
ABSTRACT
Introduction
Approved biosimilars exhibit comparable efficacy, safety, and immunogenicity to reference products. This report provides perspectives on the societal value of biosimilars within Europe and potential factors that have influenced market dynamics.
Methods
An independent, self-administered survey or one-on-one in-depth interview was used to collect viewpoints about the impact of biosimilar medicines within European markets. Key insights were also sought from an expert panel of European stakeholders.
Results
Survey respondents were clinicians, pharmacists, and payers from Europe (N = 103). Perceived benefits of biosimilars included increased access to innovative medicines (73% of respondents) or biologic treatments (66%). Biosimilar competition was thought to expand access to biologics (~50% of respondents) or drug combinations (~36%) and reduce biologic access time (34%). Key drivers of biologic access after biosimilar competition included increased biologic awareness (51%) and changes to prescribing guidelines (37%) and/or treatment paradigms (28%). The expert panel developed a market maturity framework of biosimilar adoption/opportunities comprising three stages: ‘Invest,’ ‘Expand,’ and ‘Harvest.’ Findings were supported by published literature.
Conclusions
In Europe, the perceptions of well-informed survey/interview respondents are that biosimilars have improved patient outcomes via increased access to biologics and innovative biologic products, contributing to earlier and longer treatment of a broader population.
1. Introduction
The impact of biologics has been transformative across a wide range of therapeutic areas including (but not limited to) oncology, rheumatology, gastroenterology, dermatology, endocrinology, and fertility. Biologics comprise a complex combination of purified molecules manufactured and harvested from a biological source. Unlike small-molecule drugs, which are chemically synthesized with the potential for systemic off-target effects, biologics have targeted actions against disease and in some therapeutic areas are associated with relatively fewer side effects than conventional therapeutics [Citation1]. Biologics now account for 34% of the pharmaceutical expenditure in Europe (€78.6 billion in 2021), and for more than a decade have prevailed as the dominant pharmaceutical market segment [Citation2]. However, biologics are inherently costly, and their price can pose a barrier that may more profoundly restrict accessibility to patients in need of disease-modifying therapies for chronic and disabling conditions. The introduction of biosimilars following the loss of exclusivity for originator products (reference drugs) can ease such an economic burden [Citation3]. Biosimilars are structurally close, highly similar versions of originator biologic products and have proven to be clinically equivalent to the approved reference medicine in terms of biological function, efficacy, safety, and immunogenicity profile [Citation4,Citation5].
The regulation of biosimilars was pioneered in the European Union (EU) in 2006 with the approval of a recombinant human growth hormone (somatropin), which was rapidly followed by an erythropoiesis-stimulating agent (epoetin) in 2007 and a granulocyte colony-stimulating factor (G-CSF; filgrastim) in 2008. Biosimilars were rolled out more comprehensively in 2013 when the first biosimilar monoclonal antibody (infliximab) and recombinant human luteinizing hormone (follitropin alpha) were approved [Citation6,Citation7]. Since then, numerous biosimilars have been approved in Europe across multiple drug classes and indications, and as of September 2022 the European Medicines Agency (EMA) and Heads of Medicines Agencies (HMA) jointly announced that once approved in the EU, biosimilars are interchangeable with their reference product (or vice versa) or another biosimilar with the same international nonproprietary name [Citation8].
The aggregate or holistic value of biosimilars in Europe has continued to evolve in parallel with their increased adoption. Although the total cost of healthcare will always matter, biosimilar competition can offer benefits beyond short-term cost reductions. Collectively, based on the sources accessed for this review, these may include:
increased access to biologics where biosimilars are available (cost-savings are recycled to allow healthcare systems to afford to treat more patients within a population, or treat patients for longer, using the same budget);
expanded access to biologics where biosimilars are available (cost-savings enable healthcare systems to afford to initiate treatment with biologics earlier in treatment paradigm or in combinations that were previously not possible due to the cost of reference products);
increased access to innovative medicines (cost-savings allow healthcare systems to afford to purchase [typically higher-cost] innovative medicines either within or outside of the therapy area with biosimilar competition); and
increased incremental innovation surrounding biologic products (resulting from manufacturer competition, including reformulations and/or delivery devices) or innovations relating to value offerings.
Using insights gathered from surveys, interviews, and panelists in the field, as well as published literature, this article (Part 1 of two; Part 2 will be prospective) aims to evaluate these hypotheses and provide payers – who weigh the potential for long-term cost-containment and increased value from biosimilars – with insights about the perceived and actual impact of biosimilars on costs, patient outcomes, market access dynamics, and innovation. A market maturity framework for the adoption of biosimilars and associated opportunities is also proposed.
2. Methods
2.1. Study design and procedures
An independent survey comprising 54 questions was conducted by Charles River Associates (CRA) to collect information about the impact of biosimilar medicines within European markets (Supplementary Materials). Eligibility to participate was determined using third-party recruiters who prescreened applicants based on prespecified criteria. Participants with relevant expertise in their fields were identified based on their occupational role, years of experience in their role, and their responsibilities relative to procurement of biosimilar therapies or management of formularies that include them. The survey was either self-administered (online survey link received via e-mail) or conducted via a one-on-one in-depth telephone interview. The survey was used to collect first-hand perspectives about how biosimilar medicines have impacted patient outcomes, access to biologics, and the broader benefits resulting from biosimilar competition. The survey comprised multiple-choice questions, with the possibility to respond using free text if the choices provided were not applicable (‘other’). No time limit was set for completion of the online survey, which could be taken in the respondent’s local language or English. Responses remained anonymous, and collected data were analyzed in aggregate.
The perspectives and key insights from an expert panel of eight diverse European stakeholders were also collected. Panel members were recruited for their expertise in the field, as follows: [Jorge Mestre-Ferrandiz], Economics and Policy Consultant; [Josef S. Smolen], Rheumatologist; [Silvio Danese], Gastroenterologist; [Matti S. Aapro], Oncologist; [Arnold G. Vulto], Hospital Pharmacist/Policy Advisor; Paul Cornes, Policy Advisor, Pharma Economist, and Oncology Clinician; Margaret Kyle, Economist/Health Economist; and Marcin Czech, Policy Advisor and Health Economics and Outcomes Research Expert. The panelists had longstanding knowledge of how the biosimilar landscape has evolved in European markets since inception, and their combined experience spans multiple therapeutic areas. Panel members developed a market maturity framework that characterizes stages relating to the adoption and access to biosimilars over time and the potential opportunities to derive benefit, across European markets.
Survey respondents, interviewees, and panel experts were selected based on their comprehension of the mechanisms and incentives related to biologics in their market at the clinical, regional, and national level. Where applicable, evidence from published biosimilar studies and reviews was used to contextualize the insights gathered from the observations and experiences of survey respondents/interviewees and the expert panel, about the impact of biosimilars in Europe. Relevant peer-reviewed literature and other sources were identified based on the experience and knowledge of the expert panel. A literature review of biosimilar studies (systematic or non-systematic) was not conducted.
3. Results
3.1. Survey respondents
The survey was administered in the second quarter of 2020 to 103 respondents from across six European markets (Czech Republic, France, Germany, Italy, Sweden, and the UK): 64 clinicians (22 oncologists, 21 rheumatologists, 21 gastroenterologists), 22 hospital pharmacists, and 17 subnational payers, all with >5 years’ experience in their current role. Most respondents (64/103; 62%) had a high level of experience with the funding mechanisms and procurement of biosimilar therapies.
