
ABSTRACT
Introduction It is now accepted that Parkinson’s disease (PD) is not simply due to dopaminergic dysfunction, and there is interest in developing non-dopaminergic approaches to disease management. Adenosine A2A receptor antagonists represent a new way forward in the symptomatic treatment of PD.
Areas covered In this narrative review, we summarize the literature supporting the utility of adenosine A2A antagonists in PD with a specific focus on istradefylline, the most studied and only adenosine A2A antagonist currently in clinical use.
Expert opinion: At this time, the use of istradefylline in the treatment of PD is limited to the management of motor fluctuations as supported by the results of randomized clinical trials and evaluation by Japanese and USA regulatory authorities. The relatively complicated clinical development of istradefylline was based on classically designed studies conducted in PD patients with motor fluctuations on an optimized regimen of levodopa plus adjunctive dopaminergic medications. In animal models, there is consensus that a more robust effect of istradefylline in improving motor function is produced when combined with low or threshold doses of levodopa rather than with high doses that produce maximal dopaminergic improvement. Exploration of istradefylline as a ‘levodopa sparing’ strategy in earlier PD would seem warranted.
1. Introduction: moving forward from a dopaminergic approach to treating Parkinson’s disease
The treatment of Parkinson’s disease (PD) has been dominated by dopamine replacement therapy for over 60 years. Following the introduction of levodopa in the 1960s, a range of dopamine agonist drugs (e.g. apomorphine, bromocriptine, cabergoline, pergolide, ropinirole, pramipexole, rotigotine) were added to (and some subsequently removed from) the PD armamentarium [Citation1,Citation2]. The use of levodopa as the gold standard drug has been supported by the introduction of various classes of enzyme inhibitors (peripheral dopa decarboxylase inhibitors, catechol O-methyl transferase inhibitors, monoamine oxidase B [MAO-B] inhibitors) that potentiate its actions [Citation1,Citation2]. Many of these dopaminergic agents are now available in a range of formulations and delivery systems. While no one doubts the efficacy of dopaminergic medications in controlling motor symptoms and improving quality of life, there are significant limitations regarding the benefits and adverse events that inevitably occur with their use. They do not completely reverse motor deficits in PD, their chronic use results in a high incidence of motor complications (response fluctuations and dyskinesia) and they produce undesirable adverse effects, particularly in the elderly and advanced patient populations [Citation2]. Specific efficacy-related problems that occur and that remain unsolved are the control of motor fluctuations in the form of OFF episodes and the onset and avoidance of the involuntary movements that make up dyskinesia [Citation3,Citation4]. As a consequence, a range of attempts have been made to manipulate non-dopaminergic pathways in the brain to try and find alternative drug classes that might control both motor and non-motor symptoms of PD while avoiding the dopaminergic side-effect profile [Citation5,Citation6].
One of the most scientifically based alternative approaches is adenosinergic modulation of the indirect striatal output pathway. In this narrative review, we summarize the literature supporting the utility of adenosine A2A antagonists in PD with a specific focus on istradefylline, the best studied and only adenosine A2A antagonist currently in routine clinical use.
2. Developing a non-dopaminergic therapy for the treatment of motor and non-motor symptoms
The complex pathology of PD in the brain involves neuronal systems that use a range of transmitters other than dopamine (including noradrenaline, serotonin [5-HT], glutamate, acetylcholine, and gamma-aminobutyric acid [GABA]) and these are involved in the expression of both motor and non-motor symptoms [Citation7,Citation8]. In addition, a range of adaptive changes takes place in the basal ganglia in response to the loss of dopaminergic input and these also affect the activity of a wide range of non-dopaminergic neuronal pathways and their receptors [Citation9,Citation10]. Multiple attempts have been made to exploit the alterations in neurotransmission occurring both inside and outside of the basal ganglia. To date, most of these have been unsuccessful, although a number of different approaches remain under investigation. Translation from the laboratory to the clinic has been limited, and the therapeutic window is often narrower in humans than in animals, with adverse events occurring more frequently [Citation11].
Only a few non-dopaminergic approaches have found utility in the treatment of motor components of PD (). Anticholinergics affecting muscarinic transmission were an early attempt to control motor symptoms and they have utility in the treatment of some aspects of PD, for example tremor, but there is little modern randomized clinical trial data to show efficacy, and their use is limited by side-effects, particularly in the elderly population [Citation12,Citation13]. Amantadine is generally referred to as a weak N-methyl-D-aspartate (NMDA) glutamate receptor antagonist to explain its anti-dyskinetic efficacy [Citation14]. However, its mild-moderate efficacy as monotherapy [Citation15] and against OFF time [Citation16] more likely reflects its multimodal pharmacology, including enhanced dopamine transmission through both pre-synaptic [Citation17,Citation18]) and post-synaptic mechanisms [Citation19,Citation20]). In addition, the widespread expression of NMDA receptors in the brain leads to a range of unacceptable adverse events, and amantadine is poorly tolerated by a high proportion of patients (although the recent introduction of an extended release form may reduce their incidence) [Citation21]. Lastly, zonisamide, which was originally developed as an antiepileptic, has been repurposed for PD and shown to produce symptomatic improvement and to suppress dyskinesia [Citation22,Citation23]. While zonisamide has multiple pharmacological actions, including MAO-B inhibition [Citation24], its ability to inhibit GABAergic transmission is considered to affect the activity of the striatal output pathways and so influence voluntary movement [Citation25]. Outside of these agents, non-dopaminergic approaches to PD have been limited to specific symptoms. For example, the noradrenaline precursor L-threo-dihydroxyphenylserine may have benefit on gait/ataxia [Citation26], the 5-HT2A inverse agonist pimavanserin is effective against psychosis in PD [Citation27,Citation28] and cholinesterase inhibitors may have benefit against falls [Citation29,Citation30].
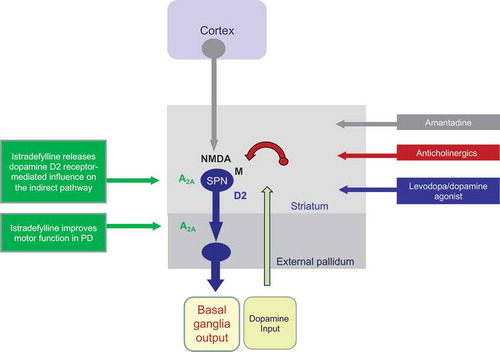
Figure 1. A schematic representation of the mode of action of commonly used dopaminergic and non-dopaminergic treatments for Parkinson’s disease compared with the adenosine A2A antagonist istradefylline
The reasons for the relative failure of non-dopaminergic approaches in treating motor and non-motor symptoms are complex. A failure to translate from preclinical models of PD may reflect the limited relevance of these paradigms to the complexity of change in the human disease. The organization of the neuronal pathways involved in motor control and in non-motor behaviors may be different among rodents, primates and humans, as will be the localization and subtype of neuronal receptor population involved [Citation31]. One particular problem has been identifying a receptor population in the brain that is relevant to the changes in neurotransmission that occur in PD and where target engagement leads to a selectivity of effect that acts to restore normal function [Citation6,Citation32]. This is what differentiates the adenosine A2A antagonists from other non-dopaminergic approaches to PD, as will be detailed below.
3. Adenosine, adenosine receptors, and A2A antagonists
Although recognized as a regulator of nervous tissue function for more than 50 years, adenosine is not a classical neurotransmitter and there are no neuronal populations or neuronal tracts that use adenosine as their neurotransmitter in the classical sense. In the PD arena, the role of adenosine is not well understood and its function in the basal ganglia is still poorly recognized by most physicians. Extracellular adenosine is formed from the dephosphorylation of ATP, ADP and AMP. It functions as a ubiquitous neuromodulator throughout the nervous system. Since the brain is bathed in adenosine, it is not surprising that adenosine receptors are found on almost every cell type – neurons and glia [Citation33,Citation34]. There are four distinct adenosine receptors (A1, A2A, A2B, A3) with different anatomical localizations and different functions [Citation35,Citation36]. In the late 1980s/early 1990s, the adenosine A2A receptor was cloned and biochemically and pharmacologically characterized and shown to have a unique and highly specific distribution in the basal ganglia. Numerous studies in the rat and human brain showed high expression of adenosine A2A receptors in the caudate nucleus, putamen, nucleus accumbens, olfactory tubercles, and external globus pallidus (GPe) [Citation37–40]. In the striatum, A2A receptor messages are generally localized together with message for dopamine D2 receptors and preproenkephalin, but segregated from dopamine D1 receptors and substance P messages [Citation41]. This clearly demonstrates a preponderance for localization of the indirect GABAergic striatal output pathway to GPe as opposed to the direct GABAergic output to the internal pallidum/substantia nigra [Citation42–44]. In addition, although A2A receptor mRNA is not detected in the GPe, immunohistochemical study of the rat brain showed that nerve terminals located in this region have A2A receptor protein localized to the axon terminal of the striato-pallidal spiny projection neurons that make up the indirect output pathway [Citation45]. Importantly, these neurons also contain the specific machinery for the production of extracellular adenosine from adenosine monophosphate (AMP; ecto-5ʹ-nucleotidase, the major enzyme able to convert extracellular AMP into adenosine, colocalizes with A2A receptors in the basal ganglia [Citation46,Citation47]) and so can be looked upon as being ‘adenosinergic’ in nature.
As a consequence of the selective localization of A2A receptors to the indirect output pathway, there has been considerable interest in developing A2A receptor antagonists for the treatment of the motor symptoms of PD. A range of compounds with different structural characteristics have been identified and examined to varying degrees in preclinical models of PD and in different clinical trial phases in PD. Those that reached clinical development are discussed in the following sections and their characteristics are summarized in . While these have all shown proof of concept in the laboratory, only istradefylline has so far completed its clinical development for treating OFF episodes in PD and been successfully registered in Japan and the USA.
Table 1. Adenosine A2A antagonists that have been examined in preclinical models of PD and those that underwent clinical development
4. Targeting the indirect striatal output pathway
Adenosine A2A receptors in the basal ganglia are uniquely positioned to selectively modulate the activity of the indirect output pathway at the level of the striatum and GPe. At the functional level, A2A receptor activation increases the excitability of the indirect output pathway through an enhancement of GABAergic synaptic transmission from the striatum to the GPe. At the pharmacological, neurochemical and electrophysiological levels, A2A receptors act to increase GABA release in the GPe and increase GABAergic inhibitory tone onto neurons in the GPe [Citation59–62].
