
ABSTRACT
Background
Enzyme replacement therapy in Fabry disease has been available in Japan since 2004. Two post-authorization safety studies were conducted to evaluate agalsidase beta in Japanese patients with Fabry disease in real-world practice.
Research Design and Methods
The Special Drug Use Investigation monitored the long-term safety and efficacy of agalsidase beta, and the Drug Use Investigation monitored safety in patients not participating in the Special Drug Use Investigation. Safety and efficacy evaluations included adverse drug reactions (ADRs), infusion-associated reactions and hypersensitivity reactions, and change in blood GL-3 level over time.
Results
Of 396 patients in the aggregated data set, safety and efficacy analysis sets comprised 307 and 196 patients, respectively. ADRs occurred in 93 (30.3%) patients and serious ADRs occurred in 25 (8.1%) patients, with general disorders and administration site conditions (n=55, 17.9%), nervous system disorders (n=30, 9.8%) and skin and subcutaneous tissue disorders (n=23, 7.5%) the most common. Reductions in blood GL-3 levels occurred over the study, irrespective of age or disease phenotype.
Conclusions
Agalsidase beta demonstrated acceptable safety and tolerability, with sustained reductions in blood GL-3 levelsin Japanese patients with Fabry disease in real-world clinical practice
Clinical Trial Registration
NCT00233870/AGAL03004 (Special Drug Use Investigation of Agalsidase beta)
1. Introduction
Fabry disease is a rare, X-linked, inherited lysosomal storage disorder caused by mutations in the GLA gene, which results in partial or complete deficiency of the lysosomal enzyme, α-galactosidase A (α-GAL) [Citation1]. Deficiency of this enzyme leads to progressive accumulation of cellular glycosphingolipids, predominantly globotriaosylceramide (GL-3), in plasma and various body tissues, and severe multisystem effects [Citation2,Citation3]. Premature death occurs due to end-stage renal disease and cardiovascular and cerebrovascular complications [Citation2].
Clinical manifestations of Fabry disease are variable, depending on the degree of deficiency [Citation4], and may be used to characterize the disease into three distinct phenotypes [Citation5]. The more severe classical phenotype, which arises due to total or near-total loss of α-GAL function [Citation6], typically presents in childhood, and most commonly affects males [Citation7]. Neuropathic pain in the extremities, sweating abnormalities, cornea verticillata, and angiokeratoma represent the earliest obvious manifestations of the disease [Citation3,Citation8,Citation9]. However, some degree of underlying irreversible organ damage is already expected at this point [Citation10,Citation11]. Severe, life-threatening complications begin to emerge in adulthood, including renal failure, cardiac disorders (cardiomyopathy, arrhythmia), and cerebrovascular events (e.g. stroke) [Citation12,Citation13].
In contrast, patients with Fabry disease other than the classic phenotype, also referred to as late-onset or atypical variant, possess residual enzyme activity, and tend to lack the early classic symptoms of the disease [Citation14]. Rather, symptoms typically develop later in life, with predominant cardiac and/or renal manifestations [Citation5]. Although Fabry disease is X-linked, heterozygous females may also be affected. However, symptoms are particularly variable due to random X inactivation, and may range from asymptomatic disease to a more severe, classic disease course [Citation5].
The number of therapies available for patients with lysosomal storage diseases has expanded considerably in the past several years [Citation15,Citation16]. Enzyme replacement therapy (ERT) represents the mainstay of treatment for Fabry Disease, with two different human recombinant α-GAL ERTs developed for Fabry disease. Agalsidase beta (Fabrazyme®), a recombinant form of human α-GAL, has been approved for the treatment of Fabry disease in the European Union and United States since 2001 [Citation17] and 2003 [Citation18], respectively, and in Japan since 2004 [Citation19]. The other ERT, agalsidase alfa, has been available in the European Union since 2001 [Citation20] and Japan since 2006 [Citation21]. Administration of these agents has been shown to result in marked increases in α-GAL A activity in human Fabry cells and Fabry mouse tissues; however, enzymatic activity in cultured fibroblasts, kidneys, heart, and spleen was higher for agalsidase beta compared with agalsidase alfa [Citation22]. More recently, pharmacological chaperone therapies have become available, and substrate reduction therapies and gene therapy approaches are also in development, which are expected to improve outcomes for patients with Fabry disease [Citation23].
