s relative effectiveness assessment were identified in evidentiary requirements. These domains were based on Shaping European Early Dialogues (SEED) and included population, comparator, endpoints, trial design and data sources, and statistical analysis [Citation11,Citation15]. Even though SEED also identified a sixth domain, namely economic modeling, it was excluded because this study focuses on relative effectiveness assessments [Citation16]. It is expected that the possibilities of realizing improved alignment in relative effectiveness assessments are promising. The domains were further divided into 19 subdomains based on Tafuri et al. 2016 [Citation2] (Appendix 2). PFS was separated from ‘other surrogate endpoints,’ because it is widely used as a primary endpoint in oncology trials, despite HTA bodies generally favoring evidence on OS [Citation11,Citation17]. Structured qualitative interviews were conducted with stakeholders involved in drug assessments from six national HTA bodies. These interviews took place in the form of digital meetings between August and September 2019 and included current and former members, and consultants [Citation11]. An overview of the respondents can be found in Appendix 3. Follow-up questions were asked via e-mail. Prior to the interviews, an interview guide was designed to elicit the evidentiary requirements for assessment for oncology products and recommendations on potential further alignment to minimize and potentially manage differences in evidentiary requirements according to the authorities. The interview guide contained, in most cases, one interview question per subdomain. The exception to this were the domains ‘PFS as an endpoint,’ and ‘quality of life/health-related quality of life and other patient-reported outcomes.’ These domains were combined into one question (Appendix 4) [Citation11].
2.2. Data analysis
Answers containing the evidentiary requirements were summarized and categorized based on similarities between the evidentiary requirements of the HTA bodies. Interviewees were asked to validate the categories that emerged from their interviews. The validation process has been described in a previous publication [Citation11].
To analyze the data on comparative degrees of evidence acceptability and alignment among the HTA bodies, the evidentiary requirements were labeled as ‘accepted,’ ‘often accepted,’ ‘case dependent,’ ‘often not accepted,’ or ‘not accepted.’ Labeling was performed by two authors (S.W. and C.J.), and disagreements were resolved through consensus. When the evidentiary requirement stated that a certain type of evidence was (not) accepted, it was labeled as ‘accepted’ or ‘not accepted.’ When the evidentiary requirement stated that a certain type of evidence was ‘accepted, if,’ or ‘not accepted, unless’ it was labeled as ‘often accepted’ or ‘often not accepted.’ When the evidentiary requirement state that a certain type of evidence was ‘accepted, but,’ it was generally labeled as ‘accepted’ as no conditions are attached to its acceptance, although some considerations on the evidence may apply. An exception to this is the inclusion of a condition that influences the chance of acceptance, resulting in labeling it as ‘often accepted.’ When the probability of (non-) acceptability seemed to vary, an evidentiary requirement was labeled as ‘case dependent’ [Citation11]. The degrees of acceptability and alignment were quantified as follows:
The degree of acceptability of the evidence was quantified by calculating the ratio of subdomains labeled ‘accepted’ or ‘often accepted,’ and the total number of subdomains was expressed as a percent per HTA body.
The degree of alignment on evidentiary requirements was quantified by dividing the number of matching acceptability labels (except for requirements labeled as ‘case dependent’ as the implication with this label can differ per HTA body) by the total number of acceptability labels, expressed as a percent. The degree of alignment was presented in three ways:
Alignment per subdomain: Alignment among HTA bodies per subdomain.
Relative alignment on group level: Alignment per HTA body on the 19 subdomains relative to 1) no other HTA body (no alignment), 2) some of the other HTA bodies (partial alignment), 3) all HTA bodies (complete alignment), presented in bar graphs.
Relative alignment on individual level: Alignment on the 19 subdomains among the individual HTA bodies, presented in a matrix.
An overview of the above-described analyses of the degrees of acceptability and alignment among HTA bodies is presented in . Example calculations are provided in Appendix 5. The recommendations of the interviewees were divided into two categories: ‘recommendations to HTA bodies’ and ‘recommendations to manufacturers.’
Figure 1. Outline of the methods of the secondary analysis of this research project.

3. Results
The evidentiary requirements of the HTA bodies are summarized in .
Table 1. Evidentiary requirements for oncology drug assessments of six HTA bodies, labeled for acceptability (symbols).