There has been intense debate about precisely how, at the neuronal level, A2A receptors modulate GABAergic output in the indirect pathway. There is evidence that A2A receptors and dopamine D2 receptors may form receptor dimers, which would suggest that A2A receptors are involved in the function of D2 receptors [Citation63,Citation64]. However, it is not clear how an interaction between A2A and D2 receptor signaling would occur at a second messenger level. In addition, both A2A agonists and antagonists clearly affect motor function in dopamine D2 receptor-deficient mice, demonstrating that A2A receptors work independently of D2 receptors [Citation59,Citation65]. In humans, positron emission tomography (PET) imaging studies have shown opposing changes with striatal A2A receptor density increased [Citation66,Citation67] and D2 receptor density decreased [Citation67] in PD. Presynaptic regulation of dopamine release by A2A receptors has similarly been proposed, but this concept is also difficult to support as there are no striatal nerve terminals that co-express A2A receptors and tyrosine hydroxylase [Citation62,Citation68].
There is also the question as to whether A2A receptors affect the function of other non-dopaminergic components of the basal ganglia pathways, such as glutamatergic and cholinergic transmission. This appears unlikely. Pharmacological blockade and genetic knockout of A2A receptors does not affect basal cortico-striatal glutamate synaptic properties or produce a consistent effect of A2A receptor-mediated change on striatal extracellular glutamate levels [Citation62,Citation69,Citation70]. A few studies have detected A2A receptor mRNA-containing cholinergic interneurons in striatum [Citation71,Citation72] and a mouse immunofluorescence analysis showed choline acetyl transferase-positive neurons labeled with A2A receptors [Citation73]. However, the majority of in situ hybridization studies have not detected the expression of A2A receptors on cholinergic neurons in rodents or primates [Citation45,Citation74–77].
5. A2A receptor function in PD – the role of A2A receptor antagonists
In PD, the balance between the direct and indirect GABAergic output pathways changes as nigrostriatal neurons degenerate and dopaminergic transmission declines with the indirect pathway to GPe becoming overactive [Citation9,Citation78]. In addition, the balance between the excitatory effect of A2A receptors and the inhibitory effect of D2 receptor stimulation is changed as dopaminergic tone is lost [Citation41,Citation79]. Changes take place in the density of A2A receptors as shown by an increase of adenosine A2A receptor mRNA and binding sites in the striatum and GPe in postmortem brain tissue in PD [Citation80]. This increase is particularly marked in individuals exhibiting dyskinesia [Citation80], which may be relevant to the use of adenosine antagonists, such as istradefylline, in later stages of PD (see later). These findings have been verified by PET imaging studies [Citation66,Citation67,Citation81].
Administration of levodopa or a D2 agonist restores dopaminergic stimulation of the indirect pathway leading to a reversal of the enhanced GABAergic tone to the GPe and, as a consequence, an improvement in motor function. However, this effect is opposed by the excitatory effects of adenosine through A2A receptor stimulation that act to increase GABAergic transmission and limit improvements in movement. Using an adenosine A2A antagonist, such as istradefylline, prevents the excitatory effects of adenosine and lowers GABAergic tone to the GPe. This results in improvement in motor control by removing a ‘brake’ from the indirect striatal pathway output that would otherwise limit the effectiveness of dopamine replacement therapy. The overall effect is to further normalize the function of the striato-thalamo-cortical loops that control voluntary movement and which are impaired by PD.
The ability of adenosine A2A antagonists, such as istradefylline, to selectively alter GABAergic tone in the indirect output pathway distinguishes this drug class from all others currently used in PD. A2A antagonists act through a mechanism beyond the damaged dopaminergic system that avoids an interaction with the direct output pathway, which is implicated in the expression of dyskinesia. This differentiates A2A antagonists from both levodopa and dopamine agonist drugs. They are also distinct from other non-dopaminergic drugs used in the treatment of PD, such as amantadine and anticholinergics, since there is little evidence to suggest that their effects on motor function are mediated by an action related to either glutamatergic or cholinergic neuronal function (). This, however, does not negate a positive therapeutic interaction when used in conjunction with dopaminergic medications as already described above, or with drugs acting on glutamatergic, cholinergic or GABAergic systems.
6. A2A antagonists and motor function in PD – the preclinical science
The selective localization of A2A receptors on the indirect output pathway focuses attention on the ability of A2A antagonists to normalize basal ganglia function and to improve motor function in PD. In experimental models of PD, the actions of this drug class are clear and consistent. Adenosine A2A receptor antagonists, such as istradefylline, KF17837, and preladenant, reverse motor impairments in a variety of rodent models of PD caused by either reserpine-induced dopamine depletion or by administration of dopamine receptor antagonists, such as haloperidol [Citation82–87] and increase locomotor activity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated or reserpinized mice [Citation83]. In addition, istradefylline, KF17837, preladenant and another A2A receptor antagonist, ST1535, potentiated the rotational behavior produced by levodopa or dopamine agonist administration to 6-hydroxy-dopamine (6-OHDA)-lesioned rats [Citation88–90]. All of this contrasts with the effect of administration of an A2A agonist, such as CGS21680, in rodent models, which leads to a decline in motor function [Citation91].
A similar consistent effect of A2A adenosine antagonists is seen in the MPTP-treated non-human primate. When administered alone to MPTP-treated common marmosets or cynomolgous monkeys, ST1535, preladenant and istradefylline produced modest dose-related increases in locomotor motor activity and reversal of motor disability with no evidence of tolerance on repeated administration [Citation92–95]. The specificity of these effects was shown by pharmacological manipulation as the effects of istradefylline in MPTP-treated common marmosets were reversed by an adenosine A2A agonist, and no improvement in motor function occurred after administration of a selective adenosine A1 receptor antagonist. These observations indicate that monotherapy with A2A receptor antagonists might produce an improvement in motor function in PD, which needs further exploration in clinical studies.
The effects of A2A antagonists administered in conjunction with levodopa have also been extensively evaluated in MPTP-treated primates. In the MPTP-treated common marmoset and cynomolgus monkey, the combination of preladenant, istradefylline or ST1535 with levodopa had an additive effect in reducing motor disability and in increasing locomotor function compared with the effects of levodopa alone [Citation92,Citation94,Citation96]. Perhaps importantly, istradefylline markedly improved the effects of a suboptimal dose of levodopa indicating that a ‘levodopa sparing’ strategy might be a potential use for the drug [Citation97]. Similarly, administration of istradefylline with the dopamine agonists ropinirole or pergolide resulted in an increase in reversal of motor disability and increase in ON time compared with that produced by either istradefylline or the dopamine agonists alone [Citation98].
The effects of A2A antagonists have similarly been examined in preclinical models of dyskinesia in PD using both rodents and non-human primates. In 6-OHDA-lesioned rodents, chronic administration of three A2A receptor antagonists, vipadenant, caffeine or istradefylline, did not produce abnormal involuntary movements (AIMs) [Citation99,Citation100]. These data suggest that A2A receptor antagonists are less likely to induce dyskinesia compared with levodopa. However, they do not prevent the onset or expression of dyskinesia induced by levodopa [Citation99]. Indeed, when istradefylline was administered to 6-OHDA-lesioned rats previously primed to exhibit AIMs, istradefylline administered alone did not elicit abnormal movements, but neither did it reduce the severity of dyskinesia seen when it was co-administered with levodopa [Citation101].
Similarly, adenosine A2A antagonists, including preladenant, istradefylline and ST1535, did not induce dyskinesia in levodopa-primed, MPTP-treated primates when administered alone [Citation92,Citation93,Citation95–97]. Also, when A2A antagonists were co-administered with levodopa, they enhanced and/or prolonged the antiparkinsonian activity of levodopa, but without exacerbating dyskinesia intensity [Citation92–94,Citation97,Citation98]. Istradefylline is the adenosine A2A antagonist most extensively evaluated in the dyskinetic MPTP-treated primate model. Acute or repeated oral administration of istradefylline for 21 days induced little or no dyskinesia [Citation96]. Co-administration of istradefylline daily for 21 days with a suboptimal dose of levodopa produced an enhancement in motor performance, but the severity of dyskinesia did not increase and, in fact, dyskinesia was significantly decreased by the end of the study. When the animals were subsequently challenged with an acute administration of an optimal higher dose of levodopa, severe dyskinesia was observed [Citation96]. The combination of preladenant and suboptimal doses of levodopa in MPTP-treated primates produced an additive effect over those seen with a suboptimal dose of levodopa alone, again without worsening dyskinesia, supporting the findings with istradefylline [Citation95]. There are some tantalizing data on the potential effects of A2A antagonists in preventing or delaying the induction of levodopa-induced dyskinesia. The chronic co-administration of istradefylline with apomorphine completely prevented the induction of dyskinesia normally seen in MPTP-lesioned cynomolgus monkeys after apomorphine administration without affecting the improvement in motor function [Citation102].
The results obtained from MPTP models of PD indicate that A2A antagonists do not themselves induce dyskinesia in primates primed by repeated treatment with levodopa and that the chronic co-administration of an A2A antagonist with levodopa does not worsen the severity of existing dyskinesia while improving motor function. A2A antagonists might also attenuate the induction of dyskinesia.
7. Pharmacodynamics and pharmacokinetics of istradefylline
The istradefylline receptor profile shows remarkable selectivity of effect. Istradefylline is a potent, competitive A2A receptor antagonist exhibiting a high affinity for A2A receptors in humans, monkeys, dogs and rodents [Citation103,Citation104]. The affinity of istradefylline for A1, A2B and A3 adenosine receptors is lower than that for the A2A receptor and it has no significant affinity for 51 other receptors, transporters and channels against which it was examined in vitro [Citation105]. From the perspective of PD, its relative lack of affinity for the A1 receptors [Citation105] is important because evidence suggests that endogenous adenosine mechanisms are active in tremor (e.g. the nonselective antagonist caffeine can exacerbate tremor) [Citation106]. Further, istradefylline showed no relevant activity at dopamine receptor subtypes or interaction with MAO-A, MAO-B, or catechol-O-methyltransferase. Istradefylline was shown not to inhibit canine phosphodiesterase enzymes I–V [Citation105] differentiating it from the nonselective adenosine antagonist caffeine [Citation107]. This purity of action supports the mechanism of action of the drug to the A2A receptors located on the indirect striatopallidal output pathway, which are proposed to explain its efficacy in PD.