Evidence for the safety and efficacy of agalsidase beta is based on the positive results from several trials, including the multinational Phase 3 double-blind and open-label extension studies [Citation24,Citation25], Phase 2 Japanese bridging study [Citation26], and Phase 4 double-blind study [Citation27]. Results of these studies demonstrated that agalsidase beta decreased tissue deposition of GL-3, improved clinical symptoms, and reduced the frequency of, and delayed the time to, major clinical events such as renal, cardiac, and cerebrovascular events, and death [Citation24–27]. However, only 13 patients with Fabry disease were enrolled in registration clinical trials in Japan [Citation26]. We therefore conducted two post-authorization safety studies (the Special Drug Use Investigation of agalsidase beta, for evaluation of long-term use, and the Drug Use Investigation of agalsidase beta, as all-case surveillance for patients who were not registered in the long-term safety study) to evaluate the long-term safety and efficacy of agalsidase beta in Japanese patients with Fabry disease in real-world practice.
2. Patients and Methods
2.1. Study Design, Patients, and Data Collection
Two post-authorization safety studies, comprising the Special Drug Use Investigation of agalsidase beta (Registry number: AGAL03004) and Drug Use Investigation of agalsidase beta (Registry number: AGAL02904), were conducted between June 2004 and March 2011, with safety data collected every 6 months. Japanese patients with Fabry disease, diagnosed by a physician according to deficient α-GAL activity or the identification of GLA gene mutation, were eligible for inclusion.
The Special Drug Use Investigation of agalsidase beta analyzed the safety and efficacy of long-term agalsidase beta use in Japanese patients with Fabry disease. The Drug Use Investigation of agalsidase beta was conducted using an all-case surveillance method and analyzed the safety profile in all Japanese patients with Fabry disease who were not included in the Special Drug Use Investigation of agalsidase beta.
These safety studies were conducted in accordance with the Japanese regulatory requirements of the Good Post-Marketing Study Practice, ethical principles of the Declaration of Helsinki and the Institutional Review Board (IRB) regulations. The need for informed consent was waived as these safety studies were mandated by the Japanese regulatory authorities in accordance with the Law for Ensuring the Quality, Efficacy, and Safety of Drugs and Medical Devices (Pharmaceutical and Medical Device Act). The clinical study protocol and other study-related documents were reviewed and approved by the local or central IRBs of study sites. Each investigator conducted the study according to applicable local or regional regulatory requirements and in accordance with the responsibilities listed in the protocol.
2.2. Data Collection and Assessments
Patient data were added into case report forms using patient medical records at baseline, and at 6-monthly intervals until the end of the study.
Upon entry to the study, baseline data were collected on demographics (sex, age, history of allergy), disease-related characteristics (disease phenotype, clinical symptoms, complications) and medication data (history of ERT, concomitant medication/s). Concomitant medications were defined as any medication that the patient was taking for any reason other than the treatment of adverse events during the study. Complications were defined as any disease that the investigator deemed insufficient to infer causal correlation with the primary disease. Clinical symptoms of Fabry disease included extremity pain, angiokeratoma, dyshidrosis, temperature paresthesias, corneal opacity, abdominal pain, diarrhea, and liver, kidney, and cerebrovascular disorders. Disease-related assessments and blood GL-3 level were collected at baseline and 6-monthly intervals. Details regarding agalsidase beta treatment, including the duration of treatment, total number of doses, mean dosing interval, time of administration, and average dose, were collected at 6-monthly intervals.
Blood samples were collected for antibody titer measurements and GL-3 levels at baseline and prior to study drug administration every 6 months, until the end of the study. The production of anti-agalsidase beta antibodies (IgG) was determined by the enzyme-linked immunosorbent assay (ELISA) and radioimmunoprecipitation (RIP) assay at the Genzyme Clinical Specialty Laboratory in Cambridge, MA, USA.
2.3. Safety
Safety assessment items were the incidence of adverse events (AEs), including serious AEs, adverse drug reactions (ADRs), infusion-associated reactions (IARs), and hypersensitivity reactions. AEs were defined as any untoward medical occurrence in a patient administered study treatment, including those that did not necessarily have a causal relationship with study treatment. Serious AEs were defined as those AEs that were life-threatening or resulted in death, permanent or significant disability, inpatient or prolonged hospitalization, or congenital abnormality. ADRs were defined as any AE for which a causal relationship to agalsidase beta could not be denied. IARs were defined as any adverse event occurring during any infusion or on the day reported after the infusion. The decision to discontinue treatment, as well as subsequent management of AEs (including switching to other treatments) was made by the individual treating investigator. Agalsidase beta-related hypersensitivity reactions were defined according to the investigators’ assessment, with adverse events defined as hypersensitivity reactions where the immune response was exaggerated or inappropriate against an antigen, allergen, or another substance. The time to onset of ADRs was also recorded. ADRs, IARs and hypersensitivity reactions were evaluated and categorized by system organ class (SOC) and preferred term (PT) using MedDRA/J version 16.1.