3.1. Degree of evidence acceptability
A summary of the degree of evidence acceptability among the HTA bodies is presented in . Poland showed the highest degree of acceptability (79%), followed by England and Wales (68%), Sweden (58%), the Netherlands (47%), Italy (42%), and finally Portugal, with the lowest degree of acceptability (37%). Low degrees of evidence acceptability were specifically observed for the subdomains ‘absence of statistical significance’ (0%), ‘post-hoc subgroup analyses’ (0%), ‘target population as authorized by EMA’ (17%), ‘class effects’ (17%), and ‘clinical relevance of effect size as assessed by EMA’ (17%).
. Evidentiary requirements for oncology drug assessments of six HTA bodies, labeled for acceptability (symbols).
3.2. Alignment among HTA bodies
All six HTA bodies were aligned on the acceptability of evidence with regard to the use of biomarkers for population selection, the selected comparator, and the use of real-world evidence as a clinical data source (100% alignment), as presented in . None of the HTA bodies were aligned on the use of other surrogate outcomes and the clinical relevance of effect size as assessed by EMA (0% alignment). The alignment among the HTA bodies regarding the acceptability of the target population as authorized by EMA, extrapolation to other populations, class effects, and the use of cross-over in trials was limited (33%). The alignment on considering indirect treatment comparisons, single-arm trials, PFS as an endpoint, absence of QoL data, network meta-analysis, novel trial designs, and short trial duration was 50%.
Several subdomains labeled as ‘often accepted’ were aligned among the HTA bodies, albeit under distinct conditions. For instance, the HTA body’s acceptance of a ‘selected comparator’ required it to be either the standard of care or the drug used in the clinical trial. The subdomain ‘novel trial design’ was labeled ‘often accepted’ when ‘if methodology is well described,’ ‘if controlled,’ and ‘if plausible biological mechanism.’ Although the HTA bodies tend to accept novel trial designs, and were therefore aligned, the conditions that the novel trial designs must meet may differ. The same is observed for the subdomain ‘evidence from small populations:’ if the evidence is satisfactory, the evidence will be accepted by four HTA bodies, but the conditions for satisfactory evidence may differ. For the subdomain ‘short time period,’ three HTA bodies accept ‘the longer the better,’ on which these three HTA bodies align. However, conditions differ as two of the three HTA bodies seemed concerned that a short period creates uncertainty, and one of those two HTA bodies appears to have requested that a convincing mean OS should be demonstrated.
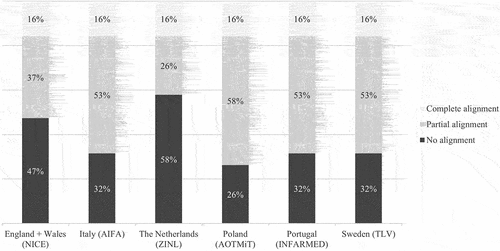
Relative alignment on the group level is presented in . Among the subdomains, 16% exhibited alignment among all HTA bodies, while 38% showed no alignment with any of the other HTA bodies. Poland was most often aligned with at least one other HTA body, with only 26% of the subdomains not in alignment with any of the HTA bodies. The Netherlands had the largest proportion of subdomains not aligned with any other HTA body, accounting for 58% of the subdomains. Relative alignment on the individual level showed that Italy and Portugal had the highest degree of alignment with each other on the 19 subdomains (58%). For all other individual comparisons, the alignment was below 50% (see Appendix 6).
Figure 2. Relative alignment on group level: alignment on subdomains for each HTA body relative to the other HTA bodies from complete to no alignment*.
3.3. Recommendations
The recommendations of the respondents on how to reduce differences and address the remaining ones in evidentiary requirements were categorized into recommendations for HTA bodies and recommendations for manufacturers.
Recommendations for HTA bodies encompassed the following:
International cooperation – If one international body were to assess the effectiveness and safety of a new drug against a few pre-agreed comparators, with distant participation of other HTA bodies who can (but are not obliged to) use the same assessment, duplication of effort among HTA bodies could be prevented. In addition, it was recommended that HTA bodies join forces with others when assessing the implications of innovations in assessment methodologies, for example on how to assess novel trial designs or new endpoints.
Use of access agreements – According to the respondents, the use of immature data for the oncology drugs assessment is increasing, creating more uncertainty in the assessment. HTA bodies must make a trade-off between uncertainty and early patient access. Access agreements such as outcome-based pricing and financing proposals were recommended to help overcome payers’ concerns; they could be used to deal with the uncertainty by increasing flexibility in accepting some uncertainty while maintaining a reasonable time for patient access.