3.2. Survey findings
3.2.1. Cost-savings and uptake drivers for biosimilars
Survey results revealed that there is no ‘one-size-fits-all’ approach to biosimilar-generated cost-savings, and the chosen policy options dictate how cost-savings can be recycled. Across Europe, numerous mechanisms designed to promote biologic cost-savings have been established (). These include mechanisms for price control; policies governing the initiation, switching, and substitution of biologics; and prescription guidance, quotas, and monitoring, as well as financial incentives. Price control mechanisms are primarily directed toward manufacturers, but can also be directed to dispensers (i.e. pharmacies); the other mechanisms are directed primarily toward prescribers and patients.
Table 1. Summary of instruments used to drive price evolution and uptake of biosimilar medicines in Europea.
3.2.2. Effect of biosimilars on patient outcomes
The generation of cost-savings via biosimilar competition has indirectly improved patient outcomes by allowing increased or expanded access to biologics. The extent to which the surveyed European stakeholders perceived biosimilar competition as benefiting patient outcomes is shown in .
Figure 1. Surveyed European stakeholders’ perception of (a) patient benefits resulting from biosimilar competition; (b) changes to access of biologics after biosimilar competition; and (c) the drivers causing changes to access of biologics after biosimilar competition.
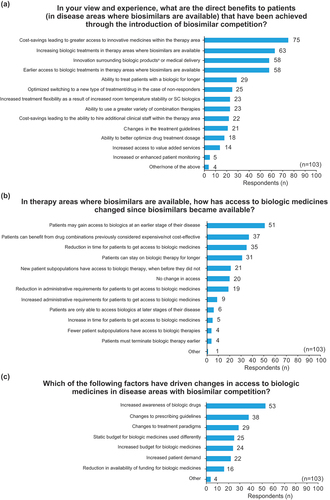
The four benefits most frequently identified by respondents broadly align with those hypothesized as the key potential benefits to patients: i) increased access to innovative medicines within the therapy area (73% of respondents); ii) increased access to biologic treatments where biosimilars are available (66%); iii) innovation surrounding biologic products (56%); and iv) earlier access to biologics in therapy areas where biologics are available (56%).
3.2.3. Effect of biosimilars on access to biologics
Stakeholders’ perceptions of changes to access of biologics after biosimilar competition is depicted in . The five most frequently reported observations all related to increased, earlier, or expanded access to biologics. Specific results included 50% of respondents reporting access to biologics in earlier stages of the disease, 36% reporting increased access to drug combinations, 34% reporting a reduction in time for patients to access biologics, 30% reporting that patients can remain on biologic therapies for a longer time, and 20% reporting new patient subpopulations having access to biologic therapies. Interestingly, a small minority of respondents (6%) noted a reduction in access to biologics since the introduction of biosimilars but did not provide specific comments to explain this. Furthermore, 19% of respondents reported they had not observed any changes to access to biologics since biosimilar competition.
3.2.4. Drivers of change in access to biologics after biosimilar competition
shows stakeholders’ perceptions of the key drivers causing changes to the access of biologics after biosimilar competition. The three most frequently reported drivers were increased awareness in biologic drugs (51%), changes to prescribing guidelines (37%), and changes to treatment paradigms (28%).
3.3. Expert panel insights
3.3.1. Pan-European observations and the biosimilar market maturity framework
The expert panel devised a market maturity framework for biosimilars across European markets. This built on initial research in this area and characterizes the different stages of biosimilar market development and the opportunities for stakeholders in each stage [Citation10]. Biosimilar markets can be thought of in three stages of ‘maturity’ broadly correlated to the level of access to biologics within the therapy area; therefore, it is possible for a country to be in different stages of maturity for different molecules or therapy areas. At each stage of ‘maturity’ there are unique opportunities for stakeholders to capture the broad range of benefits that biosimilars can deliver. The methods by which these benefits can be captured will depend on their market landscape and the specific goals of stakeholders ().
Table 2. Summary of the three stages of the market maturity framework for biologic benefit.
At Stage 1, ‘Invest,’ markets typically have no or little access to reference biologics, primarily because of their high cost. This provides an opportunity to invest and present individual patients, or specific subpopulations, with an initial opportunity to access biosimilars as lower cost biotherapeutics. At Stage 2, ‘Expand,’ some level of market access to reference biologics exists within a disease area, but some patients/subpopulations who could benefit from biologics do not qualify for reimbursement or are not routinely prescribed them. At this stage, an opportunity arises to expand the availability of low-cost biosimilars via increased, earlier, or more inclusive levels of access. At Stage 3, ‘Harvest,’ the level of access to biotherapeutics is high and covers reference biologics and biosimilars. Benefits can be realized in two ways: i) by recycling cost-savings outside of the therapy area with biosimilar competition and using this competition to maintain cost-savings; and ii) driving innovation in formulation, patient experience, and other value-adding offerings.
3.3.2. Financial flow of healthcare systems
Financial flows have also been modeled, particularly on securing three types of benefits (). ‘Provider benefit’ occurs when the cost-savings are retained by the hospital department or specialist centers where biosimilars are prescribed. ‘Purchaser benefit’ is when healthcare payer organizations or institutions procuring cost-savings retain them and healthcare budgets are not projected to be impacted, so local providers do not have any particular incentive to prescribe lower-cost options. ‘Ultimate payer benefit’ is when cost-savings are retained by the statutory health insurer, or the national or regional organization providing the healthcare budget. In this scenario, broader incentives to prescribe low-cost biologics can be eroded, particularly when resources are redirected from the healthcare sector to elsewhere within government budgets.
Table 3. Financial flow archetypes.
4. Discussion
4.1. Key results/interpretation
4.1.1. Cost-savings and uptake drivers for biosimilars
The primary benefit of biosimilar use is arguably generation of cost-savings. Not including discounts and confidential rebates, the cumulative list price savings versus the pre-biosimilar cost of the originator by 2020 were €5.7 billion in Europe (noting that the true savings may have been higher had net prices been considered). Beyond discernible list price savings, confidential rebate and discount agreements impact the total drug budget in many European countries [Citation11]. However, the magnitude of savings has been disproportionally distributed across European healthcare systems and therapeutic areas, with many countries missing out on significant savings realized by other nations. For example, in a review of the uptake of 13 biologic drugs with recently launched biosimilars among 37 Organisation for Economic Co-operation and Development (OECD) nations, three nations achieved ≥ 90% biosimilar infliximab use by year three from launch, against the OECD average of 30%—a pattern repeated for other classes of biosimilars with striking differences revealed [Citation12]. This disparity was highlighted by the stakeholders responding to our survey, and is further supported by an IQVIA White Paper [Citation11], which has been a long-standing source for primary market research on biosimilars prepared for the European Commission. These data, presented in , show considerable variability of the percentage change in price per treatment day (maximum differential of 76% for the oncology biosimilars of rituximab, trastuzumab, and bevacizumab) and the market share of biosimilars (maximum differential of 92% for anti-TNF biosimilars of adalimumab, infliximab, and etanercept) where biosimilars are available [Citation11].
Figure 2. Evolution of price per treatment days (biosimilars and reference product) and biosimilar market share for anti-TNF and oncology biologics in selected European countries, from the year before biosimilar entry to June 2020 (data extracted from, the impact of biosimilar competition in Europe, 2020 [Citation11]).
![Figure 2. Evolution of price per treatment days (biosimilars and reference product) and biosimilar market share for anti-TNF and oncology biologics in selected European countries, from the year before biosimilar entry to June 2020 (data extracted from, the impact of biosimilar competition in Europe, 2020 [Citation11]).](/cms/asset/b05d75e1-2a31-4863-94c4-286d9555017a/ierp_a_2297926_f0002_oc.jpg)
In health systems with high rates of biosimilar uptake the financial savings can be considerable. For example, in 4 years between 2015 and 2019 the Netherlands achieved savings of €320.6 million from among five key biologics, with an average increase in patient access at the same time of more than 20% [Citation13], while National Health Service (NHS) England saved £800 Million (€925 m Euros) in 2019–2020 alone [Citation14]. Even during the height of the COVID-19 pandemic, when European health systems prioritized acute care over chronic non-communicable diseases, the volume of biosimilar prescribing in 2020 generated record high savings [Citation2]. The long-term view on yearly list price savings from biosimilar competition is shown in [Citation2].