2.3.1. Efficacy
For the evaluation of efficacy, the change in blood GL-3 levels over time with agalsidase beta was assessed. Subgroup comparisons were performed for patient background factors (age, disease type, Fabry disease complications, concomitant medications and antibody production).
2.4. Statistical Analysis
The safety analysis sets comprised all patients who were eligible for these two studies. The efficacy analysis set included all patients in the Special Drug Use Investigation of agalsidase beta who had at least one GL-3 measurement available before and after treatment with agalsidase beta.
Baseline demographics were summarized using descriptive statistics. The mean (standard deviation [SD] and 95% CI), and median (range) were calculated for continuous variables, and the frequency number and proportion were calculated for categorical variables.
The frequency of adverse events, including ADRs, were summarized descriptively overall, and for each individual event (by SOC and PT).
The Kaplan–Meier method was used to estimate the cumulative ADR event rate throughout the study overall, and by age and phenotype subgroups.
3. Results
3.1. Patient Disposition
Details regarding patient disposition are presented in . A total of 332 patients in the Special Drug Use Investigation and 64 patients in the Drug Use Investigation were enrolled, comprising 396 patients in the aggregated data set (). After removal of duplicate entries at different study sites (n = 15), a total of 381 patients were enrolled. Of these, 307 patients provided permission for publication and were included in the safety analysis set (261 patients in the Special Drug Use Investigation and 46 patients in the Drug Use Investigation).
Figure 1. Disposition of patients in the (a) special drug use investigation of agalsidase beta for evaluation of long-term use and (b) drug use investigation of agalsidase beta as all-case surveillance
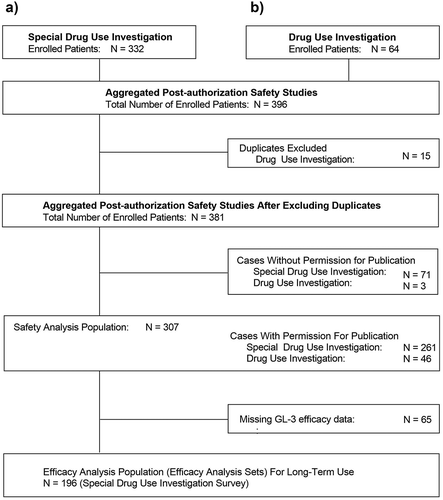
For the efficacy analysis set, a total of 65 patients in the Special Drug Use Investigation safety analysis set did not have blood GL-3 measurements available and were excluded; therefore, 196 patients were included.
Across the safety analysis sets, a total of 171 patients discontinued treatment with agalsidase beta. Reasons for discontinuation included: insufficient effect (n = 6), adverse events (n = 8), death (n = 26), transfer to a different hospital (n = 32), and other reasons (n = 103), including switching to other drugs due to the supply shortage of agalsidase beta that occurred in 2009 [Citation28].
3.2. Patient Characteristics
Baseline demographic and clinical characteristics of patients included in the safety analysis set, overall, in the Special Drug Investigation and Drug Investigation, and in specific subgroups (defined by age category and phenotype) are presented in .
Table 1. Demographic and baseline clinical characteristics
The majority of patients had a classic phenotype (56.1%), and a smaller proportion were heterozygous females (34.4%), or had renal (5.6%) and cardiac (3.9%) variants. The mean (95% CI) age was 39.5 (37.7 − 41.3) years, and more than half (65.5%) of patients were male. Most patients had not received prior enzyme replacement therapy (76.2%), nor had a history of allergy to medications in general or atopic conditions (82.1%). Renal impairment was present in 37.5% of patients at baseline, and 40.4% had cardiac dysfunction. Cardiac dysfunction was more common in patients enrolled in the Drug Use Investigation population compared with the Special Drug Use Investigation population (58.7% vs 37.2%, respectively). Over half (55.2%) of heterozygous females had cardiac dysfunction at baseline. Pain in the extremities was the most common symptom (56.7%) followed by dyshidrosis (44.6%), and angiokeratoma (30.9%). The mean (95% CI) blood GL-3 level at baseline was 8.5 (7.7 − 9.4) μg/mL (the upper limit of normal: 7.03 μg/mL). A higher proportion of patients aged ≤15 years (19.0%) had a blood GL-3 level ≥11.3 at baseline compared with those aged >15 years (11.2%). A similar finding was also observed in patients with classic disease (18.7%) compared with patients with cardiac (8.3%) or renal (5.9%) variants and heterozygous females (1.0%).