Centralization of the processes – HTA bodies should align on which requirements can be assessed nationally and which should be assessed locally, and consequently, aligning when evidence is considered sufficient. Where parts of the regional HTA body assessment are or can be aligned, unnecessary/superfluous work can be prevented. The assessment of the need for the new therapy, the available comparator in the country, the added benefit of the therapy, and the quality of the evidence can possibly be centralized.
Recommendations related to manufacturers include:
Alignment between clinical trials and the requested indication – This should be guaranteed to avoid the need for post-hoc subgroup analysis, which creates uncertainty. Therefore, the manufacturer should ensure that the population in the clinical trials is the same as the population for which reimbursement is requested.
Early dialogues with HTA bodies – Early dialogues could be used to establish alignment regarding the evidence required for each HTA assessment, including the trial design, patient population (trial population aligned with the indication), endpoints (use of PFS and surrogate endpoints), and the comparator for the trial. The HTA body is responsible for facilitating the opportunity for an early dialogue and adhering to the agreed upon requirements. Nevertheless, the manufacturer should request an early dialogue and implement the requirements to avoid submitting insufficient evidence and, consequently, minimize delay. Where the acceptability of evidence varies among the HTA bodies, early dialogues to identify the differences in evidentiary requirements should be held with each HTA body. Moreover, a joint early dialogue with European HTA bodies could prove beneficial.
4. Discussion
This study showed that the degree of evidence acceptability in HTA assessments for oncology drugs ranges between 79% (Poland) and 37% (Portugal). In addition, the degree of alignment – comprising complete and partial alignment – among HTA bodies ranges between 74% (Poland) and 42% (the Netherlands). This broad range in evidence acceptability and alignment indicates that there is room for further alignment of evidentiary requirements among HTA bodies. Stricter demands for more robust evidence potentially result in the need for new evidence generation if the demands were unknown upfront, which could, in turn, prolong the time to patient access to oncology drugs. This demonstrates how important it is for the manufacturers to be aware of the evidentiary requirements of every HTA body at an early stage when designing and undertaking clinical trials. Early dialogues with HTA bodies could help identify the evidence required for a given HTA assessment. Relative alignment on group level was shown by the proportion of subdomain on which HTA bodies had complete alignment and no alignment. Complete alignment was observed in 16% of the subdomains. No alignment with any other HTA body varied from approximately a quarter of the subdomains for Poland to over half of the subdomains for the Netherlands. This difference in the proportion of subdomains with no alignment with any other HTA body shows that, for some countries, alignment with at least one other country should be possible.
No trends were observed in terms of differences in the degree of evidence acceptability and alignment in relation to geographic location, healthcare system, time between market authorization and patient access, as well as HTA orientation of the included HTA bodies. However, the inclusion of a select number of countries with high heterogeneity may result in missing potential correlations. Moreover, other system-specific factors may contribute to the differences, for example the role of the HTA body, the involvement of stakeholders, and if the HTA body performs the assessment versus the assessment and appraisal [Citation8].
Portugal had the lowest degree of evidence acceptability but also the best alignment with another country (Italy). This demonstrates that stricter evidentiary requirements do not necessarily result in increased misalignment. The difference in evidentiary requirements further emphasizes the importance of early dialogues with HTA bodies, such as the joint scientific advice that was offered by EMA and the European Network of Health Technology Assessment (EUnetHTA) and will be offered as joint scientific consultation via the EU HTA regulation. It also reflects the challenges an international assessment body, such as the member state coordination group on HTA for the EU HTA regulation [Citation16,Citation18], may need to overcome in order to harmonize European evidentiary requirements. Such an international assessment body could use the identified differences as a basis for harmonization.
The evidentiary requirements were repeatedly labeled as ‘case dependent,’ which increased the uncertainty for the manufacturer. In the absence of clearly defined requirements, there is a risk of submitting insufficient evidence, which consequently necessitates amending the dossier or even the conducting a new trial. Moreover, the representatives of the HTA bodies in Italy, the Netherlands, Poland, England and Wales, and Sweden defined their requirements for several subdomains as ‘could be accepted,’ ‘creates uncertainty,’ or ‘can be accepted, creates uncertainty.’ As a result, the degree of uncertainty in the evidence need not necessarily affect its acceptability. Clinical and economic restrictions, unmet medical need, and pricing or financing agreements could help compensate of the uncertainty [Citation19]. Moreover, HTA bodies could request additional information [Citation20].