Figure 3. Long-term view on list price savings from biosimilar competition. Yearly savings from biosimilar competition (cost for post-biosimilar volume at pre-biosimilar list prices) (reproduced from, the impact of biosimilar competition in Europe, Troein, P., Newton, M., Scott, K. © IQVIA Inc. 2021; by permission of IQVIA Inc [Citation2])a.
![Figure 3. Long-term view on list price savings from biosimilar competition. Yearly savings from biosimilar competition (cost for post-biosimilar volume at pre-biosimilar list prices) (reproduced from, the impact of biosimilar competition in Europe, Troein, P., Newton, M., Scott, K. © IQVIA Inc. 2021; by permission of IQVIA Inc [Citation2])a.](/cms/asset/69485af2-a0d6-4b14-ad04-b525af4500ec/ierp_a_2297926_f0003_oc.jpg)
4.1.1.1. Influence of procurement methods on uptake
The procurement method can certainly affect how competition evolves in the market, and several have been used for biosimilars. In particular, the procurement of biosimilars used in the hospital setting is subject to variable tendering processes across Europe [Citation15], where four criteria can be key in determining the functioning and effectiveness of the method: number of winners allowed, criteria used to decide the winner(s), geographical scope, and duration and frequency of tenders. Playing fields structured to elicit rapid, deep price discounts using national, single-winner tenders have preferred near-100% price weighting. The downside of this approach, particularly when switching is enforced (i.e. the prescriber decides to exchange one medicine for another with the same therapeutic intent), is the threat to long-term market sustainability [Citation4,Citation16–18]. When ‘winner-takes-all’ tenders lead to a single manufacturer dominating the market as the reference product, competitors may be dissuaded from participating in further marketplace procurement rounds. Nevertheless, tenders are frequently updated annually, therefore only companies not wishing to ‘take it all’ will opt out of future rounds and the respective market.
Single-winner tendering, in countries such as France, the Czech Republic, and Poland, has been used less broadly at the regional or hospital level to retain some level of competition and prompt marginally improved market sustainability as more manufacturers remain prepared to vie for a market share. Subnational single and multi-winner tenders reward multiple biosimilar developers with smaller contracts while maintaining several marketplace competitors [Citation16,Citation19,Citation20]. Another alternative is to increase the weight of non-price-winning criteria, which may yield benefits because of incremental product innovation (e.g. subcutaneous patient-friendly buffers or more stable post-reconstitution formulations), or innovation in manufacturers’ value package offerings (e.g. provision of therapeutic drug monitoring, adherence improvement measures, or home care). Price reductions have been shown to be substantial for single national multi-winner tenders [Citation21]. Subnational reductions, comparable to single-winner tender price reductions, have occurred in Sweden. Each scenario transpired without jeopardizing the sustainability of prices or market formularies over time in other regions. In Denmark and Norway, biosimilar contracting permits several manufacturers to compete in the market, but with less intense direct price competition.
4.1.1.2. Other uptake drivers and alignment tools
Mechanisms for price control, policies governing the initiation, switching, and substitution of biologics, prescription guidance, quotas, monitoring, and financial incentives can all be used to promote biologic cost-savings. National controls on the list price of biosimilars can be mandated or expected by negotiators in certain countries [Citation22]. Discounts are divergent across European countries, and in some cases their depth depends upon the timing of a biosimilar to market [Citation22]. Some countries that use sequential price cuts noticed delays in entry of the third and subsequent biosimilar entrants relative to countries not doing so. Such circumstances potentially limit treatment options because competition among manufactures declines, along with the need to differentiate a product based on value-added offerings. The risk of supply shortages and reduced patient access then increases, and payers can miss out on capturing the total value of a product.
Although unconventional in most of Europe, price evolution of biosimilars can be prompted by mandating initiation of treatment-naïve patients to the lowest-cost biological option and by requiring currently treated patients to switch to the same [Citation22], which is now further supported by the recommendations of the EMA and HMA [Citation8]. These approaches have driven rapid and near-exclusive uptake of certain biosimilars in Norway, Denmark, and Poland. Less prescriptive recommendations have persisted in most other European markets. In the UK, the choice of either of these approaches is a decision made by the clinician, who must discuss and obtain their patient’s support [Citation23]. Pharmacy level substitution (i.e. substituting a biosimilar for branded reference biotherapeutic on a physicians’ prescription, unless explicitly prohibited by them) is not generally practiced in Europe [Citation24], and interchangeability (i.e. the possibility of exchanging one medicine for another medicine that is expected to have the same clinical effect) is now supported by the EMA and HMA [Citation8]. Prescription guidance, quotas, and monitoring are used variably throughout Europe to direct the uptake of preferred biosimilars. In Germany, these approaches are specifically tied to clinician renumeration, with financial retribution imposed when justification is lacking for prescribing higher-cost options. Although no financial penalties have been tied to prescribing targets in some other countries, CRA interviews with 30 key opinion leaders and decision-makers from six European markets revealed the perception of a tacit ethical responsibility to use drug budgets sparingly. Interviews also revealed an ethical responsibility to take advantage of the cost-saving benefits and initiate negotiations for additional benefits such as therapeutic drug monitoring to ensure drugs are used effectively or access to extra nursing staff.
Financial-based incentives are designed so that when an agreed target level of a preferred biologic option has been prescribed, a share of the cost-savings (gain- or benefit-sharing) can be obtained, particularly by a healthcare payer organization (purchasers), as in Belgium, France, Germany, Italy, the Netherlands and the UK; or it may be divided between purchasers and providers (e.g. at the hospital of care) [Citation25]. This can promote reinvestment of savings but may be administratively burdensome. Also, financial penalties that incentivize biosimilar prescribing may be viewed as limiting the autonomy of physicians. While there are multiple instances across Europe of benefit-sharing practices, striving for more timely and transparent disclosure of factors such as, the quality indicators of success, pathways for reinvestment of savings and communication of the outcomes with patients may help in realizing their full value [Citation26].
Different cost-saving strategies at the disposal of decision-makers across different European markets reveal disparate uptake patterns for biosimilars and potential risks (i.e. long-term stability of the market and security of supply may not be optimized) over time, as shown for the uptake of infliximab biosimilars (in treatment days) since launch [Citation11].
As an example of disparity in biosimilar uptake, shows uptake of infliximab biosimilars across various European markets [Citation27]. Data are depicted as percentage of treatment days, with substantial variability. The impact of strategy selection was demonstrated by exploring the outcomes for two markets at the opposite extremes of uptake, Denmark and Germany.
Figure 4. Uptake of infliximab biosimilar since its launch across European markets (reproduced from, leveraging biosimilars for better access and lower cost, Troein, P. Source: IQVIA MIDAS restricted MTH Oct 2017, © IQVIA Inc. 2017, by permission of IQVIA Inc [Citation27]).