3.3. Treatment
In the Safety Analysis Set, the mean (SD) total duration of study drug administration was 1363.7 (772.4) days, the mean (SD) total number of doses was 83.6 (50.4), and the mean dose was 0.93 mg/kg.
3.4. Safety
3.4.1. Incidence of Adverse Events
The incidence of AEs (with a PT incidence of ≥n = 3) in the safety analysis set, overall and by subgroup are presented in Table S1.
Overall, AEs occurred in 171 (55.7%) patients, including 155 (59.4%) patients in the Special Drug Use Investigation and 16 (34.8%) patients in the Drug Use Investigation. A slightly higher proportion of patients aged ≤15 years experienced AEs compared with those aged >15 years. Similarly, a higher proportion of patients with cardiac and classic disease experienced AEs compared with patients with renal variants and heterozygous females.
The most common AEs were general disorders and administration site conditions (n = 63, 20.5%), nervous system disorders (n = 62, 20.2%), and infections and infestations (n = 52, 16.9%).
3.4.2. Incidence of Adverse Drug Reactions
The incidence of ADRs (with a PT incidence of ≥n = 3) in the safety analysis set, overall and by subgroup are presented in , and the incidence of ADRs classified by SOC and PT, overall and by subgroup are presented in Table S2.
Table 2. Occurrence of adverse drug reactions (with a PT Incidence ≥n = 3a) overall and by subgroup
Overall, ADRs occurred in 93 (30.3%) patients, including 85 (32.6%) patients in the Special Drug Use Investigation and 8 (17.4%) patients in the Drug Use Investigation. A higher proportion of patients aged ≤15 years experienced ADRs compared with those aged >15 years. Similarly, a higher proportion of patients with classic disease experienced ADRs compared with patients with cardiac or renal variants and heterozygous females.
Serious ADRs were experienced in 25 (8.1%) patients, and 8 deaths (2.6%) occurred during the study where the causality to agalsidase beta or ERT could not be denied by the investigator. The causes of these deaths were completed suicide (n = 1), cachexia (n = 1), pulmonary edema (n = 1), disseminated intravascular coagulation with pneumonia (n = 1), cardio-respiratory arrest (n = 1), sudden death (n = 1), and unexplained death (n = 2).
A total of 27 (8.8%) patients discontinued treatment due to ADRs, 25 (92.6%) of whom were older than 15 years and 2 (7.4%) of whom were pediatric patients. Common ADR events (occurring with an incidence of ≥2 events) which led to treatment discontinuation included chills (7 events), pyrexia (2 events), dyspnea (5 events), rash (3 events), urticaria (3 events), tremor (2 events), cardiac failure (2 events), rhinorrhea (2 events), wheezing (2 events), abdominal pain (2 events), abdominal pain upper (2 events), and pruritus (2 events).
The most common ADRs were general disorders and administration site conditions (n = 55, 17.9%), nervous system disorders (n = 30, 9.8%) and skin and subcutaneous tissue disorders (n = 23, 7.5%).
ADR occurrence, stratified by patient background characteristics, is presented in Table S3. A higher proportion of patients with a classic phenotype experienced ADRs compared with cardiac or renal variants and heterozygous females. No difference in the incidence of ADR was observed between patients with a presence of complications, including hepatic dysfunction and renal impairment, compared with patients without these complications. There was a numerically higher incidence of ADRs in patients with a blood GL-3 level ≥7.2 μg/mL.
3.4.3. Incidence of Adverse Drug Reactions Throughout the Course of the Study Period
The incidence of ADRs occurring throughout the course of the study period overall and by age and phenotype subgroups is presented in .
Figure 2. Incidence of adverse drug reactions throughout the course of the study period A) Overall and by age and B) Phenotype subgroup
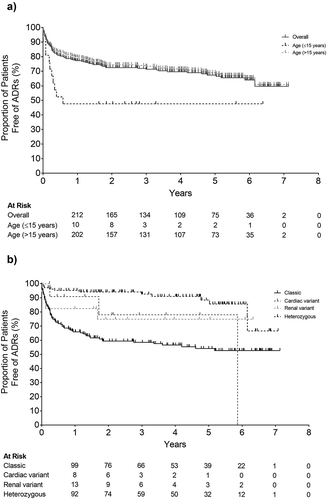
The incidence of ADRs was 22.3% within the first year of ERT and the cumulative incidence for the 7 years was 40.0%. When examined by disease phenotype, 33.7% of patients with classic disease experienced ADRs in the first year of ERT. In contrast, the incidence of ADRs in patients with cardiac variants was 9.1% in the first year and 22.1% in the first 2 years. The incidence of ADRs in patients with renal variants was 17.6% and 25.1% in the corresponding time periods, respectively. The incidence of ADRs was lower in heterozygous females compared with other phenotypes.