This analysis indicates that the areas in which HTA bodies are least aligned are the acceptability of extrapolation to other populations, class effects, PFS as an endpoint, surrogate endpoints, absence of QoL data, single-arm trials, cross-over in trials, short trial period, and clinical relevance of the effect size. These findings are consistent with those of a previous study, in which the majority of these subdomains were also the least aligned with EMA [Citation11]. However, subdomains ‘class effects,’ ‘absence of QoL data,’ and ‘single-arm trial’ subdomains were not misaligned with EMA. This suggests that HTA bodies could be more aligned on the acceptability of these subdomains, showing the importance of collaboration between HTA bodies.
Recommendations proposed by the interviewees included initiation of early dialogues (per country or even jointly), intensification of collaboration and exchange between countries, alignment on which domains can be assessed centrally and which by local and regional HTA bodies, and the use of access agreements. These recommendations for improving alignment have already been described in previous studies [Citation2,Citation20,Citation21]. Since the interviews in 2019, several international and local initiatives have been developed to improve cooperation between countries. For example, the European Network for Health Technology Assessment (EUnetHTA 21) performs joint relative effectiveness assessments, which can be used as an alternative or addition or be incorporated into the national submission [Citation22]. Moreover, as of 2025, the new EU HTA regulation also plans to begin joint clinical assessments for new molecular entities in oncology [Citation16]. Following the joint clinical assessment, the final appraisal and the subsequent reimbursement decision remain within the remit of each member state. The EU HTA regulation has the potential to enhance alignment on evidentiary requirements of oncology drugs through discussions with all member states. Via these discussions all differences can be identified and agreements on acceptability of evidentiary requirements can be made. Enhanced alignment between HTA bodies continues to be crucial for improving equity in patient access. Challenges of the EU HTA Regulation are the areas where alignment during the joint clinical assessments cannot be reached or when there is an unwillingness to cooperate due to the feeling of underrepresentation of the countries’ evidentiary requirements. These challenges can result in an additional request for information. It is therefore of the utmost importance that the joint clinical assessment incorporates as much perspectives as possible, i.e. both in the inclusion of various evidentiary requirements in case of irreconcilable differences as well as in various HTA body perspectives. Examples of smaller international collaborations are BeNeLuxA, FINOSE, and the Valletta Declaration. They represent HTA collaborations that perform joint pricing negotiations horizon scanning and share knowledge [Citation23]. An additional benefit of a joint assessment is the use of one language for dossiers rather than different languages for individual countries [Citation24].
Results from a European retrospective study of reimbursement recommendations for conditionally approved drugs suggested that the use of uncontrolled trials or the use of controlled trials did not influence on the HTA decision [Citation25]. Our study, however, showed that uncontrolled trials were assessed as less strong evidence in Poland, whereas uncontrolled trials were often not accepted in Portugal. A reason for this deviation could be the difference in country selection, as the retrospective study only included Western European countries, whereas our study also includes Eastern and Southern European countries. Another European study demonstrated a varying relevance of PFS among the HTA bodies and the missing QoL data in the dossiers [Citation3], as found in our study.
The analysis of six European countries is a limitation of this research, even though we purposefully included countries from different European locations, which are representative of all European national HTA systems. Still, more research with a larger sample of European countries is needed to identify a complete overview of evidentiary requirements in Europe. In addition, the use of a highly heterogeneous sample can give an overestimation of the average level of disagreement between European HTA bodies [Citation26]. On the other hand, countries using cost-effectiveness are overrepresented thereby underreporting the differences in evidentiary requirements for countries without using cost-effectiveness. As no official HTA guidelines regarding the relative effectiveness of oncology drugs were available, interviews were conducted with stakeholders in drug assessments. To reduce the potential for personal bias/subjectivity, interviews with individuals representing the HTA bodies were carried out to the greatest extent feasible. Members and advisors represented the HTA bodies. The consultants were professors in HTA of the respective countries and were considered to give an objective representation. No bias was expected between countries represented by two respondents versus countries represented by one respondent. When two respondents represented the same country, the interview was carried out together. The respondents had no disagreement between the answers.