![Figure 4. Uptake of infliximab biosimilar since its launch across European markets (reproduced from, leveraging biosimilars for better access and lower cost, Troein, P. Source: IQVIA MIDAS restricted MTH Oct 2017, © IQVIA Inc. 2017, by permission of IQVIA Inc [Citation27]).](/cms/asset/bb55bcce-415e-44ee-89d0-d5be55ad1730/ierp_a_2297926_f0004_oc.jpg)
4.1.2. Effect of biosimilars on innovation and patient outcomes
The stakeholder survey responses suggested a wide range of benefits resulting from biosimilar competition, such as increased and/or expanded access to biologics and innovative medicines, and incremental innovations in the products, all of which were thought to contribute to improved patient outcomes overall. For example, biosimilar savings from the NHS England multiple-winner tenders for adalimumab enabled reimbursement for 9000 more patients a month to access this treatment [Citation28]. Patient outcomes can improve indirectly when more patients can afford biologics for longer periods and/or earlier in the treatment paradigm [Citation29,Citation30], in combination with other previously unaffordable therapies, and/or because accessibility to medicines that are typically costlier or more innovative improves with the availability of biosimilars. Patient outcomes can also improve when incremental innovations or value offerings occur with manufacturing competition. According to our survey, the most important patient benefits resulting directly from biosimilar competition were cost-savings leading to greater access to innovative medicines within the therapy area and increasing biologic treatments in therapy areas where biosimilars are available. The most important perceived changes to biologic medicine access after biosimilar competition were improved patient accessibility to biologics at earlier stages of disease, and potential benefit from drug combinations previously considered too expensive or not cost-effective.
4.1.2.1. Benefits of biosimilars for driving innovative medicines
Increased access to innovative medicines can occur after recycling biosimilar-generated cost-savings to fund budgetary reallocations, or via the uptake of lower-cost biosimilar medicines for use in combination with innovative medicines (because of cost-effectiveness, reduced budgetary impact, and improved affordability). Evidence for the first scenario is found in several regions in Italy, where 50% of the savings generated from biosimilar uptake are reallocated to supplement 20% of the budget dedicated to coverage of innovative medicines [Citation31]. In 2016, NHS England established a specific Cancer Drugs Fund with an annual budget of £340 million (€393 m) to reimburse new and often expensive breakthrough treatments [Citation32]. The success of this program has enabled a second Innovative Medicines Fund to be created in 2021 with an additional annual budget of £340m (€393 m) for non-cancer medicines innovation, including those for rare and genetic diseases, to reimburse early access to the most clinically promising treatments [Citation33]. These funds may be supported by contributory savings from patent expiry of medicines including biologics and savings generated from biosimilar use. For the second scenario, the innovative medicine pertuzumab became cost-effective in combination with trastuzumab because of access to biosimilar trastuzumab [Citation34]. Overall, biosimilar competition in Europe has contributed to improved access to biologics and improved patient outcomes, but accessibility remains variable in some market segments [Citation2].
4.1.2.2. Other types of innovation arising from biosimilar competition and benefits for patients and payers
Although price remains a key differentiator in terms of the competitiveness of biosimilars with biologics that are clinically equivalent, undifferentiated biosimilar competition has advanced the drive for manufacturers of the reference product to innovate in several ways, including: improving a drug’s formulation, providing an alternative mode of administration or delivery system (also encouraged for biosimilar manufacturers), and offering value-based innovations designed to encourage product use and improve the patient’s experience.
Undifferentiated biosimilar competition has been a stimulus for innovation. Points of differentiation for biologic products (e.g. device design and citrate-free formula for subcutaneous administration) can offer patients additional benefits. Indeed, in Europe, 60% of the whole adalimumab market has been dominated by high-concentration formulations, and over 90% of the originator standard-concentration adalimumab market has been replaced by citrate-free high-concentration originator adalimumab [Citation2,Citation35]. CT-P17 became the first adalimumab biosimilar with a high-concentration, low-volume, and citrate-free formulation [Citation36]. Biosimilar developers have sponsored trials to show that intravenous administration costs can be cut, for example with co-infusion of pre-mixed biosimilar trastuzumab and reference pertuzumab in the Lavender trial [Citation37]. Innovation of biologics to develop alternatives to intravenous administration (e.g. subcutaneous versions of originator products trastuzumab, rituximab, tocilizumab, and natalizumab as well as infliximab biosimilar CT-P13) provides numerous advantages [Citation38–40].
Findings from a Belgian case study of intravenous biosimilar trastuzumab and subcutaneous reference trastuzumab in the treatment of adjuvant HER2-positive breast cancer suggest awareness of complex and multiple factors beyond drug costs is necessary to fully understand such cost comparisons between formulations [Citation41]. Patient choice is largely contingent upon a more convenient route of administration that is less time-consuming and administration facility. Treatment satisfaction may also be enhanced by potential benefits such as in-home treatment or the option to self-administer [Citation42].
Differentiation can be achieved if more extensive evidence about a product’s stability, also after reconstitution, is provided, even when it is not a marketing authorization requirement. Biosimilar manufacturers continue to confer benefit by providing more robustly supported predictions about expiration dates beyond which disposal must occur under a variety of real-world conditions that in practice may not have previously been assessed over longer periods [Citation43–45]. The availability of data beyond standardized conditions over extended time points has implications for nurse and pharmacy workloads, as well an important reduction in drug wastage when drugs are reconstituted/diluted in advance of patient visits. For example, the stability of trastuzumab post-dilution/reconstitution was limited to 24–48 hours depending on storage temperature, whereas the trastuzumab biosimilar PF-05280014 can be stored for 30 days post dilution [Citation46]. However, in situations when an originator biologic is reaching the end of its lifecycle, sizable investments to provide more extensive evidence or further innovate a device can sometimes be difficult to justify.
A competitive advantage can be leveraged in an otherwise undifferentiated biologics market when biosimilar manufacturers present their products as a value proposition that transcends certain monetary considerations such as ever-deeper discounts [Citation47]. These include supplementary services (e.g. extra staff programs to improve patient adherence, physician education and support, therapeutic drug monitoring, homecare services) or innovative contracts designed to relieve the financial burden payers contend with in relation to biologic budgets [Citation15,Citation48]. Therapeutic drug monitoring of biosimilars warrants highlighting because it contributes to effective drug use, which can circumvent financial losses associated with inappropriate dosing regimens [Citation49].
Value offerings can help sustain the price of biologics and have been shown to influence decision-makers awarding contracts in the tendering process in some European markets [Citation50]. Creative strategies designed to add value can facilitate the delivery of high-quality care.
4.1.3. Effect of biosimilars on access to biologics
Empirical data from across Europe support the perception of many stakeholders responding to the survey, who believed that biosimilar competition improved access to biologics. Increased, earlier/expanded access to anti-TNFs in Europe almost doubled the average per capita treatment days (from ~0.5 to just under 1 day), and with a few exceptions, access by country aligned with economic prosperity [Citation11].
Evidence supporting earlier biologic access was exemplified by a marked decrease in the average time from diagnosis to first receiving biologic disease-modifying anti-rheumatic drugs (infliximab, etanercept, adalimumab and rituximab) in Germany; it was 7.4 years before biosimilar competition reduced the costs, and only 16 weeks after biosimilar entry, potentially averting years of inflammatory damage for most patients with chronic disease [Citation51].
In general, the accessibility of biologics tends to be inversely related to price, although non-financial factors are also important, and the balance of that relationship tends to vary depending on historical market access, therapeutic area, and time on the market [Citation11]. With some exceptions, markets with historically high levels of access to biological medicines appear to have increased access by a larger magnitude than markets with historically lower access to them. The magnitude of accessibility is also affected to a lesser extent by a price reduction for oncology medicines compared with those used to treat chronic conditions such as inflammatory diseases (e.g. anti-TNFs). This is because many cancers can severely impact life expectancy, so the impact of competition on access can only be assessed over a comparatively short period. This has resulted in payers and others being more willing to reimburse for expensive/innovative biologics in oncology after national reimbursement is agreed [Citation52]. Lower uptake volumes of biosimilars can also occur because short survivorship limits the window of opportunity for patients to switch treatments, and because deeper discounts for newer oncology biosimilars can take time. This scenario contrasts with anti-TNF biologics, for which the barrier to accessibility is more heavily cost-related. Finally, manufacturer’s (both originators and biosimilar) pricing strategies can also affect the level of confidential net price discounting, which we have seen varies among biologics prescribed in different therapeutic areas.