3.4.4. Timing of the Occurrence of Infusion-Associated Reactions Throughout the Course of the Study Period
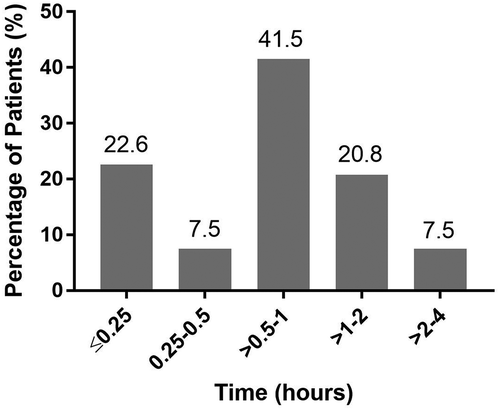
The majority of infusion-associated reactions (92.5%) occurred within 2 h after administration of agalsidase beta. Infusion-associated reactions occurred within the first 15 min after administration in 22.6% of patients, and within 1 h after infusion in 71.7% of patients ().
Figure 3. Timing of infusion-related reactions following initiation of agalsidase beta
3.4.5. Correlation Between Antibody Production and the incidence of Adverse Drug Reactions and Infusion-Associated Reactions
The correlation between antibody production and the incidence of adverse drug reactions and infusion-associated reactions is presented in . Overall, a significantly higher proportion of patients who were antibody-positive experienced ADRs and IARs compared with those who were antibody-negative.
Table 3. Correlation between antibody production and adverse drug reactions and infusion-associated reactions
A significantly higher proportion of patients with a classic phenotype who experienced ADRs and IARs were antibody-positive (ADRs: 58.8%; IARs: 54.1%) compared with patients who were antibody-negative (ADRs: 25.0%; IARs: 12.5%). However, it is difficult to compare antibody-positive patients with cardiac or renal variants and heterozygous females who experienced ADRs and IARs against antibody-positive patients who experienced ADRs and IARs in the overall population due to the limited number of patients in these subgroups.
Regarding prior exposure to ERT, there was a greater incidence of ADRs and IARs in the antibody-positive population with previous ERT use (ADRs: 63.0%; IARs: 59.3%) compared with the antibody-positive population without previous ERT use (ADRs: 53.0%; IARs: 45.6%). Of those who were antibody-positive, patients with a maximum antibody titer of <800 experienced ADRs and IARs with a lower incidence (ADRs: 22.7%; IARs: 18.2%) than patients with a maximum antibody titer of 800─<3,200 (ADRs: 54.2%; IARs: 45.8%), and a substantially lower incidence than those with a maximum antibody titer of 3,200-<6,400 (ADRs: 78.6%; IARs: 71.4%).
The relationship between AEs and ADR incidence and antibody status, including maximum IgG antibody titer value, was also examined separately throughout the course of the studies (). Compared with patients who were antibody-negative, a significantly higher proportion of patients who were antibody-positive experienced AEs (P = 0.015), SAEs (P = 0.021), ADRs (P < 0.001), IARs with and without a causal relationship (P = 0.006), IARs with a causal relationship (P < 0.001), serious IARs with and without a causal relationship (P = 0.033) and hypersensitivity reactions with and without a causal relationship (P < 0.001), including severe hypersensitivity reactions (P = 0.025). In contrast, no significant difference was observed between antibody-positive and antibody-negative patients experiencing serious ADRs (P = 0.498) and serious IARs with a causal relationship (P = 0.102).
Table 4. Relationship between adverse events and IgG antibody titer
3.5. Efficacy
3.5.1. Change in Blood GL-3 Level Throughout the Course of the Study Period
The change in mean blood GL-3 level throughout of the course of the study period in the 196 patients in the efficacy analysis set is presented in . In the overall population, reductions in blood GL-3 levels were observed over the course of the study, from 8.7 ± 5.3 μg/mL at treatment initiation, to 5.7 ± 2.5 μg/mL at 6 months, and remained below 6.0 μg/mL at all subsequent time points. Similar findings were observed when examined by age-group (≤15 years versus >15 years) ( and ). Patients with a classic phenotype experienced a comparable reduction in the first 6 months, which was maintained throughout the study period. Reductions in blood GL-3 levels were also observed in patients with the cardiac variant in the first 6 months, and values were lower in heterozygous females during the study compared with baseline. Patient background factors associated with a greater mean reduction in blood GL-3 levels were male sex (P < 0.001), concomitant medications (P < 0.001), age >15 years (P < 0.001), and classic phenotype (P < 0.001). Similarly, large reductions in blood GL-3 were observed in patients irrespective of concomitant therapy use or the presence or absence of complications.