Istradefylline has the characteristics required for a once daily medication for PD [Citation104,Citation108,Citation109]. After oral administration, peak plasma concentrations are achieved after approximately 4 hours and the drug has a terminal half-life of approximately 83 hours. Istradefylline exhibits dose-proportional pharmacokinetics after multiple oral doses of 20–80 mg and steady state levels are reached after two weeks of once daily dosing with no differences between healthy individuals and those with PD and no differences based on age, sex, weight or race [Citation108,Citation110]. There are no clinically significant effects of food on pharmacokinetic parameters. The precise bioavailability of istradefylline after oral administration is unknown as the insolubility of the drug prevents assessment after intravenous administration. In humans, istradefylline is entirely eliminated by metabolism with six known metabolites; tracer studies show 48% of drug product in feces and 39% in urine after a 40 mg dose [Citation110]. No changes in drug exposure are seen in severe renal impairment or mild hepatic impairment but moderate hepatic impairment marginally increases drug exposure [Citation110]. Metabolism is primarily by cytochrome P450 (CYP) 1A1 and CYP3A4 and the expected drug interactions can consequently occur. Smoking decreases systemic exposure to istradefylline and higher doses are recommended. From a PD perspective, istradefylline does not alter the pharmacokinetic profile of levodopa or carbidopa [Citation110].
8. The clinical development program
The istradefylline clinical development program was initiated around 1996, eventually culminating in eight pivotal studies (). To date, the istradefylline studies in PD have explored its potential utility for:
Motor improvement as monotherapy
Additive motor improvement as levodopa adjunctive therapy
Suppression of dyskinesia as levodopa adjunctive therapy
Sustainable motor improvement with reduced levodopa (levodopa sparing effect) for patients with unacceptable levodopa side-effects.
Table 2. Summary of efficacy outcomes from randomized controlled studies (istradefylline versus placebo)
The first proof-of-concept study, termed 6002-US-001, sought to understand its utility in the management of motor complications (wearing-off and dyskinesia) [Citation111]. Key findings from this study were that (i) istradefylline showed promise for motor fluctuations, but not dyskinesia, and (ii) doses of 20 and 40 mg/day are adequate for wearing-off improvement. While this early study did not indicate a significant benefit for dyskinesia, a parallel study (6002-US-004) indicated that, when combined with low-dose levodopa infusion, istradefylline potentiated the antiparkinsonian response, but with significantly less dyskinesia compared with that induced by optimal dose levodopa alone (p< 0.05) [Citation112].
The 6002-US-001 and 6002-US-004 studies informed the istradefylline development strategy, which focused on the efficacy and safety of adjunct istradefylline in patients with levodopa-induced motor complications. At the time of study conduct, and until early 2000, most clinical studies evaluating motor complications used simple patient-reported ON/OFF diaries to assess motor fluctuations [Citation113]. However, dyskinesia was mostly assessed as an adverse event, not as an efficacy parameter, and new methods were needed. In 2000, Hauser et al. proposed a new home diary format where dyskinesia assessments are divided (according to the patient perspective) into non-troublesome dyskinesia and troublesome dyskinesia [Citation114]. These ‘Hauser diary’ assessments were employed in all eight pivotal studies of adjunct istradefylline for motor complications and, as will be reviewed later, suggest that istradefylline increases ON time without troublesome dyskinesia – something that the early proof-of-concept studies were not able to pick up.
The efficacy of istradefylline as monotherapy in early PD was explored in one Phase II study. In the 6002-US-051 study [Citation115], istradefylline 40 mg/day failed to demonstrate significant separation in the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III (motor) scores from placebo at 12 weeks (least-squares mean difference, – 1.11, p= 0.228), and no further trials of istradefylline as monotherapy have been undertaken.
9. Pivotal trials of adjunct istradefylline in levodopa-treated patients experiencing motor fluctuations
9.1. Double-blind placebo-controlled studies
Eight double-blind, placebo-controlled trials have been conducted to evaluate istradefylline as an adjunct to levodopa for the treatment of PD in patients with motor fluctuations [Citation116–123] (). These studies all enrolled patients with moderate to advanced PD (Hoehn and Yahr stages 2–4) and motor fluctuations (at least 2–3 hours of daily OFF time, depending on the study). Patients were randomized to the addition of placebo or istradefylline 10, 20, 40, or 60 mg/day, depending on the study, and multiple doses were tested in several of the studies. Treatment duration was 12 weeks except EU-007, which was a 16-week study. In each study, the primary efficacy measure was reduction in daily OFF time (in hours or as a percentage of daily awake time) from baseline, as assessed by patient home diaries.
The primary efficacy outcome of reduction in OFF time was met in five of these studies. Istradefylline provided reductions in daily OFF time relative to placebo of 0.64–0.76 hours with the 20 mg/day dose [Citation117,Citation118,Citation121,Citation122], 0.74–1.2 hours with the 40 mg/day dose [Citation116,Citation121,Citation122] and 0.77 hours with the 60 mg/day dose [Citation117]. In addition, istradefylline 40 mg/day increased ON time without troublesome dyskinesia relative to placebo by 0.96 hours (p= 0.026) in one study [Citation116] and by 0.81 hours (p= 0.004) in another [Citation122]. In one study, ON time with troublesome dyskinesia was significantly increased by 0.25 hours (p= 0.011) with istradefylline 40 mg/day relative to placebo [Citation121]; changes in this measure were not significant in the other studies.
Taken together, these trials indicate that istradefylline in doses of 20 and 40 mg/day is efficacious to reduce OFF time as an adjunct to levodopa in PD patients experiencing motor fluctuations. The key outlier study was the 6002-US-018 trial [Citation119], which evaluated istradefylline 10 mg/day, 20 mg/day and 40 mg/day, and neither reductions in OFF time nor increases in ON time without troublesome were statistically significant compared with placebo. In this study, there was a notably robust placebo response with a reduction in OFF time from baseline of 1.3 hours; this reduction in OFF time was greater than that seen with placebo treatment in the other five studies (where reductions in daily OFF time in the placebo group ranged from 0.23 to 0.9 hours) and may explain this trial’s failure to meet the primary efficacy outcome [Citation119].
9.2 Open-label long-term trials of istradefylline as adjunct to levodopa in patients with motor fluctuations
In two long-term studies, adjunctive istradefylline was demonstrated to maintain reductions in OFF time and afford a favorable safety profile through 52 weeks [Citation124,Citation125]. These trials enrolled PD patients with motor fluctuations who had previously participated in pivotal randomized, placebo-controlled, double-blind trials in North America and Japan.
The 6002-US-007 study [Citation125] enrolled participants from three prior North American randomized, placebo-controlled, double-blind trials of adjunctive istradefylline in PD patients with motor fluctuations (6002-US-001, 6002-US-005, 6002-US-006). Patients were included in Group I if they had completed double-blind treatment with istradefylline within 15 days from enrollment in the open-label study, while patients were included in Group II if they had completed double-blind treatment with istradefylline more than 15 days prior to enrollment in the open-label study (washed-out) or had previously been randomized to placebo (istradefylline-naive). Istradefylline dosing was flexible; patients completed two week’s treatment with istradefylline 40 mg/day after which the dose could be increased to 60 mg/day or decreased to 20 mg/day daily. A total of 315 patients were included in Group I and 181 patients in Group II. Due to reports of brain mineralization in rodents the study was administratively terminated early by the sponsor. However, the mean duration of istradefylline exposure was 25 weeks; 77% of subjects discontinued early due to study termination, 8% due to adverse events, 8% due to lack of efficacy, and 5% due to withdrawal of consent. For patients in Group I (who were continuing with istradefylline treatment), daily OFF time remained similar to baseline over the 52 weeks of the study (range over 52 weeks from – 0.12 hour to +0.49 hour) indicating sustained long-term efficacy. By contrast, Group II patients (who were previously on placebo or who had >15 days washout between trials) experienced sustained improvements in daily OFF time from baseline (between – 0.53 and – 1.19 hours). Likewise, ON time without troublesome dyskinesia was maintained throughout the study for Group I and was improved compared with baseline at all visits in Group II.
The Japanese 52-week study [Citation124] enrolled patients who had completed a 12-week randomized, double-blind, placebo-controlled study (6002–009) of adjunctive istradefylline. Patients were again separated into groups depending on whether they had received placebo (n = 100), istradefylline 20 mg/day (n = 101), or istradefylline 40 mg/day (n = 107) in the preceding double-blind study. In the open-label phase, all patients were initially administered istradefylline 20 mg/day, which could be increased to 40 mg/day after 4 weeks, and the dose was held constant after 8 weeks. Overall, 231 patients (75%) completed the study, with 81 receiving istradefylline 20 mg/day and 150 receiving 40 mg/day at Week 52. Efficacy results were consistent with reductions in OFF time in patients previously treated with placebo, and maintenance of response through 52 weeks in patients previously treated with placebo or istradefylline.
9.2. Safety and tolerability of istradefylline as adjunct to levodopa
During the course of the clinical development program, istradefylline was evaluated in over 3400 patients, with >1100 patients taking istradefylline for 1 year or longer [Citation126]. Across the studies, most treatment-emergent adverse events (TEAEs) experienced by study subjects were mild to moderate in severity. Dyskinesia was the most commonly reported TEAE, followed by nausea and dizziness () [Citation116–119,Citation126]. The mean onset of dyskinesia was 16 days after starting istradefylline and did not appear to be a common cause for study discontinuation (1.3% of istradefylline patients discontinued due to dyskinesia versus 0.7% with placebo) [Citation110,Citation126]. These data are supported by a Japanese post-marketing study, which found a higher frequency of dyskinesia in patients with preexisting dyskinesia compared with patients without preexisting dyskinesia (10.6% versus 1.7%) [Citation127].