This research provides an initial indication of differences in evidentiary requirements and the degree of evidence acceptability among HTA bodies. It is a snapshot, which will continue to change over time. Therefore, monitoring the requirements for oncology drugs should be continued, to map changes in (differences) in evidentiary requirements. Moreover, this study focused on the differences in relative effectiveness assessments. Differences in the evidentiary requirements of economic modeling are to be expected. Future research could use a similar analysis and approach to identify the differences and provide recommendations toward an increased alignment.
5. Conclusion
There is a broad range in evidence acceptability and alignment indicating that there is room for further alignment of evidentiary requirements among HTA bodies. The difference in evidentiary requirements reflects the challenges an international assessment body will need to overcome in order to harmonize European evidentiary requirements. Recommendations to achieve this include joint early dialogues, intensifying collaboration and exchange between countries, joint relative effectiveness assessments, and the use of access agreements.
6. Expert opinion
This research demonstrates the differences in evidentiary requirements among HTA bodies, reflecting the potential challenges that could arise during the joint clinical assessment following EU HTA regulation. This research indicates that the areas in which HTA bodies are least aligned are the acceptability of extrapolation to other populations, class-effects, PFS as an endpoint, surrogate endpoints, absence of QoL data, single-arm trials, cross-over in trials, short trial period, and clinical relevance of the effect size. The identified differences indicate which evidentiary requirements for oncology assessment in the included countries could potentially enhance alignment. Harmonization of the European evidentiary requirements is necessary to ensure a smooth and efficient assessment process. Furthermore, it would serve to prevent countries from needing to request additional information and analyses. To realize harmonization, countries must discuss their evidentiary requirements and the significance of each element for their assessment. It is essential to develop, through collaborative effort, a new set of clearly defined guidelines outlining the evidentiary requirements for a joint clinical assessment which all member states unanimously endorse. The recommendations to improve alignment provided by the respondents are realistically due to the implementation of the EU HTA regulation. To accomplish this, a subgroup of the Member State Coordination Group on Health Technology Assessment (HTACG) will perform joint scientific consultations and another subgroup will perform joint clinical assessments. The joint clinical assessments aim at a collaboration between European countries, with the key challenge being including the needs and perspectives of all of them. If this aspect is not sufficiently addressed, it might result in a reduced willingness to adopt the advice of the assessment. Discussions that involve sufficient representation from all countries could solve this challenge. The recommendation to use access agreements to manage additional uncertainty is already used in some countries, thus holding great potential for its adoption in others. This would improve the speed of HTA decision-making: as negotiations on different access agreements can be challenging and long-lasting, starting these discussions early on, before the HTA decision-making, can help speed up this process.
Although the EU HTA regulation has considerable potential, its actual benefits remain uncertain. The regulation aims to improve the availability of innovative health technologies and to guarantee the optimal utilization of resources and enhance the quality of HTA in the European Union. Its objective is to reduce duplication of efforts for all stakeholders and create predictability. While these are promising goals, the aforementioned challenges can make achieving them difficult. Doubts have emerged whether the HTA regulation will indeed improve availability and facilitate the procedure, or if it will introduce additional hurdles. Future research should therefore investigate whether patient equity has improved and how the regulation has influenced the time to HTA decision-making and patient access. In 10 years, the assessment of alignment between the individual countries and the joint clinical assessment is warranted, along with potential improvements. Based on the additional information that will be sought during each of the national assessments post joint clinical assessment can reveal any remaining redundancies among HTA bodies.
Beyond oncology assessments, joint clinical assessments will also be carried out for advanced therapy medicinal products. These medications present their own challenges and differences within evidentiary requirements. This is also a promising area to explore how the EU regulation could manage the differences and improve alignment between countries.
Within 10 years, joint clinical assessments will be performed for all medicinal products approved under the EU centralized procedure. It will hopefully improve the medication availability for patients and shorten the time to HTA decision-making and patient access without compromising careful deliberations and evidence-based decision-making of the individual countries.
Article highlights
Although the same pivotal trial data is used for the oncology reimbursement assessments of health technology assessment bodies, the duration and outcome of the assessment vary owing to a different weighing of various elements of the assessment and evidence by the health technology assessment bodies.