Figure 4. Change in blood GL-3 Level by (a) age subgroups, (b) disease phenotype, and (c) antibody status
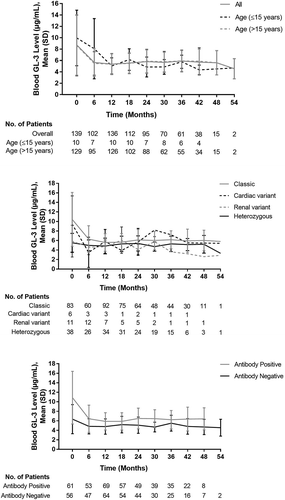
Table 5. Change from baseline in blood GL-3 level by patient background factors
4. Discussion
The aggregated data from these two post-authorization safety studies, comprising a large cohort of over 300 patients with Fabry disease, demonstrate the long-term safety and efficacy of agalsidase beta in Japanese patients with Fabry disease in real-world practice. Overall, adverse drug reactions occurred in 30.3% of patients in the safety analysis population. Importantly, no unexpected safety signals or concerns were identified, indicating that the safety profile of agalsidase beta was consistent with the established profile of this drug [Citation18].
The most common ADRs in the aggregated data set were general disorders and administration site conditions, nervous system disorders, and skin and subcutaneous tissue disorders. Respiratory, thoracic and mediastinal disorders, and gastrointestinal disorders were reported less frequently, but occurred more frequently in pediatric patients aged ≤15 years compared with the older population, and in those with classic disease compared with other phenotypes. By preferred term, the most common ADRs were pyrexia, chills, headache, rash, and urticaria. Common ADRs reported during our study were broadly consistent with AEs reported in previous clinical trials [Citation24,Citation25,Citation29,Citation30], including the Phase 2 registration clinical trial in Japan [Citation26], as well as the known safety profile of agalsidase beta [Citation18]. Rigors and fever were among the most frequently reported AEs associated with agalsidase beta in the Phase 3 study (48% and 24%, respectively) [Citation24], 54-month extension study (59% and 36%, respectively) [Citation25], and Phase 2 Japanese Bridging study (15─23% and 15%, respectively), with headache, chills, and hypertension less frequently reported [Citation26]. However, differences in the methods of reporting of adverse events and the small sample size in some of the clinical trials precluded meaningful comparisons from being drawn.
When examined the time to onset of ADRs over the course of the studies (), ADRs were predominantly reported within the first year after ERT initiation, with subsequent declines to year six. A similar but slightly more pronounced pattern was observed in the phase 3 and long-term extension studies, in which an increase in ADRs was observed in the first 2 years of ERT treatment, suggesting that only a small proportion of patients may newly experience ADRs after 1 year from ERT initiation [Citation25,Citation30].
IgG antibodies developed in 50.5% of patients overall, which is substantially lower than the rate of IgG seroconversion reported in the product information for agalsidase beta in adult (79%) and pediatric (69%) patients [Citation18], and the rate reported in the Japanese Phase 2 study (85%) [Citation26]. Overall, the incidence of ADRs and IARs was higher in patients who were antibody-positive compared with those who were antibody-negative, and generally increased with increasing antibody titer level. There was no statistically significant difference in serious ADR incidence (P = 0.498) between patients with either the presence or absence of anti-drug antibodies (ADAs). However, these results should be interpreted with caution due to the relatively small number of antibody-positive (n = 11) and antibody-negative (n = 8) patients, and warrant confirmation in larger, well-designed trials.
Although no clear guidelines exist regarding the management of antibodies in Fabry disease patients treated with agalsidase beta [Citation18], routine antibody testing in patients undergoing ERT [Citation31,Citation32], and close monitoring of patients with elevated antibody titers [Citation33], particularly during the infusion process, are recommended. More recently, the application of immune tolerance induction protocols used in other lysosomal storage diseases for Fabry disease have been suggested as a possible means of preventing ADA formation in ERT-naïve patients and decreasing or saturating existing antibody titers in antibody-positive patients, although the benefit remains to be established [Citation31,Citation34].