4.1.4. Drivers of change in access to biologics after biosimilar competition
Among the European biosimilar stakeholders surveyed, the three most frequently reported drivers considered to influence accessibility to biologics in disease areas with biosimilar competition were (i) increased awareness of biologic drugs, (ii) changes to prescribing guidelines and (iii) changes to treatment paradigms. The survey results revealed that the emergence of biosimilars stimulated discussions that were primarily perceived to raise awareness about biologics to the extent that accessibility increased to indirectly benefit patient outcomes.
Biosimilar competition has also driven expanded accessibility to biologics more widely. Approved in 2008, biosimilar G-CSF has quadrupled patient access to filgrastim in 20 European countries where cost has become less likely to preclude its use in conjunction with chemotherapy to treat potentially life-threatening neutropenic fever in cancer patients [Citation11]. Competition associated with the biosimilar infliximab (CT-P13) in the UK contributed to the increased accessibility of infliximab for an earlier line of treatment in patients with moderately active ulcerative colitis in 2015, as opposed to only for severely active ulcerative colitis in patients who had an inadequate response to conventional therapy (authorized when it was originally launched in 2006) [Citation53,Citation54]. NICE was able to expand their treatment recommendations in the same month as the UK launch of infliximab biosimilars, despite the expanded indication for originator infliximab having been authorized as early as 2011 [Citation39,Citation53,Citation55]. In June 2021 NICE enabled around 25,000 people with moderate RA that had not responded to conventional therapies to be able to be prescribed anti-TNFs with biosimilar competition (adalimumab, etanercept and infliximab) following a pragmatic review of existing guidance in response to the availability of biosimilars [Citation56]. In other examples, NICE has agreed reimbursement of rituximab for Myasthenia gravis at biosimilar prices [Citation57] while biosimilars of erythropoiesis‑stimulating agents enabled the restrictions on their use for anemia induced by chemotherapy to be lifted and reimbursement agreed in NICE technology appraisal guidance TA323 [Citation58]. These guidance updates issued by the national payer NICE had meaningful impacts on access to treatment.
4.1.5. Pan-European observations and the “market maturity” framework
Biosimilars can deliver a broad range of healthcare benefits, albeit nonuniformly across markets that are still maturing. Stakeholders have the possibility to take advantage of opportunities at all stages of market maturity, and the expert panel of respondents surveyed devised a framework that can be used to characterize those stages to take advantage of biosimilar-generated benefits in line with regional goals. The three stages of maturity identified (Invest, Expand, Harvest) align with levels of access to biologics within a disease area, and conceptually are mutually exclusive. Stakeholders should be able to optimize the benefit of biosimilars if they strive to exploit opportunities at each stage.
Supporting the theory behind the proposed framework, in analyses of regional market dynamics for biosimilars, Moorkens et al. reported that the benefits achievable with biosimilars depend on the local/regional practices and policies of key stakeholders and decision-makers who can influence aspects such as prescribing targets and market uptake trends [Citation59].
An example of the market maturity framework across European markets for adalimumab is depicted in . Over the 12 months before the first quarter of 2020, IQVIA biologic data for volume of treatment days (per capita) one year before biosimilar entry into the marketplace, plotted against the increase in treatment days (per capita) since biosimilar entry by country, showed that variability in adalimumab uptake in relation to biosimilar entry closely corresponded with what is generally known about each country’s stage of market maturity [Citation60]. For instance, the use of adalimumab in Romania was relatively low before and after the entry of biosimilars. Several policy elements appeared to result in this scenario. Although possible in this marketplace, no contracts were reopened to permit biosimilar entry, biosimilar quotas were lacking, as were guidance and reimbursement incentives for prescribers to consider biosimilars over reference biologics [Citation61]. Therefore, opportunities for stakeholders were present at the ‘Invest’ stage of the maturity framework in Romania.
Figure 5. Change in volume of adalimumab uptake from the year before biosimilar entry to the year ending Q1/2020 across various European markets (reproduced from, country scorecards for biosimilar sustainability, methodology appendix. © IQVIA and its affiliates. 2020; by permission of IQVIA Inc [Citation60]).
![Figure 5. Change in volume of adalimumab uptake from the year before biosimilar entry to the year ending Q1/2020 across various European markets (reproduced from, country scorecards for biosimilar sustainability, methodology appendix. © IQVIA and its affiliates. 2020; by permission of IQVIA Inc [Citation60]).](/cms/asset/0582848b-ada1-43de-b2da-82c1c6f17f31/ierp_a_2297926_f0005_oc.jpg)
France, on the other hand, had limited uptake of biologics before biosimilar entry, yet increased by 20‒25% following biosimilar entry. The 2018‒2022 French National Health Strategy contributed to advancing the uptake of biologics by establishing a target of 80% biosimilar penetration by 2022 [Citation62]. Furthermore, pharmacists were incentivized: they could profit from biosimilar substitutes, owing to higher margins compared with off-patent reference biotherapeutics [Citation60]. Because access to adalimumab lags behind other European markets, the capacity to expand access to optimize overall patient outcomes represents an opportunity at the ‘Expand’ stage of the market maturity framework.
In Norway, adalimumab use grew by more than 25%, positioning it in the ‘Harvest’ stage of the maturity framework. Broad acceptance of biosimilars by payers, providers, and patients was attributed to an extensive program that prepared stakeholders, invested in evidence generation, and presented incentive models that shared payer savings with hospitals [Citation63]. The benefit of biosimilars in Norway extended to improving patient outcomes through healthcare system savings that could be reinvested into innovative medicines across therapeutic areas. Every effort in this mature market is geared toward market sustainability and preserving total benefits into the future [Citation60]. Overall, opportunities to build on the value that biosimilars can bring to healthcare can be found in EU markets of all maturity levels, and the stages outlined should not be viewed as a rigid framework.
4.1.6. Financial flow of healthcare systems
The structure and flow of finances around the healthcare system can impact the magnitude and variability of cost-savings in association with procurement mechanisms and the strategies of biologic manufacturers. Moreover, financial flows can add to healthcare system complexities, making the prediction of cost-savings challenging in real-world scenarios. Alignment of stakeholder motivations has been shown to be key to optimizing cost-savings and incentivizing the lower-cost biologic options. For example, the ‘Purchaser benefit’ financial flow archetype – commonly adopted as a tool to financially incentivize sharing of biosimilar cost-savings at the hospital of care – requires the division of savings between ‘purchasers’ and ‘providers,’ as occurs with gain share agreements reported previously for key stakeholders [Citation25].
4.2. Study limitations
Key limitations of this study were that the survey chiefly focused on perceptions across six European markets (Czech Republic, France, Germany, Italy, Sweden, and the UK), the one-on-one in-person interviews were only in English, and participants were not randomly selected. Survey respondents and interviewees were screened to ensure they had a high level of experience in their role pertaining to biosimilars. In addition, the survey did not assess potential factors for encouraging or promoting the use of biosimilars in clinical practice. An ongoing justification for including clinical outcome studies as part of biosimilar development is the perceived value of the information they provide, particularly for life-saving medications in therapeutic areas such as oncology. However, inclusion of clinical outcome studies as part of biosimilar development may slow access to these medications. It would have been helpful to assess clinicians’ perception on the value of biosimilar clinical outcome studies and if such studies would impact the use of biosimilars in clinical practice. Finally, relevant peer-reviewed literature and other sources cited in this article were largely identified based on the experience and knowledge of the expert panel rather than derived from a literature review (systematic or non-systematic) of biosimilar studies.