This study identified the oncology evidentiary requirements based on the views of stakeholders in drug assessments of six national health technology assessment bodies, concerning current and former members, and consultants. Areas in which HTA bodies are least aligned are the acceptability of extrapolation to other populations, class-effects, PFS as an endpoint, surrogate endpoints, absence of QoL data, single-arm trials, cross-over in trials, short trial period, and clinical relevance of the effect size.
Some evidentiary requirements were not clearly defined, resulting in a level of uncertainty. In the absence of clearly defined requirements, there is a risk of submitting insufficient evidence, which consequently necessitates amending the dossier of even conducting a new trial.
Recommendations to increase alignment and optimally manage remaining differences include joint early dialogues, intensified collaboration and exchange between countries, joint relative effectiveness assessments, and the use of access agreements.
Evidentiary requirements concern snapshots, which will continue to change over time. Therefore, monitoring the requirements for oncology drugs should be continued, to map changes in evidentiary requirements.
The new EU HTA regulation might be able to improve alignment and enhance inequity in patient access.
Author contributions
S Wolters, C Jansen, and M J Postma participated in the concept and design of the paper. S Wolters conducted the acquisition of data. All authors participated in the analysis and interpretation of data, in the drafting of the manuscript, and in the critical revision of the paper for important intellectual content. Final approval of the manuscript was given by all authors.
Declaration of interest
S. Wolters reports grants from European Federation of Pharmaceutical Industries and Associations, during the conduct of the study. S. Wolters is an employee of Asc Academics, however, this organization had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
C. Jansen reports grants from European Federation of Pharmaceutical Industry Associations (E.F.P.I.A), during the conduct of the study; grants from European Federation of Pharmaceutical Industry Associations (E.F.P.I.A), outside the submitted work. C. Jansen is currently employed by Inbeeo ltd, consultancy that receives payments for projects in the field of market access. Inbeeo ltd was not involved in the study design, data collection and analysis, interpretation of data, decision to publish of preparation of the manuscript.
MJ. Postma reports interests in Pharmacoeconomics Advice Groningen, and in Health-Ecore, both of which are unrelated to the submitted work.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- IQVIA. EFPIA patients W.A.I.T. Indicator 2021 survey [Internet]. 2022 [cited 2023 Aug 21]. Available from: https://www.efpia.eu/media/676539/efpia-patient-wait-indicator_update-july-2022_final.pdf.
- Tafuri G, Pagnini M, Moseley J, et al. How aligned are the perspectives of EU regulators and HTA bodies? A comparative analysis of regulatory-HTA parallel scientific advice. Br J Clin Pharmacol. 2016;82:965–973.
- Kleijnen S, Lipska I, Alves TL, et al. Relative effectiveness assessments of oncology medicines for pricing and reimbursement decisions in European countries. Ann Oncol Internet. 2016 [cited 2019 Nov 26];27(9):1768–1775. doi: 10.1093/annonc/mdw233
- Angelis A, Lange A, Kanavos P. Using health technology assessment to assess the value of new medicines: results of a systematic review and expert consultation across eight European countries. Eur J Health Econ. 2018;19(1):123–152. doi: 10.1007/s10198-017-0871-0
- Wilking N, Bucsics A, Sekulovic LK, et al. Achieving equal and timely access to innovative anticancer drugs in the European Union (EU): summary of a multidisciplinary CECOG-driven roundtable discussion with a focus on Eastern and South-Eastern EU countries. ESMO Open. 2019;4(6):550. doi: 10.1136/esmoopen-2019-000550
- Akehurst RL, Abadie E, Renaudin N, et al. Variation in Health technology assessment and reimbursement processes in Europe. Value Health Internet. 2017;20:67–76. doi: 10.1016/j.jval.2016.08.725
- EFPIA. The root cause of unavailability and delay to innovative medicines the root cause of unavailability and delay to innovative medicines: reducing the time before patients have access to innovative medicines [Internet]. 2023. Available from: https://www.efpia.eu/media/602581/principles-on-the-transparency-of-evidencefrom-novel-pricing-and-payment-models.pdf.