Compared with other studies conducted globally and in Japan, a higher proportion of patients in our study had classic disease [Citation35–37]. As agalsidase beta was the first approved medication for Fabry disease in Japan, it is likely that patients with relatively severe disease preferentially received agalsidase beta due to its high dosage (1 mg/kg of body weight). There was no substantial difference in the incidence of specific clinical symptoms of Fabry disease in our studies when compared against the existing literature [Citation12,Citation38]. Pain in the extremities was the most common symptom, and was particularly common in children aged ≤15 years and those with classic disease, followed by dyshidrosis, and angiokeratoma. Predictably, cardiac dysfunction was present in over 80% of patients with the cardiac variant at baseline. Cardiac hypertrophy is a frequent observation in patients with cardiac variants of Fabry disease and is associated with lysosomal accumulation of GL-3 [Citation39]. Notably, over half of heterozygous females had cardiac dysfunction at baseline, consistent with the findings by MacDermot et al. (2001), who reported a high prevalence of cardiac manifestations in female patients, including chest pain, heart valve abnormalities, and arrhythmia [Citation38]. Although ‘carrier’ females were once thought to be clinically unaffected [Citation40], it is now recognized that heterozygous females with Fabry disease should be monitored for signs and symptoms of cardiovascular manifestations and renal impairment and considered for enzyme replacement therapy [Citation41].
The mean age of patients with Fabry disease was 39.5 years, and most patients (76.2%) had no prior exposure to enzyme replacement therapy. By this stage, 40.4% of patients had cardiac dysfunction and 37.5% had renal impairment. It is well recognized that patients who initiate enzyme replacement therapy early in the disease experience greatest benefit, including delays in kidney and cardiac involvement [Citation25,Citation42]. In contrast, patients who initiate treatment at an older age, once substantial organ damage had already likely occurred, may experience continued disease progression despite therapy [Citation42,Citation43]. Nevertheless, efficacy was demonstrated in the overall population, irrespective of age (including elderly patients) or antibody status, with patients experiencing a substantial decline in GL-3 levels throughout the course of ERT treatment.
All recombinant protein products, including agalsidase beta, have the potential to trigger immunogenicity reactions, particularly with long-term use [Citation44]. The presence of ADAs during enzyme replacement therapy has been previously reported, with concerns over loss of drug efficacy and disease progression, including increased cellular Gb-3 deposition [Citation45] and plasma lyso-Gb3 concentrations [Citation46,Citation47]. Interestingly, Lenders et al. (2016) demonstrated that, in patients who seroconverted, saturation of ADA-binding sites during agalsidase beta treatment resulted in a stable interventricular septum thickness and a significant decrease in plasma lyso-Gb3 levels over time compared with non-saturated patients, presumably as a result of free enzyme activity [Citation47]. These findings suggest that, in patients with ADAs, a saturated status may be preferential during infusion in terms of clinical outcome and management of progressive deterioration.
Monitoring of blood GL-3 levels is a clinically important tool to assess disease progression [Citation48]. However, results of our current study and others suggest that blood GL-3 may not be a sensitive enough marker [Citation49,Citation50], including in patients with cardiac and renal variants and heterozygous females, most of whom do not exhibit a high plasma GL-3 concentration [Citation51,Citation52]. Recently, lyso-Gb3 has been suggested as a more sensitive biomarker for diagnosis and monitoring of response to enzyme replacement therapy [Citation49,Citation50]. Although not specifically measured in the post-authorization safety studies, elevated levels of lyso-Gb3 correctly identified both classic and late-onset mutations in males and females in a study of 2,360 Japanese patients with suspected Fabry disease [Citation53].
Limitations were inherent to post-marketing safety studies, and included the noninterventional, observational study design, with the lack of an independent control arm, and incomplete or missing data. This included missing information regarding the normalized ratio of α-GAL activity to the standard reference value. Further, the correlation between phenotype and genotype information regarding GLA gene mutations in each patient was also not available for these studies, which precluded any associations between genotype and phenotype from being investigated. Nevertheless, the diagnosis of Fabry disease and management of ERT in all Fabry disease patients was performed in accordance with the guidance document of Fabry Disease-Diagnosis and Management Handbook for Japanese [Citation54]. Further, the study populations represent a particularly large cohort of patients when taking into consideration the relative rarity of Fabry disease, and also reflect the type of patients seen in clinical practice with a relative lack of selection bias compared with randomized controlled design types.
5. Conclusions
Results of this analysis of aggregated data from these two post-authorization safety studies, conducted in over 300 Japanese patients with Fabry Disease, demonstrate the long-term safety and efficacy of agalsidase beta over a period of up to 7 years. Overall, agalsidase beta was well tolerated, and the safety profile of adverse drug reactions, including hypersensitivity reactions and infusion-related reactions, was broadly consistent with that reported in previous global clinical trials [Citation24,Citation25], and the Phase 2 study in Japan [Citation26]. Further, no unexpected safety signals and concerns were identified. These findings add to the wealth of data supporting the long-term safety and efficacy of agalsidase beta in the treatment of Fabry disease [Citation27,Citation42].