5. Conclusions
This paper consolidates the relatively abundant evidence that demonstrates that biosimilars can deliver a wide range of benefits to healthcare systems and society. The perspectives presented here also show access to biologics has improved in Europe, at least partly because of biosimilar competition. Further, the perceptions of survey/interview respondents are that improved access to biosimilars has allowed patients to be treated earlier and for longer, and target populations can broaden. Cost-savings can be reinvested in the same and other therapeutic areas, benefiting healthcare systems and patients and biosimilars have contributed to improved patient outcomes. But there is certainly room for improvement, given all the experience we have so far in Europe with biosimilars. There is still significant variability in the level of increased access between markets. This implies the benefits of greatest value to key stakeholders and decision-makers have varied between markets and were dependent upon their specific goals; these in turn were informed by each country’s environment and the maturity of their biosimilar markets.
There is general consensus that biosimilars have generated significant cost-savings for pharmaceutical budgets across European countries; yet again further savings could have been achieved, without necessarily compromising the sustainability of the biosimilar industry, with better-designed policies. The cost and uptake of biosimilars has varied significantly among European markets, which is in part due to having different procurement methods, financial flows, and stakeholder alignment tools. Decision-makers’ strategies are limited to the bounds of these elements and their own internal and external imperatives, with no ‘one-size-fits-all’ strategy for maximizing cost-savings through use of biosimilars. Decision-makers have been tasked with balancing the advantages versus the risks of each policy or instrument available in their markets to devise a strategy based on their short- and long-term goals. Each decision-maker needs to define their objectives clearly and understand the implications of changes to the entire system.
One of the advantages of the market maturity framework devised by the expert panel to characterize the different stages of biosimilar market development and the opportunities for stakeholders at each stage, is that it allows navigation across the different stages of biosimilar market development to maximize biosimilar-generated benefits. Hence, this framework could be used going forward when designing future policies at a national level. In Part 2, we will discuss these and other topics affecting the future use of biosimilars.
Article highlights
Biologics have transformed patient outcomes across a wide range of therapeutic areas; however, the high price of biologics can create barriers to access for patients.
Biosimilars are highly similar in structure and clinically equivalent in biological function, efficacy, safety, and immunogenicity to licensed (i.e. reference or originator) biologic products.
Using insights gathered from a survey, interviews, and experts in the field, as well as from a review of published literature, this manuscript (Part 1 of 2) provides perspectives on the societal value of biosimilars within Europe and potential factors that have influenced market dynamics.
Findings from the survey and literature review provide evidence that biosimilars have generated significant cost-savings and can deliver a wide range of benefits to healthcare systems and society.
Findings also demonstrate that access to biologics in Europe has improved in part due to biosimilar competition and this may allow earlier and longer treatment of a broader population.
An expert panel describes a market maturity framework for the adoption of biosimilars comprising three stages (‘Invest,’ ‘Expand,’ and ‘Harvest’) that broadly correlate to the level of access to biologics within a therapy area and that could be used to maximize biosimilar-generated benefits.
Compliance with ethical standards
This research was considered exempt from institutional research board review because it involved survey procedures and was conducted according to the following codes of conduct: BHBIA/MRS/ESOMAR/Data Protection Act. Informed consent was obtained either online electronically or verbally only during telephone interviews.
Declaration of interest
J Mestre-Ferrandiz declares an honorarium received from Pfizer for participation in the expert panel in support of the preparatory work upon which this manuscript is based.
M Czech declares a consulting/advisory role in AbbVie, Amgen, AstraZeneca, Boehringer Ingelheim, Biogen, Eli Lilly, GSK, Janssen, MSD, Novartis, Pfizer, Sanofi, Takeda and UCB.
J S Smolen has received grants for his institution from AbbVie, AstraZeneca, Janssen, Lilly, Novartis, and Roche, and has received honoraria for consulting or speaking engagements from AbbVie, Amgen, Astro, BMS, Celgene, Celltrion, Chugai, Gilead, ILTOO, Janssen, Lilly, MSD, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi, and UCB.
P Cornes declares honoraria or support for attending meetings from Accord Healthcare, Amgen, Astro Pharma, European Association for Hospital Pharmacists, European Commission, Medicines for Europe/European Generics Association, Medscape, Mylan, Napp, Pfizer/Hospira, Sandoz and Teva.
M S Aapro declares a consulting/advisory role in Amgen, BMS, Daiichi Sankyo, Fresenius Kabi, G1 Therapeutics, Genomic Health, Helsinn Healthcare, Merck, Merck KGaA, Novartis, Pfizer, Pierre Fabre, Roche, Sandoz, Tesaro, and Vifor Pharma; speakers’ bureau at Accord Research, Amgen, Biocon, Dr Reed, Genomic Health, Helsinn Healthcare, Mundipharma, Novartis, Pfizer, Pierre Fabre, Roche, Sandoz, Taiho Pharmaceutical, Tesaro, and Vifor Pharma; research funding from Helsinn Healthcare, Novartis, Pierre Fabre, and Sandoz.
S Danese declares consultancy fees from AbbVie, Alimentiv, Allergan, Amgen, Applied Molecular Transport, AstraZeneca, Athos Therapeutics, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Celltrion, Dr Falk Pharma, Eli Lilly, Enthera, Ferring Pharmaceuticals Inc., Gilead, Hospira, Inotrem, Janssen, Johnson & Johnson, Morphic, MSD, Mundipharma, Mylan, Pfizer, Roche, Sandoz, Sublimity Therapeutics, Takeda, Teladoc Health, TiGenix, UCB Inc., Vial, Vifor and lecture fees from AbbVie, Amgen, Ferring Pharmaceuticals Inc., Gilead, Janssen, Mylan, Pfizer, Takeda.
S Deitch was a full-time employee of Charles River Associates when the work was conducted and was contracted by Pfizer in connection with the development of the manuscript.
H Tyldsley and W Foster are full-time employees of Charles River Associates and were contracted by Pfizer in connection with the development of the manuscript.
P Shah and M Latymer are full-time employees of and hold stock and stock options in Pfizer.
A G Vulto founded the KU Leuven Fund on Market Analysis of Biologics and Biosimilars following Loss of Exclusivity (MABEL Fund). He is involved in consulting, advisory work and speaking engagements for Accord Healthcare, Amgen, Biogen, Effik, Fresenius/Kabi, Medicines for Europe, Mundipharma, Novartis, Pfizer/Hospira, and Sandoz.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author contributions
All authors contributed to content development, writing, and critical review of the paper. M.L. is the overall guarantor. All authors have read and agreed to the published version of the manuscript.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Interactive summary
Download PDF (1.1 MB)Survey & Interview Questions
Download PDF (613 KB)Acknowledgments
Margaret Kyle, Professor of Economics, Centre d’économie industrielle (Cerna), Mines ParisTech, and a subject-matter expert, participated on the expert panel and endorsed the final survey report. Medical writing support was provided by Sue Reinwald, PhD, and Elyse Smith, PhD, of Engage Scientific Solutions and funded by Pfizer.
Data availability statement
All original data are contained within Figure 1 or in the article text.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14737167.2023.2297926
Additional information
Funding
References
- Shepard HM, Phillips GL, Thanos CD, et al. Developments in therapy with monoclonal antibodies and related proteins. Clin Med. 2017 Jun;17(3):220–232.
- Troein P, Newton M, Scott K, et al. The impact of biosimilar competition in Europe 2021. [ updated 2021 Dec; cited 2023 May 30]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/library/white-papers/the-impact-of-biosimilar-competition-in-europe-2021.pdf
- Giuliani J, Fiorica F, Albanese V, et al. Financial toxicity and cancer treatments: help from biosimilars - the explanatory case of bevacizumab. Eur J Cancer. 2021 Jan;143:40–42.