- Fontrier AM, Visintin E, Kanavos P. Similarities and differences in Health technology assessment systems and implications for coverage decisions: evidence from 32 countries. Pharmacoecon Open Adis. 2022;6(3):315–328. doi: 10.1007/s41669-021-00311-5
- Shah KK, Mestre-Ferrandiz J, Towse A, et al. A review of health technology appraisals: case studies in oncology. Int J Technol Assess Health Care. 2013;29(1):101–109. doi: 10.1017/S0266462312000669
- Leyens L, Brand A. Early patient access to medicines: Health technology assessment bodies need to catch up with new marketing authorization methods. Public Health Genomics. 2016;19(3):187–191. doi: 10.1159/000446537
- Wolters S, Jansman F, Postma M. Differences in evidentiary requirements between European Medicines Agency and European Health technology assessment of oncology drugs—can alignment be Enhanced? Value Health. 2022;25(12):1958–1966. doi: 10.1016/j.jval.2022.05.006.
- Jansen C, Amesz B. Improving time to patient access to innovative oncology therapies in Europe. Vintura/EFPIA; 2020. https://www.efpia.eu/media/554646/every-day-counts-improving-time-to-patient-access-to-innovative-oncology-therapies-in-europe.pdf.
- Vogler S, Haasis MA, Dedet G, et al. WHO. MEDICINES REIMBURSEMENT POLICIES in EUROPE; 2018. https://iris.who.int/handle/10665/342220
- IQVIA. EFPIA patients W.A.I.T indicator 2019 survey. 2020;
- Facey K, Granados A, Meyer F, et al. Multi-stakeholder collaboration in generating the best possible knowledge – the SEED experience. Report of HTAi 2015 panel session. Health Technology Assessment International – Canada. 2015. [Internet]. [cited 2020 Mar 20]. Available from: https://htai.org/wp-content/uploads/2018/02/HTAi_SEED_Panel_Report_final.pdf.
- Barbuto V. European Commission. Update on the implementation of regulation (EU) 2021/2282 on Health technology assessment; 2022. https://www.eunethta.eu/wp-content/uploads/2022/11/20221118-EUnetHTA-stakeholder-meeting-FINAL.pdf
- Miksad RA, Zietemann V, Gothe R, et al. Progression-free survival as a surrogate endpoint in advanced breast cancer. Int J Technol Assess Health Care. 2008;24(4):371–383. doi: 10.1017/S0266462308080495
- European Commission. Implementing the EU health technology assessment regulation; 2023. https://health.ec.europa.eu/system/files/2023-06/hta_regulation-implementation_factsheet_en.pdf
- Bloem L, Vreman R, Peeters N, et al. Associations between uncertainties identified by the European Medicines Agency and national decision making on reimbursement by HTA agencies. Clin Transl Sci Internet. 2021 [cited 2022 Apr 4];14(4):1566–1577. doi: 10.1111/cts.13027
- Wang T, McAuslane N, Liberti L, et al. Building synergy between regulatory and HTA agencies beyond processes and procedures—can we effectively align the evidentiary requirements? A survey of stakeholder perceptions. Value Health Internet. 2018;21:707–714. doi: 10.1016/j.jval.2017.11.003
- Ofori-Asenso R, Hallgreen CE, de Bruin ML. Improving Interactions between Health technology assessment bodies and regulatory Agencies: a Systematic review and cross-Sectional Survey on processes, progress, outcomes, and challenges. Front Med. Frontiers Media S.A. 2020;7:1–17. doi: 10.3389/fmed.2020.582634.
- EUnetHTA WP7 research and analysis activity 1: final report an analysis of HTA and reimbursement procedures in EUnetHTA partner countries: final report.
- Association of European Cancer Leagues (ECL). LET’S TALK ACCESS! White paper on tackling challenges in access to medicines for all cancer patients in Europe. 2018.
- European Network for health technology assessment. EUnetHTA WP7 research and analysis activity 1: annex 1 Agency data an analysis of HTA and reimbursement procedures in EUnetHTA partner countries: annex 1 Agency data.
- Vreman RA, Bouvy JC, Bloem LT, et al. Weighing of evidence by Health technology assessment bodies: retrospective study of reimbursement recommendations for conditionally approved drugs. Clin Pharmacol Ther. 2019;105(3):684–691. doi: 10.1002/cpt.1251
- Nicod E, Berg Brigham K, Durand-Zaleski I, et al. Dealing with uncertainty and accounting for social Value judgments in assessments of orphan drugs: evidence from four European countries. Value Health. 2017;20(7):919–926. doi: 10.1016/j.jval.2017.03.005
Appendix 1
– Selected countries
Selection criteria employed by the 6 countries.*
Appendix 2
– overview of the domains and subdomains
Predefined domains and subdomains representing the elements of discussion during an oncology assessment.*
Appendix 3
– Overview of HTA-body respondents*
Overview of the HTA body respondents
Appendix 4
– Interview questions*
Interview questions per domain
General
Which hurdles are seen during the assessment of anti-cancer drugs and what can be improved? (in relation to reducing the time to patient access through faster assessments and better alignment of the evidence criteria and requirements). Where in the process can we avoid duplication and reduce timelines?