Table 3. Adverse events in clinical trials of istradefylline (≥2% istradefylline-treated patients)
Of note, the incidence of other TEAEs that are known to occur with levodopa and other dopaminergic drugs (e.g. orthostatic hypotension, sleep attacks, somnolence, psychosis, confusion, and impulse control disorders) was similar for patients receiving istradefylline and those receiving placebo. The lower incidence of these prominent dopaminergic side effects, especially daytime somnolence, impulse control disorders and orthostatic hypotension is a clinically important feature of the risk-benefit profile of istradefylline.
10. The path to registration in Japan and the USA
The path to regulatory approval in the USA was not straightforward and merits commentary. In 2007, after completion of the first five pivotal studies (US-005, US-006, US-013, US-018, and EU007, which included sites in and outside of Europe), Kyowa Kirin filed a new drug application with the Food and Drug Administration (FDA). However, the FDA initially considered that the evidence of motor benefit was insufficient for approval, although no serious safety concerns were identified.
Kyowa Kirin decided to discontinue the development program outside of Japan and two additional trials (6002–0608, 600–009) were undertaken in Japan. Both studies showed significant OFF time reduction versus placebo as the primary efficacy measure and improvement on UPDRS Part III (motor) scores as one of the secondary outcomes [Citation121,Citation122]. These studies led to the Japanese approval of istradefylline for ‘improvement of wearing-off phenomenon in patients with PD on concomitant treatment with levodopa-containing preparations’ in 2013. Additionally, an interim analysis of a Japanese post-marketing surveillance study following 476 PD patients showed that istradefylline effectively reduced OFF time symptoms and improved motor dysfunction in 44.7% and 48.5% of patients, respectively, with a mean decrease of the UPDRS Part III score of about 3 points and a physician’s global assessment of the drug as effective in 61.3% of patients [Citation127].
Following approval in Japan, attention turned to seeking approval for the drug in the USA and Europe. A new study (6002–014, clinicaltrials.gov identifier: NCT01968031) was initiated using the same basic design as in previous investigations, under a special protocol assessment agreement with the US FDA (2013) (see [Citation128]). The study enrolled a PD population that was enriched for a higher dopaminergic load when compared to the other seven randomized controlled trials, due to specific study inclusion criteria that stipulated the presence of levodopa-induced dyskinesia and use of levodopa at a dose of ≥400 mg/day, plus at least one additional dopaminergic PD medication. However, around the end of 2016, study results showed that the primary outcomes at both 20 and 40 mg/day did not meet statistical significance versus placebo [Citation129]. The reasons for this are unclear, but the study requirement that patients should have both OFF time and documented dyskinesia at baseline may make demonstrating either a reduction in OFF time or an increase in ON time without troublesome dyskinesia much more difficult. The failure to show separation from placebo has also been suggested to reflect the difficulties of conducting research in multiple countries [Citation130]. Similar problems in the consistency of efficacy outcomes were reported for a Phase III study of the A2A antagonist preladenant, conducted at around the same time, which found unequal placebo effects among countries and suggested that a push for enrollment might have degraded the quality of patients that sites enrolled [Citation131].
As the regulatory authorities have noted, it is not uncommon to encounter negative studies in clinical development programs, and their presence alone does not preclude a conclusion that substantial evidence of effectiveness has been demonstrated [Citation132]. Thus, after obtaining the results of the 6002–014 study, Kyowa Kirin entered discussions with the FDA to apply for Class 2 resubmission based on eight pivotal trials, including the five studies originally submitted and three additional controlled efficacy studies (0608, 009 and 6002–014) in February 2019. Specifically, the FDA decided to re-review the data for istradefylline based on four of the eight clinical studies: US-005, US-013, 0608 and 009 [Citation132]. Final approval was given in August 2019 for use as adjunctive treatment to levodopa/carbidopa in PD patients experiencing OFF episodes [Citation132,Citation133]. Currently, the marketing authorization application for istradefylline as an ‘adjunctive treatment to levodopa-based regimens in adult patients with PD experiencing OFF time’ is under review by the European Medicines Agency [Citation134].
11. Preclinical and clinical data evaluating istradefylline for non-motor and axial symptoms
The relationships between psychiatric functions, sleep-wake control, and adenosine A2A receptors (located in the basal ganglia and limbic regions) have already been extensively studied in animal models [Citation135] and several small open-label studies have explored the potential utility of istradefylline in the management of non-motor symptoms, such as mood disorders, sleep disturbance, fatigue, and urinary disturbance, as well as difficult-to-treat axial symptoms, such as freezing of gait and posture abnormality () [Citation136–146].
Table 4. Post-marketing clinical research to evaluate Parkinson’s disease symptoms in Japan
One small-scale clinical study of the effects of istradefylline on mood disorders in PD has been undertaken [Citation137]. As expected, administration of istradefylline over 12 weeks improved UPDRS scores, consistent with its anti-parkinsonian activity. However, istradefylline also improved the Beck Depression Inventory, the Snaith-Hamilton Pleasure Scale (Japanese version) and the Apathy scale, showing its potential for treating depression in PD. Importantly, there was no correlation between the changes in motor function and the improvement in other markers, demonstrating that these occurred independently of one another.
It is well accepted that A2A receptors play a dominant role in the induction and gating of sleep, as demonstrated by both pharmacological manipulation and genetic modification [Citation147], and also reflected by the nonselective effects of caffeine. While there have been no investigations of istradefylline in preclinical models of the sleep-wake cycle to ascertain its effects on arousal and wakefulness, two small clinical studies have shown benefits in reducing excessive daytime sleepiness (as assessed by the Epworth Sleepiness Scale) without affecting nocturnal sleep (as assessed by the PD Sleep Scale) [Citation141,Citation142]. In addition, in a small study reported by Abe et al., treatment with istradefylline showed a significant improvement on fatigue over 8 weeks; fatigue severity scores improved from 62.8 ± 7.1 at baseline to 52.3 ± 9.3 (p= 0.049) [Citation136].
Axial symptoms, such as freezing of gait (FOG) and postural abnormality, are poorly responsive to dopaminergic medications. In one small case series, 14 PD patients treated with istradefylline showed significantly reduced Freezing of Gait Questionnaire (FOG-Q) scores (from 12.1 ± 5.8 at baseline to 9.8 ± 7.2 after 1 month). The authors also noted that three of the patients showed remarkable improvements in the timed up and go (TUG) test, although results were not consistent across the full group of patients [Citation141]. In another case series, four consecutive patients with postural deformities (antecollis, Pisa syndrome, and camptocormia) converted from a dopamine agonist to istradefylline showed postural improvements without deterioration of motor symptoms (istradefylline was initiated after an average of 1.3 months following dopamine agonist withdrawal) [Citation145]. Axial symptoms are hard to mimic in animal models, and it is difficult to speculate on the mechanism of A2A antagonist effects. However, the action of A2A antagonists in reducing excessive GABAergic suppression of the thalamus/brainstem, including the pedunculopontine nucleus (involved in gait and posture [Citation148]), may provide one plausible explanation.
Finally, bladder dysfunction is a major clinical issue in PD patients [Citation149]. Two small studies by Kitta et al. [Citation139,Citation140] have reported beneficial effects. In their one-year study following 14 patients, they reported significant improvements in the International Prostate Symptom Score and in the Overactive Bladder Symptom Score (p< 0.05). Nighttime urinary frequency and the nocturnal urine volume also improved significantly at 3 months’ administration (p< 0.01) [Citation139]. While the authors speculated on an action via the basal ganglia network, further work is needed to explore the potential mechanisms behind this interesting observation.
12. Demonstrating the clinical efficacy and safety of istradefylline where others have faced challenges
A key question is why istradefylline has been successfully developed and registered for use in PD in Japan and the USA where other molecules of the same class have not been approved. The first point that needs to be made is that this was a process that took over 25 years from discovery to registration of istradefylline in the USA, and required belief in the molecule and the concept of its action at all levels within Kyowa Kirin. This required appropriate financial investment and a consistent development team over many years. Much of the initial enthusiasm stemmed from a continuous investment in novel preclinical experimentation within the company and also through external collaboration that was published in peer-reviewed journals.
When faced with negative results in the preclinical and clinical development program, the studies were closely analyzed and necessary changes to the approach to studying istradefylline were made so that the drug could continue to be studied in a clinically meaningful way. For example, the cause of mineralization in the brain during rodent toxicology studies also needed explanation and detailed study showed no evidence of tissue necrosis, hemorrhage, inflammation or glial nodules [Citation126]. The drug also survived out-licensing and a period of inactivity before the molecule was returned to the parent company. What this story shows is that belief in the molecule and a carefully planned approach to study facilitated the eventual approval.
The initial lack of FDA approval for istradefylline on the grounds of insufficient evidence of efficacy to support its clinical utility could have led to the discontinuation of the clinical program. However, the decision to undertake two further Phase III studies in Japan led to its registration in that territory. It also provided the confidence to continue the registration process in the USA. The reapplication would therefore exceed the usual requirement for independent substantiation of the efficacy findings in two studies.
The failure of preladenant to show efficacy in Phase III investigations against OFF time compared with placebo either as monotherapy or in conjunction with levodopa led to Merck & Co discontinuing its clinical development program in 2013. Notably, the failure of the already approved comparator drug, the MAO-B inhibitor rasagiline, in these studies raised serious questions about the design and conduct of the clinical trials. Indeed, a number of important lessons learned were reported as a result of the negative outcomes [Citation131]. These included the importance of recruitment of an appropriate patient population; pressure to meet target enrollment numbers as difficult eligibility criteria may lead to poor patient selection; training of patients to complete accurate diary assessments; using a small number of expert sites may be better than recruiting patients from a large and diverse number of sites; smaller, better selected patient populations may be better able to demonstrate efficacy (as was seen in the US-001 study, which had less than 30 patients per arm, enrolled across ≤12 USA sites [Citation111]); care over the geographic localization of trial sites – bigger placebo responses may occur in some territories (or sites) for reasons that are not well defined. Some of these lessons may also apply to the varying success of the Phase III studies with istradefylline. It may be significant that those Phase III studies with istradefylline carried out in a single territory, namely Japan, in a smaller number of carefully chosen centers showed a consistent effect of the drug.