- European Medicines Agency and European Commission. Biosimilars in the EU: information guide for healthcare professionals 2019. [ updated 2019 Oct 10; cited 2023 May 30]. Available from: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
- Dutta B, Huys I, Vulto AG, et al. Identifying key benefits in European off-patent biologics and biosimilar markets: it is not only about price! BioDrugs. 2020 Apr;34(2):159–170. doi: 10.1007/s40259-019-00395-w
- Gimeno-Gracia M, Gargallo-Puyuelo CJ, Gomollón F. Bioequivalence studies with anti-TNF biosimilars. Expert Opin Biol Ther. 2019 Oct 03;19(10):1031–1043.
- Generics and Biosimilars Initiative (GaBI). Biosimilars approved in Europe 2011. [ updated 2023 May 12;cited 2023 May 30]. Available from: https://www.gabionline.net/biosimilars/general/biosimilars-approved-in-europe
- European Medicines Agency and Heads of Medicines Agencies. Statement on the scientific rationale supporting interchangeability of biosimilar medicines in the EU. 2023. [updated 2023 April 21; cited 2023 May 30]. Available from: https://www.ema.europa.eu/en/documents/public-statement/statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf
- Medicines for Europe. Market review - biosimilar medicines markets 2020: medicines for Europe. 2020. [updated 2021 Mar; cited 2023 May 30]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2021/03/Biosimilar%20Market%20Review-Final.pdf
- Moorkens E, Vulto AG, Kent J, et al. A look at the history of biosimilar adoption: characteristics of early and late adopters of infliximab and etanercept biosimilars in subregions of England, Scotland and Wales - a mixed methods study. BioDrugs. 2021 Jan;35(1):75–87.
- Troein P, Newton M, Scott K. The impact of biosimilar competition in Europe 2020. [updated 2020 Dec; cited 2023 May 30]. Available from: https://health.ec.europa.eu/system/files/2021-01/biosimilar_competition_en_0.pdf
- National Prescription Drug Utilization Information System. Potential savings from biosimilars in Canada 2017. [updated 2018 May 2; cited 2023 May 30]. Available from: https://www.pmprb-cepmb.gc.ca/view.asp?ccid=1304
- de Vos Burchart H, Vulto VG. Programma Implementatie Biosimilars op Maat in ziekenhuizen (BOM) - Managementsamenvatting: Instituut Verantwoord Medicijngebruik; 2021 [updated 2021 Feb; cited 2023 May 30].
- May E, Taylor K. The cost-effectiveness of biosimilars and their future potential for the NHS, 2021. [updated 2021 May 21; cited 2023 Jul 23]. Available from: https://blogs.deloitte.co.uk/health/2021/05/the-cost-effectiveness-of-biosimilars-and-their-future-potential-for-the-nhs.html
- Simoens S, Cheung R. Tendering and biosimilars: what role for value-added services? J Mark Access Health Policy. 2020 Jan 01;8(1):1705120.
- Aitken M, Rodríguez I, Diamantara J, et al. Advancing biosimilar sustainability in Europe: a multi-stakeholder assessment 2018. [updated 2018 Sep 4; cited 2023 May 30]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/advancing-biosimilar-sustainability-in-europe.pdf
- van Bodegraven AA, Süle A, Celano A, et al. How to realize the potential of off-patent biologicals and biosimilars in Europe? Guidance to policymakers. 2018 [cited 2023 May 30]. Available from: http://gabi-journal.net/how-to-realize-the-potential-of-off-patent-biologicals-and-biosimilars-in-europe-guidance-to-policymakers.html
- Mestre-Ferrandiz J, Towse A, Berdud M. Biosimilars: how can payers get long-term savings? PharmacoEconomics. 2016;34(6):609–616. doi: 10.1007/s40273-015-0380-x
- Vulto A, Cheesman S, Gonzalez-McQuire S, et al. pbi56 sustianable biosimilar procurement in europe: a review of current policies and their potential impact. Value Health. 2019;22:S427. doi: 10.1016/j.jval.2019.09.160
- Ferrario A, Dedet G, Humbert T, et al. Strategies to achieve fairer prices for generic and biosimilar medicines. BMJ. 2020 Jan 13;368:l5444.
- NHS England. NHS set to save record £300 million on the NHS’s highest drug spend 2018. [updated 2018 Dec 6; cited 2023 May 30]. Available from: https://www.england.nhs.uk/2018/11/nhs-set-to-save-record-300-million-on-the-nhss-highest-drug-spend/
- Moorkens E, Vulto AG, Huys I, et al. Policies for biosimilar uptake in Europe: an overview. PLoS One. 2017;12(12):e0190147. doi: 10.1371/journal.pone.0190147
- NHS England. Biosimilar medicines 2023. [cited 2023 May 30]. Available from: https://www.england.nhs.uk/medicines-2/biosimilar-medicines/
- The Canadian Agency for Drugs and Technologies in Health (CADTH). International policies on the appropriate use of biosimilar drugs. 2018 [updated 2018 Oct; cited 2023 May 30]. Available from: https://www.cadth.ca/sites/default/files/pdf/es0333_international-policies-on-use-of-biosimilar-drugs.pdf
- Razanskaite V, Bettey M, Downey L, et al. Biosimilar infliximab in inflammatory bowel disease: outcomes of a managed switching programme. J Crohns Colitis. 2017 Jun 1;11(6):690–6. doi: 10.1093/ecco-jcc/jjw216
- Barcina Lacosta T, Vulto AG, Turcu-Stiolica A, et al. Qualitative analysis of the design and implementation of benefit-sharing programs for Biologics Across Europe. BioDrugs. 2022 Mar;36(2):217–229.
- Troein P. Leveraging biosimilars for better access and lower cost, 2018 [updated 2021 Dec; cited 2023 Jul 11]. Available from: https://www.eahp.eu/sites/default/files/p._troein.pdf
- Bruce F. Can biosimilar orphans increase health system sustainability in the UK? 2021 [updated 2021, Jan 27; cited 2023 Jul 23]. Available from: https://pink.pharmaintelligence.informa.com/PS143672/Can-Biosimilar-Orphans-Increase-Health-System-Sustainability-In-The-UK
- Cornes P, Gascon P, Vulto AG, et al. Biosimilar pegfilgrastim: improving access and optimising practice to supportive care that enables cure. BioDrugs. 2020;34(3):255–263. doi: 10.1007/s40259-020-00411-4
- Jenkinson PW, Plevris N, Siakavellas S, et al. Temporal trends in surgical resection rates and biologic prescribing in Crohn’s disease: a population-based cohort study. J Crohns Colitis. 2020 Sep 16;14(9):1241–1247. doi: 10.1093/ecco-jcc/jjaa044
- IMS Institute for Healthcare Informatics. Delivering on the potential of biosimilar medicines: the role of functioning competitive markets 2016. [updated 2016 Mar; cited 2023 May 30]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/delivering-on-the-potential-of-biosimilar-medicines.pdf
- NHS England. NHS saves £1.2 billion on medicines over three years 2022. [updated 2022, Jul 1; cited 2023 Jul 23]. Available from: https://www.england.nhs.uk/2022/07/nhs-saves-1-2-billion-on-medicines-over-three-years/
- NHS England. Innovative Medicines Fund. 2023 [cited 2023 Jul 23]. Available from: https://www.england.nhs.uk/medicines-2/innovative-medicines-fund/
- NICE. Pertuzumab for adjuvant treatment of HER2-positive early stage breast cancer 2019. [updated 2019 Mar 20; cited 2023 May 30]. Available from: https://www.nice.org.uk/guidance/TA569/chapter/1-Recommendations
- European Pharmaceutical Review. EC approves first high concentration, low-volume adalimumab biosimilar 2021. [updated 2021 Feb 16; cited 2023 May 30]. Available from: https://www.europeanpharmaceuticalreview.com/news/143114/ec-approves-first-high-concentration-low-volume-adalimumab-biosimilar-yuflyma/
- Businesswire. Celltrion healthcare receives European Commission (EC) approval for the first high concentration, low-volume and citrate-free biosimilar adalimumab, YuflymaTM (CT-P17) 2021. [updated 2021 Feb 15; cited 2023 Feb 15]. Available from: https://www.businesswire.com/news/home/20210215005195/en/Celltrion-Healthcare-receives-European-Commission-EC-approval-for-the-first-high-concentration-low-volume-and-citrate-free-biosimilar-adalimumab-YuflymaTM-CT-P17
- Hanes V, Chow V, Stewart T, et al. A randomized, double-blind, single-dose study (LAVENDER) to assess the safety, tolerability, pharmacokinetics, and immunogenicity of a combined infusion of ABP 980 and pertuzumab in healthy subjects. Cancer Chemother Pharmacol. 2021 Nov;88(5):879–886.