Population
Could the target population differ from the population identified by EMA and for what reasons?
Is the use of biomarkers accepted and are there any conditions?
Is extrapolation to other populations accepted and under what conditions?
Comparator
Which comparators are acceptable in the assessment, and do they differ from those required by EMA? (Whatever was used in the RCT/best possible care/best standard of care/placebo/other)
Are indirect comparisons accepted, and under what conditions?
Are class effects accepted?
Endpoints
Do the accepted endpoints and HRQoL measures for oncolytics differ from the ones accepted by EMA and how?
Are pathologic complete response and minimal residual disease also accepted as surrogate endpoints, and under what conditions are surrogate endpoints accepted?
Trial design
Is real world evidence/real world data accepted, and under what conditions?
Are network meta-analyses accepted, and under what conditions?
Are single-arm trials accepted, and what best practices apply when only single-arm trials are submitted?
Would novel trial designs (for example master protocols and enrichment strategies) be accepted, and under what conditions?
Is crossover in a trial accepted, and what constitutes best practices when crossover is included?
If the evidence for a small target population is accepted by EMA, is it also accepted by the HTA body?
What are the usual time periods for measuring endpoints, and what is the minimal time to follow-up?
Statistical analyses
How decisive is statistical significance?
Are (post-hoc) subgroup analyses accepted, and under what conditions?
If EMA considers an endpoint (for example OS and PFS) to be of large enough magnitude/have a clinically relevant effect size, does the HTA body always concur?
* Obtained from:Wolters S, Jansman F, Postma M. Differences in Evidentiary Requirements Between European Medicines Agency and European Health Technology Assessment of Oncology Drugs – Can Alignment Be Enhanced? Value in Health 2022; S1098-3015: 01987–8.
Appendix 5
– examples calculation of the degrees of acceptability and alignment
Evidentiary requirements for oncology drug assessments, labeled for the acceptability (symbols)
Degree of evidence acceptability:
The degree of evidence acceptability for Italy is 33%, whereas 1 out of the 3 evidentiary requirements were labeled as ‘accepted’ or ‘often accepted.’
The degree of evidence acceptability for the Netherlands is 33%, whereas 1 out of the 3 evidentiary requirements were labeled as ‘accepted’ or ‘often accepted.’
The degree of evidence acceptability for Poland is 67%, whereas 2 out of the 3 evidentiary requirements were labeled as ‘accepted’ or ‘often accepted.’
Alignment
1) Alignment per subdomain
The alignment of the HTA bodies on the subdomain ‘target population as authorized by EMA’ is 50%, whereas 2 (Italy and Poland) out of the 3 countries had the same acceptability label
The alignment of the HTA bodies on the subdomain ‘use of biomarkers’ is 100%, whereas all countries had the same acceptability label.
The alignment of the HTA bodies on the subdomain ‘extrapolation to other populations’ is 0%, whereas none of countries had the same acceptability label.
2) Relative alignment on group level:
Italy is for 33% of the subdomains aligned with all the other countries: Italy had the same acceptability label as the other 2 countries on 1 of the 3 subdomains, namely ‘use of biomarkers.’
Italy is for 33% of the subdomains aligned with one other country: Italy had the same acceptability label as 1 other country on 1 of the 3 subdomains, namely ‘target population as authorized by EMA.’
Italy is for 33% of the subdomains aligned with none of the other countries: Italy had no matching acceptability label with any of the other country on 1 of the 3 subdomains, namely ‘extrapolation to other populations.’
3) Relative alignment on individual level:
Italy is aligned with Poland on 67% of the subdomains: 2 out of the 3 subdomains had matching acceptability labels.
Italy is aligned with the Netherlands on 33% of the subdomains: 1 out of the 3 subdomains had matching acceptability labels
Poland is aligned with the Netherlands on 33% of the subdomains: 1 out of the 3 subdomains had matching acceptability labels