The development of adenosine A2A antagonists for the treatment of OFF periods has largely followed the same protocols and designs that have been used in the past for most dopaminergic drugs and adjuncts to levodopa therapy. This was supposed to ensure regulatory consistency and enable comparison across the spectrum of available treatments through the use of comparable markers and endpoints. However, the adenosine antagonists represent a novel non-dopaminergic approach to the treatment of PD and that may require a different approach to their clinical evaluation. Indeed, while the initial development and adoption of the UPDRS as a unified scale were largely driven by dopamine agonist development [Citation150], the istradefylline clinical program was one of the first to adopt the ‘Hauser’ patient diaries, which differentiate between troublesome and non-troublesome dyskinesia.
The expectation that the full potential of adenosine antagonists will be seen in those later stage patients who are already optimized to any of the available dopaminergic treatments, but show suboptimal clinical benefit, goes in the face of the preclinical profile of these compounds. Preclinical studies clearly indicate that the greatest effect on motor function is seen when A2A adenosine antagonists are administered in combination with low or threshold doses of levodopa and this may represent the most appropriate design for subsequent clinical studies and the most effective use of A2A adenosine antagonists in the general PD patient population.
13. Expert opinion
The use of istradefylline in the treatment of PD is currently limited to the approved indication of the treatment of ‘wearing-off’, as demonstrated by the results of randomized clinical trials and evaluation by Japanese and US regulatory authorities and our experience thus far is that it provides a useful option in the PD armamentarium for this purpose. Since istradefylline is a first-in-class molecule, only experience of using the drug in real-world clinical practice will reveal the overall effectiveness of an adenosine A2A antagonist in the treatment of PD. Indeed, experience from Japan shows that the drug may have efficacy in the treatment of both motor and non-motor components of PD that are currently poorly controlled by current therapies. Examples cited above include gait and ataxia, neuropsychiatric components of PD and sleep disturbance as well as urinary dysfunction [Citation136–146]. While the signals of effect in these specific areas are tantalizing, there needs to be specific, large-scale, randomized and blinded clinical evaluations of istradefylline in a series of Phase IV studies to confirm the results of early, small-scale open studies. However, it is not unlikely that this novel drug class will bring additional benefit to the PD patient population considering the localization of the target A2A receptor population and the specificity of the pharmacological action of A2A receptor antagonists.
Evaluation of istradefylline so far has taken place in classically designed studies of the effect of the drug on later stage PD patients with motor fluctuations optimized for all currently available dopaminergic medications using classical and regulatory necessary endpoints. At this point, there has yet to be exploration of the effects of istradefylline in earlier PD where it might be used to avoid increasing levodopa dosage and the onset of common dopaminergic side-effects, as suggested by findings in preclinical studies with the drug. In animal models, there is a consensus that a more robust effect of istradefylline in improving motor function is produced when it is combined with low or threshold doses of levodopa rather than with high doses producing maximal dopaminergic improvement. Examination in a ‘levodopa sparing’ strategy in early PD would seem warranted.
13.1. Five-year view
Further real-world clinical experience with istradefylline will identify the point in the illness where it exerts most effect, the nature of the patient population in which it is most effective and the signs and symptoms of PD that are most amenable to treatment with an adenosine A2A antagonist. In addition, it is highly probable that within 5 years, istradefylline and other adenosine antagonists will have been studied in disorders of movement other than PD and for a range of neuro-psychiatric disorders, including cognition, and anxiety and depression, based on the extensive preclinical literature that exists.
So far, there has been relatively little experience using istradefylline and other A2A antagonists as a monotherapy treatment for the early stages of PD disease and istradefylline did not show significant improvement in UPDRS Part III (motor scores) [Citation115]. Thus, it is unlikely that this will be explored further using istradefylline itself. However, another molecule (KW-6356) has already advanced through its preclinical stages showing a high affinity and selectivity for A2A receptors and has entered clinical development. In early Phase II studies undertaken in Japan, this molecule has shown effectiveness both as a monotherapy [Citation134] and in combination with levodopa [Citation151], showing an improvement in MDS-UPDRS part III scores. Therefore, there may be the potential for early use and expanded utility of adenosine A2A antagonists in PD. A 5-year view would be that istradefylline will be used for the treatment of the later stages of PD as an adjunct to dopaminergic therapy while it is anticipated that other adenosine A2A antagonists in development will be tested at different stages of the disease.
Perhaps the most intriguing step forward that could potentially occur in the next 5 years would the development of adenosine A2A antagonists as neuroprotective or disease-modifying therapies for PD. This represents the greatest challenge and unmet need for PD. To date, nothing has been shown to be effective in controlled clinical trials despite encouraging preclinical evidence of effect. Based on the increasingly clear evidence that caffeine intake reduces the risk of developing PD [Citation152], there is significant interest in a neuroprotective effect of adenosine A2A antagonists. This should be nuanced by pointing out that caffeine is a nonselective adenosine receptor antagonist, having actions on both A1 and A2 receptors and that it also exerts other biochemical effects, for example inhibition of phosphodiesterase. However, preclinical studies have consistently linked A2A receptors to the neurodegenerative process in both in vitro and in vivo models of PD using genetic knockouts and pharmacological manipulation with a range of A2A antagonists [Citation153–157]. These have also suggested that the mechanism through which A2A receptor blockade leads to neuroprotection is different from that involved in controlling motor function. The exact mechanism (or mechanisms) through which neuroprotection is induced remains to be resolved. Among those mechanisms which appear relevant are the control of glutamate release to limit excitotoxicity [Citation158], effects on glial cells and inflammatory mechanisms [Citation159–161], alterations in mitochondrial function and actions affecting protein handling at the level of the proteasome and autophagy pathways [Citation162–165] and regulation of the handling and toxicity of α-synuclein via synuclein phosphorylation [Citation166]. These observations may also have implications beyond PD. For example, postmortem studies of Alzheimer’s disease indicate that increases in astrocytic A2A receptor levels correlate with cognitive impairment and pathological change, and this finding is also observed in aging mice expressing human amyloid precursor protein (hAPP) [Citation167]. In this mouse model of the disease, istradefylline treatment enhanced spatial memory and habituation, suggesting that A2A receptor antagonists might help counteract memory problems with amyloid pathology [Citation168].
There is already some preclinical evidence to link adenosine A2A antagonism to a potential neuroprotective effect in PD. In MPTP-treated mice, istradefylline attenuated the toxin-induced decrease in striatal dopamine content [Citation153,Citation169,Citation170] – an effect not seen following administration of an A1 antagonist [Citation170]. In addition, there was a preservation of striatal presynaptic dopaminergic terminals and tyrosine hydroxylase activity. Similarly, in 6-OHDA lesioned rats, istradefylline reduced the loss of striatal dopaminergic terminals, but was also shown to protect tyrosine positive nigral cells against toxin action [Citation153]. These actions mirror those seen with caffeine and a range of other adenosine A2A antagonists and warrant further study in both animals and in humans.
The introduction of istradefylline as a first-in-class drug for use in PD has opened a new era in the pharmacological treatment of the illness. With this comes the promise of a change in expectations of the outcome of therapy and a lessening of side-effect potential that will be seen over the next 5 years – as a non-dopaminergic approach avoids the inevitability of the classic dopaminergic profile. In particular, it is evident that istradefylline has a favorable risk-benefit profile since the safety and adverse event profiles of the drug are unremarkable. The long and slow development of istradefylline has also yielded valuable lessons on how to develop a novel non-dopaminergic molecule for PD and shown that this should not necessarily follow the classic pathway used for dopaminergic therapies as these do not encompass the pharmacology and range of action specific to adenosine A2A antagonists. These experiences will guide the design of clinical trials for other novel molecules in PD and may of necessity lead to a change in the requirements and expectations of regulatory authorities.
Article highlights
The translation of laboratory experience, based on the emerging understanding of motor pathways downstream from the striatum, led to the development of selective adenosine A2A receptor antagonists for advanced Parkinson disease
Istradefylline has high selectivity for blocking the adenosine A2A receptor and exerts virtually no clinically relevant off-target neurochemical effects
The istradefylline clinical trial development program focused on lessening OFF time when used as an adjunct to levodopa with or without other anti-parkinsonian drugs.
Based on multiple clinical trials, istradefylline has been approved for marketing in Japan and the USA for the management of motor fluctuations (‘wearing-off’ and ‘OFF episodes’, respectively).
Reviewer disclosures
Peer reviewers in this manuscript have no relevant financial or other relationships to disclose.
Declaration of interest
Peter Jenner has received honoraria for consultancy and advisory boards from Kyowa Kirin Co. Akihisa Mori is employed by Kyowa Kirin Co., Ltd. Robert A Hauser reports consultancy/advisory boards for Kyowa Kirin Pharmaceuticals and has been a principal investigator in clinical trials of istradefylline. Stephen Aradi has nothing to report in relation to this work. No author has received specific remuneration for this work.
Acknowledgments
The authors thank Tomoyuki Kanda and Mayumi Saki (Kyowa Kirin) for their assistance with the literature review as well as Anita Chadha-Patel and Elizabeth Hocking of ACP Clinical Communications Ltd (funded by Kyowa Kirin Inc.) for medical writing support (literature searching, referencing and editing) in the development of this report.
Additional information
Funding
References
- Fox SH, Katzenschlager R, Lim SY, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of parkinson’s disease. Mov Disord. 2018;33:1248–1266.
- Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology. 2009;72:S1–136.
- Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of parkinson’s disease. N Engl J Med. 2004;351:2498–2508.
- Aquino CC, Fox SH. Clinical spectrum of levodopa-induced complications. Mov Disord. 2015;30:80–89.
- Gonzalez-Latapi P, Bhowmick SS, Saranza G, et al. Non-dopaminergic treatments for motor control in parkinson’s disease: an update. CNS Drugs. 2020;34:1025-1044.
- Hung AY, Schwarzschild MA. Treatment of parkinson’s disease: what’s in the non-dopaminergic pipeline? Neurotherapeutics. 2014;11:34–46.
- Lang AE, Obeso JA. Time to move beyond nigrostriatal dopamine deficiency in parkinson’s disease. Ann Neurol. 2004;55:761–765.
- Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis. 2017;7:S71–S85.
- Obeso JA, Marin C, Rodriguez-Oroz C, et al. The basal ganglia in parkinson’s disease: current concepts and unexplained observations. Ann Neurol. 2008;64(Suppl 2):S30–46.
- Kalia LV, Brotchie JM, Fox SH. Novel nondopaminergic targets for motor features of parkinson’s disease: review of recent trials. Mov Disord. 2013;28:131–144.