- Celltrion Healthcare. Remsima®SC is the world’s first SC formulation of biosimilar infliximab developed by Celltrion. It was approved by the European Medicines Agency(EMA), Korean Ministry of Food and Drug Safety(MFDS) and Health Canada. 2019 [cited 2023 May 30]. Available from: https://www.celltrionhealthcare.com/en-us/products/remsimasc/
- European Medicines Agency. Remsima. Summary Of Product Charateristics. 2013 [updated 2023 May 30; cited 2023 May 30]. Available from: https://www.ema.europa.eu/en/documents/product-information/remsima-epar-product-information_en.pdf
- Liu J, Sylwestrzak G, Ruggieri AP, et al. Intravenous versus subcutaneous anti-TNF-alpha agents for Crohn’s disease: a comparison of effectiveness and safety. J Manag Care Spec Pharm. 2015 Jul;21(7):559–566.
- Simoens S, Vulto AG, Dylst P. Simulating costs of intravenous biosimilar trastuzumab vs. subcutaneous reference trastuzumab in adjuvant HER2-positive breast cancer: a Belgian case study. Pharmaceuticals. 2021;14(5):450. doi: 10.3390/ph14050450
- Epstein RS. Payer perspectives on intravenous versus subcutaneous administration of drugs. Clinicoecon Outcomes Res. 2021;13:801–807. doi: 10.2147/CEOR.S317687
- Weiser S, Burns C, Zartler ER. Physicochemical stability of PF-05280014 (trastuzumab-qyyp; Trazimera(TM)), a trastuzumab biosimilar, under extended in-use conditions. J Oncol Pharm Pract. 2023 Apr;29(3):590–600. doi: 10.1177/10781552221074649
- Yun J, Kim J, Chung J, et al. Extended stability of reconstituted and diluted SB3 (trastuzumab biosimilar) assessed by physicochemical and biological properties. Adv Ther. 2019 Jul;36(7):1700–1714.
- Paul M, Astier A, Vieillard V. Extended stability of a biosimilar of trastuzumab (CT-P6) after reconstitution in vials, dilution in polyolefin bags and storage at various temperatures. GaBi J. 2018 [cited 2023 May 30]; 7 (3):101–110. doi: 10.5639/gabij.2018.0703.022
- European Medicines Agency. Trazimera. Summary Of Product Characteristics. 2018 [updated 2022 Oct 17; cited 2023 May 30]. Available from: https://www.ema.europa.eu/en/documents/product-information/trazimera-epar-product-information_en.pdf
- de Mora F. Biosimilars: a value proposition. BioDrugs. 2019 Aug;33(4):353–356. doi: 10.1007/s40259-019-00360-7
- NHS England. NHS England board paper. Best Value Biologicals: Adalimumab 2018. [updated 2018 Nov 28; cited 2023 May 30]. Available from: https://www.england.nhs.uk/wp-content/uploads/2018/11/08-pb-28-11-2018-best-value-adalimumab-product-in-nhs.pdf
- Gils A. Combining therapeutic drug monitoring with biosimilars, a strategy to improve the efficacy of biologicals for treating inflammatory bowel diseases at an affordable cost. Dig Dis. 2017;35(1–2):61–68. doi: 10.1159/000449085
- Raffaelli EA, Massimino F. Reference biological medicines (‘originators’) and ‘biosimilars’: competition and patient protection. Antitrust & Public Policies. 2018;4(2):128–177.
- Guntern R. 15 years of biosimilar access in Europe PharmaTimes online 2021. [updated 2021 May; cited 2023 May 30]. Available from: https://www.pharmatimes.com/magazine/2021/may_2021/15_years_of_biosimilar_access_in_europe
- Cherny N, Sullivan R, Torode J, et al. ESMO European consortium study on the availability, out-of-pocket costs and accessibility of antineoplastic medicines in Europe. Ann Oncol. 2016 Aug;27(8):1423–1443.
- NICE. Infliximab, adalimumab and golimumab for treating moderately to severely active ulcerative colitis after the failure of conventional therapy 2015. [updated 2015 Feb 25; cited 2023 May 30]. Available from: https://www.nice.org.uk/guidance/ta329/chapter/1-Guidance
- Samaan MA, Irving PM. The impact of updated NICE guidelines on biologic treatment of ulcerative colitis: reflections on past practices, the changing present and implications for the future. Expert Opin Biol Ther. 2016 Aug;16(8):975–977. doi: 10.1080/14712598.2016.1189529
- Quintiles IMS. The impact of biosimilar competition in Europe. 2017 [cited 2023 May 30]. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2017/05/IMS-Biosimilar-2017_V9.pdf
- Taylor PC, Askari A, Choy E, et al. Approaches to optimising access to NICE-approved biologic anti-TNFs for patients with rheumatoid arthritis with moderately active disease. BMC Med. 2023 Feb 14;21(1):55. doi: 10.1186/s12916-023-02746-5
- NHS England. Clinical commissioning policy statement: rituximab bio-similar for the treatment of myasthenia gravis (adults). 2018 [updated 2021 Mar 25; cited 2023 Jul 23]. Available from: https://www.england.nhs.uk/wp-content/uploads/2021/04/Rituximab-biosimilar-for-the-treatment-of-myasthenia-gravis-adults-v2.pdf?UNLID=47063793820225187102
- NICE. Erythropoiesis‑stimulating agents (epoetin and darbepoetin) for treating anaemia in people with cancer having chemotherapy, 2014 [updated 2018 April 26; cited 2023 July 23]. Available from: https://www.nice.org.uk/guidance/ta323
- Moorkens E, Simoens S, Troein P, et al. Different policy measures and practices between Swedish counties influence market dynamics: part 2-biosimilar and originator etanercept in the outpatient setting. BioDrugs. 2019 Jun;33(3):299–306.
- IQVIA. Country scorecards for biosimilar sustainability: methodology appendix 2020. [updated 2020 Jun; cited 2023 May 30]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/country-scorecards-for-biosimilar-sustainability/iqvia-institute-scorecards-appendix-orb2520.pdf
- IQVIA. Biosimilar Scorecard 2020: Romania. 2020. [cited 2023 May 30]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/country-scorecards-for-biosimilar-sustainability/iqvia-biosimilar-scorecard-romania.pdf?la=en&_=1621259055784
- Ministère des solidarites et de la Santé. Stratégie nationale de santé 2018-2022. 2017 [cited 2023 May 30]. Available from: https://sante.gouv.fr/IMG/pdf/dossier_sns_2017_vdef.pdf
- IQVIA. Biosimilar scorecard 2020: Norway. 2020 [cited 2023 May 30]. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/country-scorecards-for-biosimilar-sustainability/iqvia-biosimilar-scorecard-norway.pdf?la=en