- Duty S, Jenner P. Animal models of parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol. 2011;164:1357–1391.
- Compta Y, Tolosa E. Anticholinergic medications. In: Koller WC, Melamed E, editors. Handbook of Clinical Neurology. Vol. 84. Amsterdam, Netherlands. Elsevier; 2007. p. 121–125.
- Crispo JAG, Willis AW, Thibault DP, et al. Associations between anticholinergic burden and adverse health outcomes in Parkinson disease. PLoS One. 2016;11:e0150621.
- Kong M, Ba M, Ren C, et al. An updated meta-analysis of amantadine for treating dyskinesia in parkinson’s disease. Oncotarget. 2017;8:57316–57326.
- Amantadine and other antiglutamate agents: management of Parkinson’s disease. Mov Disord. 2002;17(Suppl 4):S13–22.
- Pahwa R, Tanner CM, Hauser RA, et al. ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (ease lid study): a randomized clinical trial. JAMA Neurol. 2017;74:941–949.
- Takahashi T, Yamashita H, Zhang YX, et al. Inhibitory effect of MK-801 on amantadine-induced dopamine release in the rat striatum. Brain Res Bull. 1996;41:363–367.
- Mizoguchi K, Yokoo H, Yoshida M, et al. Amantadine increases the extracellular dopamine levels in the striatum by re-uptake inhibition and by N-methyl-D-aspartate antagonism. Brain Res. 1994;662:255–258.
- Peeters M, Page G, Maloteaux JM, et al. Hypersensitivity of dopamine transmission in the rat striatum after treatment with the NMDA receptor antagonist amantadine. Brain Res. 2002;949:32–41.
- Gianutsos G, Chute S, Dunn JP. Pharmacological changes in dopaminergic systems induced by long-term administration of amantadine. Eur J Pharmacol. 1985;110:357–361.
- Elkurd MT, Bahroo LB, Pahwa R. The role of extended-release amantadine for the treatment of dyskinesia in parkinson’s disease patients. Neurodegener Dis Manage. 2018;8:73–80.
- Bermejo PE, Anciones B. A review of the use of zonisamide in parkinson’s disease. Ther Adv Neurol Disord. 2009;2:313–317.
- Iwaki H, Tagawa M, Iwasaki K, et al. Comparison of zonisamide with non-levodopa, anti-parkinson’s disease drugs in the incidence of parkinson’s disease-relevant symptoms. J Neurol Sci. 2019;402:145–152.
- Sonsalla PK, Wong LY, Winnik B, et al. The antiepileptic drug zonisamide inhibits MAO-B and attenuates MPTP toxicity in mice: clinical relevance. Exp Neurol. 2010;221:329–334.
- Yamamura S, Ohoyama K, Nagase H, et al. Zonisamide enhances delta receptor-associated neurotransmitter release in striato-pallidal pathway. Neuropharmacology. 2009;57:322–331.
- Tohgi H, Abe T, Takahashi S. The effects of L-threo-3,4-dihydroxyphenylserine on the total norepinephrine and dopamine concentrations in the cerebrospinal fluid and freezing gait in parkinsonian patients. J Neural Transm. 1993;5:27–34.
- Sahli ZT, Tarazi FI. Pimavanserin: novel pharmacotherapy for parkinson’s disease psychosis. Expert Opin Drug Discov. 2018;13:103–110.
- Kianirad Y, Simuni T. Pimavanserin, a novel antipsychotic for management of parkinson’s disease psychosis. Expert Rev Clin Pharmacol. 2017;10:1161–1168.
- Li Z, Yu Z, Zhang J, et al. Impact of rivastigmine on cognitive dysfunction and falling in parkinson’s disease patients. Eur Neurol. 2015;74:86–91.
- Henderson EJ, Lord SR, Close JC, et al. The respond trial–rivastigmine to stabilise gait in parkinson’s disease a phase II, randomised, double blind, placebo controlled trial to evaluate the effect of rivastigmine on gait in patients with parkinson’s disease who have fallen. BMC Neurol. 2013;13:188.
- Milardi D, Quartarone A, Bramanti A, et al. The cortico-basal ganglia-cerebellar network: past, present and future perspectives. Front Syst Neurosci. 2019;13:61.
- Nutt JG, Bohnen NI. Non-dopaminergic therapies. J Parkinsons Dis. 2018;8:S73–S78.
- Haskó G, Pacher P, Vizi ES, et al. Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci. 2005;26:511–516.
- Parkinson FE, Xiong W, Zamzow CR. Astrocytes and neurons: different roles in regulating adenosine levels. Neurological Res. 2005;27:153–160.
- Klotz KN, Hessling J, Hegler J, et al. Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1–9.
- Fredholm BB, Abbracchio MP, Burnstock G, et al. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143–156.
- Jarvis MF, Williams M. Direct autoradiographic localization of adenosine A2 receptors in the rat brain using the A2-selective agonist, [3H]CGS 21680. Eur J Pharmacol. 1989;168:243–246.
- Rosin DL, Robeva A, Woodard RL, et al. Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J Comp Neurol. 1998;401:163–186.
- Svenningsson P, Hall H, Sedvall G, et al. Distribution of adenosine receptors in the postmortem human brain: an extended autoradiographic study. Synapse. 1997;27:322–335.
- Ishiwata K, Mishina M, Kimura Y, et al. First visualization of adenosine A2A receptors in the human brain by positron emission tomography with [11C]TMSX. Synapse. 2005;55:133–136.
- Ferré S, Fredholm BB, Morelli M, et al. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997;20:482–487.
- Mori A, Shindou T. Modulation of Gabaergic transmission in the striatopallidal system by adenosine A2A receptors: a potential mechanism for the antiparkinsonian effects of A2A antagonists. Neurology. 2003;61:S44–8.
- Jenner P, Mori A, Hauser R, et al. Adenosine, adenosine A2A antagonists, and parkinson’s disease. Parkinsonism Relat Disord. 2009;15:406–413.
- Cui G, Jun SB, Jin X, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242.
- Schiffmann SN, Jacobs O, Vanderhaeghen JJ. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J Neurochem. 1991;57:1062–1067.
- Ena SL, De Backer JF, Schiffmann SN, et al. FACS array profiling identifies ecto-5ʹ nucleotidase as a striatopallidal neuron-specific gene involved in striatal-dependent learning. J Neurosci. 2013;33:8794–8809.
- Augusto E, Matos M, Sévigny J, et al. Ecto-5ʹ-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci. 2013;33:11390–11399.
- Kuwana Y, Shiozaki S, Kanda T, et al. Antiparkinsonian activity of adenosine A2A antagonists in experimental models. Adv Neurol. 1999;80:121–123.
- Kanda T, Jackson MJ, Smith LA, et al. Combined use of the adenosine A2A antagonist KW-6002 with L-DOPA or with selective D1 or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys. Exp Neurol. 2000;162:321–327.
- Hodgson RA, Bertorelli R, Varty GB, et al. Characterization of the potent and highly selective A2A receptor antagonists preladenant and SCH 412348 [7-[2-[4-2,4-difluorophenyl]-1-piperazinyl]ethyl]-2-(2-furanyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine] in rodent models of movement disorders and depression. J Pharmacol Exp Ther. 2009;330:294–303.
- Hauser RA, Cantillon M, Pourcher E, et al. Preladenant in patients with parkinson’s disease and motor fluctuations: a phase 2, double-blind, randomised trial. Lancet Neurol. 2011;10:221–229.
- Factor SA, Wolski K, Togasaki DM, et al. Long-term safety and efficacy of preladenant in subjects with fluctuating Parkinson’s disease. Mov Disord. 2013;28:817–820.
- Michel A, Downey P, Van Damme X, et al. Behavioural assessment of the A2A/NR2B combination in the unilateral 6-OHDA-lesioned rat model: a new method to examine the therapeutic potential of non-dopaminergic drugs. PLoS One. 2015;10:e0135949.
- Michel A, Nicolas JM, Rose S, et al. Antiparkinsonian effects of the “Radiprodil and Tozadenant” combination in MPTP-treated marmosets. PLoS One. 2017;12:e0182887.
- Hauser RA, Olanow CW, Kieburtz KD, et al. Tozadenant (SYN115) in patients with parkinson’s disease who have motor fluctuations on levodopa: a phase 2b, double-blind, randomised trial. Lancet Neurol. 2014;13:767–776.
- Papapetropoulos S, Borgohain R, Kellett M, et al. The adenosine A2A receptor antagonist BIIB014 is effective in improving ON-time in parkinson’s disease (PD) patients with motor fluctuations. Mov Disord. 2010;25(Suppl 2):S305.
- Pinna A, Pontis S, Borsini F, et al. Adenosine A2A receptor antagonists improve deficits in initiation of movement and sensory motor integration in the unilateral 6-hydroxydopamine rat model of parkinson’s disease. Synapse. 2007;61:606–614.
- Pinna A. Adenosine A2A receptor antagonists in parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs. 2014;28:455–474.
- Aoyama S, Kase H, Borrelli E. Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist. J Neurosci. 2000;20:5848–5852.
- Ochi M, Shiozaki S, Kase H. Adenosine A2A receptor-mediated modulation of GABA and glutamate release in the output regions of the basal ganglia in a rodent model of parkinson’s disease. Neuroscience. 2004;127:223–231.
- Mori A. Chapter four - mode of action of adenosine A2a receptor antagonists as symptomatic treatment for parkinson’s disease. In: Mori A, editor. International Review of Neurobiology. Vol. 119. London, UK. Academic Press; 2014. p. 87–116.
- Mori A. How do adenosine A(2A) receptors regulate motor function? Parkinsonism Relat Disord. 2020;80(Suppl 1):S13–s20.
- Canals M, Marcellino D, Fanelli F, et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem. 2003;278:46741–46749.
- Fuxe K, Ferre S, Canals M, et al. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J Mol Neurosci. 2005;26:209–220.
- Chen JF, Moratalla R, Impagnatiello F, et al. The role of the D2 dopamine receptor (D2R) in A2A adenosine receptor (A2AR)-mediated behavioral and cellular responses as revealed by A2A and D2 receptor knockout mice. Proc Natl Acad Sci USA. 2001;98:1970–1975.
- Ramlackhansingh AF, Bose SK, Ahmed I, et al. Adenosine 2A receptor availability in dyskinetic and nondyskinetic patients with Parkinson disease. Neurology. 2011;76:1811–1816.
- Mishina M, Ishiwata K, Naganawa M, et al. Adenosine A2A receptors measured with [C]TMSX PET in the striata of parkinson’s disease patients. PLoS One. 2011;6:e17338–e17338.
- Hettinger BD, Lee A, Linden J, et al. Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of gabaergic neurons in rat striatum. J Comp Neurol. 2001;431:331–346.
- Corsi C, Melani A, Bianchi L, et al. Striatal A2A adenosine receptor antagonism differentially modifies striatal glutamate outflow in vivo in young and aged rats. Neuroreport. 2000;11:2591–2595.
- Corsi C, Melani A, Bianchi L, et al. Striatal A2A adenosine receptors differentially regulate spontaneous and K+-evoked glutamate release in vivo in young and aged rats. Neuroreport. 1999;10:687–691.
- Preston Z, Lee K, Widdowson L, et al. Adenosine receptor expression and function in rat striatal cholinergic interneurons. Br J Pharmacol. 2000;130:886–890.
- Richardson PJ, Dixon AK, Lee K, et al. Correlating physiology with gene expression in striatal cholinergic neurones. J Neurochem. 2000;74:839–846.
- Tozzi A, de Iure A, Di Filippo M, et al. The distinct role of medium spiny neurons and cholinergic interneurons in the D₂/A₂A receptor interaction in the striatum: implications for parkinson’s disease. J Neurosci. 2011;31:1850–1862.
- Schiffmann SN, Libert F, Vassart G, et al. Distribution of adenosine A2 receptor mRNA in the human brain. Neurosci Lett. 1991;130:177–181.
- Fink JS, Weaver DR, Rivkees SA, et al. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res. 1992;14:186–195.
- Augood SJ, Emson PC. Adenosine A2A receptor mRNA is expressed by enkephalin cells but not by somatostatin cells in rat striatum: a co-expression study. Brain Res Mol Brain Res. 1994;22:204–210.
- Svenningsson P, Le Moine C, Aubert I, et al. Cellular distribution of adenosine A2A receptor mRNA in the primate striatum. J Comp Neurol. 1998;399:229–240.
- Obeso JA, Rodriguez-Oroz MC, Rodriguez M, et al. Pathophysiology of the basal ganglia in parkinson’s disease. Trends Neurosci. 2000;23:S8–19.
- Schiffmann SN, Fisone G, Moresco R, et al. Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol. 2007;83:277–292.
- Calon F, Dridi M, Hornykiewicz O, et al. Increased adenosine A2A receptors in the brain of parkinson’s disease patients with dyskinesias. Brain. 2004;127:1075–1084.
- Mishina M, Ishiwata K. Adenosine receptor PET imaging in human brain. Int Rev Neurobiol. 2014;119:51–69.
- Correa M, Wisniecki A, Betz A, et al. The adenosine A2A antagonist KF17837 reverses the locomotor suppression and tremulous jaw movements induced by haloperidol in rats: possible relevance to parkinsonism. Behav Brain Res. 2004;148:47–54.
- Shiozaki S, Ichikawa S, Nakamura J, et al. Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacol (Berl). 1999;147:90–95.
- Hauber W, Neuscheler P, Nagel J, et al. Catalepsy induced by a blockade of dopamine D1 or D2 receptors was reversed by a concomitant blockade of adenosine A2A receptors in the caudate-putamen of rats. Eur J Neurosci. 2001;14:1287–1293.
- Salamone JD. Preladenant, a novel adenosine A2A receptor antagonist for the potential treatment of parkinsonism and other disorders. Drugs. 2010;13:723–731.
- Collins LE, Sager TN, Sams AG, et al. The novel adenosine A2A antagonist Lu AA47070 reverses the motor and motivational effects produced by dopamine D2 receptor blockade. Pharmacol Biochem Behavior. 2012;100:498–505.
- Kanda T, Shiozaki S, Shimada J, et al. KF17837: a novel selective adenosine A2A receptor antagonist with anticataleptic activity. Eur J Pharmacol. 1994;256:263–268.
- Rose S, Ramsay Croft N, Jenner P. The novel adenosine A2A antagonist ST1535 potentiates the effects of a threshold dose of l-dopa in unilaterally 6-OHDA-lesioned rats. Brain Res. 2007;1133:110–114.
- Koga K, Kurokawa M, Ochi M, et al. Adenosine A2A receptor antagonists KF17837 and KW-6002 potentiate rotation induced by dopaminergic drugs in hemi-Parkinsonian rats. Eur J Pharmacol. 2000;408:249–255.
- Fenu S, Pinna A, Ongini E, et al. Adenosine A2A receptor antagonism potentiates L-DOPA-induced turning behaviour and c-fos expression in 6-hydroxydopamine-lesioned rats. Eur J Pharmacol. 1997;321:143–147.
- Ferré S, Rubio A, Fuxe K. Stimulation of adenosine A2 receptors induces catalepsy. Neurosci Lett. 1991;130:162–164.
- Rose S, Jackson MJ, Smith LA, et al. The novel adenosine A2A receptor antagonist ST1535 potentiates the effects of a threshold dose of L-DOPA in MPTP treated common marmosets. Eur J Pharmacol. 2006;546:82–87.
- Grondin R, Bedard PJ, Hadj Tahar A, et al. Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys. Neurology. 1999;52:1673–1677.
- Hodgson RA, Bedard PJ, Varty GB, et al. Preladenant, a selective A2A receptor antagonist, is active in primate models of movement disorders. Exp Neurol. 2010;225:384–390.
- Kanda T, Tashiro T, Kuwana Y, et al. Adenosine A2A receptors modify motor function in MPTP-treated common marmosets. Neuroreport. 1998;9:2857–2860.
- Kanda T, Jackson MJ, Smith LA, et al. Adenosine A2A antagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol. 1998;43:507–513.
- Uchida S, Tashiro T, Kawai-Uchida M, et al. Adenosine A2A-receptor antagonist istradefylline enhances the motor response of L-DOPA without worsening dyskinesia in MPTP-treated common marmosets. J Pharmacol Sci. 2014;124:480–485.
- Uchida S, Soshiroda K, Okita E, et al. The adenosine A2A receptor antagonist, istradefylline enhances anti-parkinsonian activity induced by combined treatment with low doses of L-DOPA and dopamine agonists in MPTP-treated common marmosets. Eur J Pharmacol. 2015;766:25–30.
- Jones N, Bleickardt C, Mullins D, et al. A2A receptor antagonists do not induce dyskinesias in drug-naive or L-dopa sensitized rats. Brain Res Bull. 2013;98:163–169.
- Tronci E, Simola N, Borsini F, et al. Characterization of the antiparkinsonian effects of the new adenosine A2A receptor antagonist ST1535: acute and subchronic studies in rats. Eur J Pharmacol. 2007;566:94–102.
- Lundblad M, Vaudano E, Cenci MA. Cellular and behavioural effects of the adenosine A2A receptor antagonist KW-6002 in a rat model of l-DOPA-induced dyskinesia. J Neurochem. 2003;84:1398–1410.
- Bibbiani F, Oh JD, Petzer JP, et al. A2A antagonist prevents dopamine agonist-induced motor complications in animal models of Parkinson’s disease. Exp Neurol. 2003;184:285–294.
- Jenner P. Istradefylline, a novel adenosine A2A receptor antagonist, for the treatment of Parkinson’s disease. Expert Opin Invest Drugs. 2005;14:729–738.
- Dungo R, Deeks ED. Istradefylline: first global approval. Drugs. 2013;73:875–882.
- Saki M, Yamada K, Koshimura E, et al. In vitro pharmacological profile of the A2A receptor antagonist istradefylline. Naunyn Schmiedebergs Arch Pharmacol. 2013;386:963–972.
- Bekar L, Libionka W, Tian GF, et al. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med. 2008;14:75–80.
- Boswell-Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. Brit J Pharmacol. 2006;147(Suppl 1):S252–S257.
- Knebel W, Rao N, Uchimura T, et al. Population pharmacokinetic analysis of istradefylline in healthy subjects and in patients with parkinson’s disease. J Clin Pharmacol. 2011;51:40–52.
- Müller T. Suitability of the adenosine antagonist istradefylline for the treatment of parkinson’s disease: pharmacokinetic and clinical considerations. Expert Opin Drug Metab Toxicol. 2013;9:1015–1024.
- NOURIANZ™ (istradefylline) tablets, for oral use. US Prescribing information. Available at https://www.nourianz.com/assets/pdf/nourianz-full-prescribing-information.pdf. Last accessed February 1, 2021.
- Hauser RA, Hubble JP, Truong DD. Randomized trial of the adenosine A2A receptor antagonist istradefylline in advanced PD. Neurology. 2003;61:297–303. .
- Bara-Jimenez W, Sherzai A, Dimitrova T, et al. Adenosine A2A receptor antagonist treatment of parkinson’s disease. Neurology. 2003;61:293–296.
- Poewe W. The role of COMT inhibition in the treatment of parkinson’s disease. Neurology. 2004;62:S31–8.
- Hauser RA, Friedlander J, Zesiewicz TA, et al. A home diary to assess functional status in patients with parkinson’s disease with motor fluctuations and dyskinesia. Clin Neuropharmacol. 2000;23:75–81.
- Fernandez HH, Greeley DR, Zweig RM, et al. Istradefylline as monotherapy for Parkinson disease: results of the 6002-US-051 trial. Parkinsonism Relat Disord. 2010;16:16–20.
- LeWitt PA, Guttman M, Tetrud JW, et al. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol. 2008;63:295–302.
- Stacy M, Silver D, Mendis T, et al. A 12-week, placebo-controlled study (6002-US-006) of istradefylline in Parkinson disease. Neurology. 2008;70(23):2233–2240. .
- Hauser RA, Shulman LM, Trugman JM, et al. Study of istradefylline in patients with parkinson’s disease on levodopa with motor fluctuations. Mov Disord. 2008;23:2177–2185.
- Pourcher E, Fernandez HH, Stacy M, et al. Istradefylline for Parkinson’s disease patients experiencing motor fluctuations: results of the KW-6002-US-018 study. Parkinsonism Related Disord. 2012;18(2):178–184. .
- Kyowa Hakko Kirin UK L. A 16-week, Double-Blind, Placebo-Controlled, Randomised, Parallel-Group, Multicentre, International Study to Evaluate the Efficacy and Safety of 40 mg/Day KW-6002 (Istradefylline) and That of Entacapone Versus Placebo as Treatment for Parkinson’s Disease in Patients With Motor Response Complications on Levodopa Therapy. 2005. Available from: https://clinicaltrials.gov/ct2/show/study/NCT00199394. Last accessed February 1, 2021.
- Mizuno Y, Hasegawa K, Kondo T, et al. Clinical efficacy of istradefylline (KW-6002) in parkinson’s disease: a randomized, controlled study. Mov Disord. 2010;25:1437–1443.
- Mizuno Y, Kondo T. Japanese istradefylline study g. adenosine A2A receptor antagonist istradefylline reduces daily OFF time in parkinson’s disease. Mov Disord. 2013;28:1138–1141.
- Kyowa Hakko Kirin Pharma I. A Phase 3, 12-week, Double-Blind, Placebo-Controlled, Randomized, Multicenter Study to Evaluate the Efficacy of Oral Istradefylline 20 and 40 mg/Day as Treatment for Subjects With Moderate to Severe Parkinson’s Disease 2016. Available from: https://www.clinicaltrials.gov/ct2/show/NCT01968031. Last accessed February 1, 2021.
- Kondo T, Mizuno Y. A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin Neuropharmacol. 2015;38:41–46.
- Factor S, Mark MH, Watts R, et al. A long-term study of istradefylline in subjects with fluctuating parkinson’s disease. Parkinsonism Related Disord. 2010;16:423–426.
- Center for drug evaluation and research. Clinical Review: NDA 022075. Available at https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/022075Orig1s000MedR.pdf. Last accessed November 2020. Last accessed February 1, 2021.
- Takahashi M, Fujita M, Asai N, et al. Safety and effectiveness of istradefylline in patients with parkinson’s disease: interim analysis of a post-marketing surveillance study in japan. Expert Opin Pharmacother. 2018;19:1635–1642.
- Kyowa Hakko Kirin Co. L. Kyowa Hakko Kirin Initiates a Global Phase 3 Trial of Istradefylline (KW-6002) for Parkinson’s Disease 2013. Available from: https://www.kyowakirin.com/media_center/news_releases/2013/e20131121_01.html. Last accessed February 1, 2021.
- Kyowa Hakko Kirin Announces Top-Line Results of Global Phase 3 Trial of KW-6002 (Istradefylline) for Parkinson’s Disease [Internet]. 2016; December 13. Available from: https://www.kyowakirin.com/media_center/news_releases/2016/pdf/e20161213_01.pdf. Last accessed Jan 2021.
- Lewitt P, Aradi S, Hauser RA, et al. The challenge of developing adenosine A2 A antagonists for Parkinson disease: istradefylline, preladenant, and tozadenant. Parkinsonism Relat Disord. 2020;80(Supp 1):S54–S63.
- Hauser RA, Stocchi F, Rascol O, et al. Preladenant as an adjunctive therapy with levodopa in Parkinson disease: two randomized clinical trials and lessons learned. JAMA Neurol. 2015;72:1491–1500.
- Center for drug evaluation and research. Summary Review: 022075Orig1s000. Available at https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/022075Orig1s000SumR.pdf. Last accessed February 1, 2021.
- Kyowa Kirin Announces FDA Approval of NOURIANZ™ (istradefylline) for Use in Parkinson’s Disease [Internet]. 2019; August 28. Available from: https://www.kyowakirin.com/media_center/news_releases/2019/e20190828_01.html. Last accessed Jan 2021.
- Kyowa Kirin Announces Marketing Authorisation Application for Istradefylline Validated by European Medicines Agency. Available at https://www.kyowakirin.com/media_center/news_releases/2020/e20200106_01.html. Last accessed February 1, 2021.
- Jenner P, Mori A, Kanda T. Can adenosine A2A receptor antagonists be used to treat cognitive impairment, depression or excessive sleepiness in Parkinson’s disease? Parkinsonism Relat Disord. 2020;80(Supp 1):S28–S36.
- Abe K, Fujita M, Yoshikawa H. Effectiveness of istradefylline for fatigue and quality of life in parkinson’s disease patients’ and of their caregivers’. Adv Parkinsons Dis. 2016;5:24–28.
- Nagayama H, Kano O, Murakami H, et al. Effect of istradefylline on mood disorders in Parkinson’s disease. J Neurol Sci. 2019;396:78–83.
- Ito H, Fukutake S, Kamei T, et al. Clinical efficacy of istradefylline for depression in parkinson’s disease. J Neurol Neurosci. 2018;9:261.
- Kitta T, Yabe I, Kanno Y, et al. Long-term outcome of adenosine A2a receptor antagonist on lower urinary tract symptoms in male Parkinson disease patients. Clin Neuropharmacol. 2018;41:98–102.
- Kitta T, Yabe I, Takahashi I, et al. Clinical efficacy of istradefylline on lower urinary tract symptoms in parkinson’s disease. Int J Urol. 2016;23:893–894.
- Matsuura K, Kajikawa H, Tabei KI, et al. The effectiveness of istradefylline for the treatment of gait deficits and sleepiness in patients with parkinson’s disease. Neurosci Lett. 2018;662:158–161.
- Suzuki K, Miyamoto M, Miyamoto T, et al. Istradefylline improves daytime sleepiness in patients with parkinson’s disease: an open-label, 3-month study. J Neurol Sci. 2017;380:230–233.
- Suzuki K, Miyamoto T, Miyamoto M, et al. Could istradefylline be a treatment option for postural abnormalities in mid-stage parkinson’s disease? J Neurol Sci. 2018;385:131–133.
- Iijima M, Orimo S, Terashi H, et al. Efficacy of istradefylline for gait disorders with freezing of gait in parkinson’s disease: a single-arm, open-label, prospective, multicenter study. Exp Opin Pharmacother. 2019;20:1405–1411.
- Fujioka S, Yoshida R, Nose K, et al. A new therapeutic strategy with istradefylline for postural deformities in parkinson’s disease. Neurol Neurochir Pol. 2019;53:291–295.
- Kataoka H, Sugie K. Does istradefylline really have a dystonic mechanism? J Neurol Sci. 2018;388:233–234.
- Huang ZL, Zhang Z, Qu WM. Roles of adenosine and its receptors in sleep-wake regulation. Int Rev Neurobiol. 2014;119:349–371.
- French IT, Muthusamy KA. A review of the pedunculopontine nucleus in parkinson’s disease. Front Aging Neurosci. 2018;10:99.
- Chaudhuri KR, Yates L, Martinez-Martin P. The non-motor symptom complex of parkinson’s disease: a comprehensive assessment is essential. Curr Neurol Neurosci Rep. 2005;5:275–283.
- Mitchell SL, Harper DW, Lau A, et al. Patterns of outcome measurement in parkinson’s disease clinical trials. Neuroepidemiology. 2000;19:100–108.
- Kyowa Kirin Announces Positive Phase 2b Results for KW-6356 in Patients with Parkinson’s Disease [Internet]. 2020. Available from: https://www.kyowakirin.com/media_center/news_releases/2020/pdf/e20201021_01.pdf. Last accessed February 1, 2021.
- Hong CT, Chan L, Bai C-H. The effect of caffeine on the risk and progression of parkinson’s disease: a meta-analysis. Nutrients. 2020;12:1860.
- Ikeda K, Kurokawa M, Aoyama S, et al. Neuroprotection by adenosine A2A receptor blockade in experimental models of parkinson’s disease. J Neurochem. 2002;80:262–270.
- Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Sig. 2005;1:111–134.
- Kachroo A, Schwarzschild MA. Adenosine A2A receptor gene disruption protects in an alpha-synuclein model of parkinson’s disease. Ann Neurol. 2012;71:278–282.
- Hu QD, Ren XP, Liu Y, et al. Aberrant adenosine A2A receptor signaling contributes to neurodegeneration and cognitive impairments in a mouse model of synucleinopathy. Exp Neurol. 2016;283:213–223.
- Chen JF, Schwarzschild MA. Do caffeine and more selective adenosine A2A receptor antagonists protect against dopaminergic neurodegeneration in parkinson’s disease? Parkinsonism Relat Disord. 2020;80(Supp 1):S45–S53.
- Dai SS, Zhou YG, Li W, et al. Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. J Neurosci. 2010;30:5802–5810.
- Brambilla R, Cottini L, Fumagalli M, et al. Blockade of A2A adenosine receptors prevents basic fibroblast growth factor-induced reactive astrogliosis in rat striatal primary astrocytes. Glia. 2003;43:190–194.
- Boison D, Chen JF, Fredholm BB. Adenosine signaling and function in glial cells. Cell Death Diff. 2010;17:1071–1082.
- Hui CW, Zhang Y, Herrup K. Non-neuronal cells are required to mediate the effects of neuroinflammation: results from a neuron-enriched culture system. PLoS One. 2016;11:e0147134.
- Webb JL, Ravikumar B, Atkins J, et al. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013.
- Cuervo AM, Stefanis L, Fredenburg R, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295.
- Sinha RA, Farah BL, Singh BK, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59:1366–1380.
- Liu YW, Yang T, Zhao L, et al. Activation of Adenosine 2A receptor inhibits neutrophil apoptosis in an autophagy-dependent manner in mice with systemic inflammatory response syndrome. Sci Rep. 2016;6:33614.
- Ferreira DG, Batalha VL, Vicente Miranda H, et al. Adenosine A2A receptors modulate alpha-synuclein aggregation and toxicity. Cereb Cortex. 2017;27:718–730.
- Orr AG, Hsiao EC, Wang MM, et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat Neurosci. 2015;18:423–434.
- Orr AG, Lo I, Schumacher H, et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis. 2018;110:29–36.
- Pierri M, Vaudano E, Sager T, et al. KW-6002 protects from MPTP induced dopaminergic toxicity in the mouse. Neuropharmacology. 2005;48:517–524.
- Chen JF, Xu K, Petzer JP, et al. Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of parkinson’s disease. J Neurosci. 2001;21:Rc143.