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Declaration of interest
All authors are employees of Sanofi K.K. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Data sharing statement
Data supporting the findings of this study are available in the supplementary material of this article.
Supplemental Material
Download MS Word (117.2 KB)Acknowledgments
The authors thank all clinicians for their involvement and contribution to the study. The authors also thank Jordana Campbell, BSc, CMPP of inScience Communications, Springer Healthcare, for writing the outline and the first draft of the manuscript. This medical writing assistance was funded by Sanofi K.K.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Gal A, Schafer E, Rohard I. Chapter 33: the genetic basis of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Accessed 2020 Jul 17. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11574/
- Waldek S, Patel M, Banikazemi M, et al. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Gen Med. 2009;11:790–796.
- Mehta A, Widmer U. Chapter 19: natural history of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors Fabry Disease: perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Accessed 2020 Jul 17. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11572/
- Oder D, Üçeyler N, Liu D, et al. Organ manifestations and long-term outcome of Fabry disease in patients with the GLA haplotype D313Y. BMJ Open. 2016;6(4):e010422. .
- Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. 2017(28);1631–1641.
- Tsukimura T, Nakano S, Togawa T, et al. Plasma mutant α-galactosidase A protein and globotriaosylsphingosine level in Fabry disease. Mol Gen Metab Rep. 2014;1:288–298.
- Rozenfeld PA. Fabry disease: treatment and diagnosis. IUBMB Life. 2009;61(11):1043–1050.
- Laney DA, Peck DS, Atherton AM, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med. 2015;17(5):323–330. .
- Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123:416–427.
- Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr Res. 2008;64:550–555.
- Salviati A, Burlina AP, Borsini W. Nervous system and Fabry disease, from symptoms to diagnosis: damage evaluation and follow-up in adult patients, enzyme replacement, and support therapy. Neurol Sci. 2010;31:299–306.
- MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38:750–760.
- Sims K, Politei J, Banikazemi M, et al. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009;40:788–794.
- Germain DP, Brand E, Cecchi F, et al. The phenotypic characteristics of the p.N215S Fabry disease genotype in male and female patients: a multi-center Fabry Registry study. Mol Genet Metab. 2017;120:S51–S52.
- Macauley SL. Combination therapies for lysosomal storage diseases: a complex answer to a simple problem. Pediatr Endocrinol Rev. 2016;13(Suppl 1):639–648.
- Sestito S, Ceravolo F, Falvo F, et al. Pathobiological insights into the newly targeted therapies of lysosomal storage disorders. J Pediatr Biochem. 2016;6:1.
- European Medicines Agency. EPAR summary for the public: fabrazyme. 2013. Available from: https://www.ema.europa.eu/en/documents/overview/fabrazyme-epar-summary-public_en.pdf. Cited 2020 Jun 15.
- Fabrazyme [prescribing information]. Genzyme Corporation, 2010. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103979s5135lbl.pdf. Cited 2020 Jul 16.
- Uyama E. Fabry disease in light of recent review. Brain Nerve. 2008;60:1235–1244.
- European Medicines Agency. EPAR summary for the public: replagal. 2015. Available from: https://www.ema.europa.eu/en/documents/overview/replagal-epar-summary-public_en.pdf. Cited 2020 Nov 6
- Pharmaceutical and Medical Devices Agency, Japan. List of approved products (FY 2006). Available from: https://www.pmda.go.jp/files/000153730.pdf. Cited 2020 Nov 6.
- Sakuraba H, Murata-Ohsawa M, Kawashima I, et al. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J Hum Genet. 2006;51:180–188.
- Felis A, Whitlow M, Kraus A, et al. Current and investigational therapeutics for Fabry Disease. Kidney Int Rep. 2020;5:407–413.
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16.
- Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase β therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18:1547–1557.
- Eto Y, Ohashi T, Utsunomiya Y, et al. Enzyme replacement therapy in Japanese Fabry disease patients: the results of a Phase 2 bridging study. J Inherit Metab Dis. 2005;28:575–583.
- Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86.
- Pisani A, Bruzzese D, Sabbatini M, et al. Switch to agalsidase alfa after shortage of agalsidase beta in Fabry disease: a systematic review and meta-analysis of the literature. Genet Med. 2017;19:275–282.
- Wraith JE, Tylki-Szymanska A, Guffon N, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr. 2008;152:563–570.e561.
- Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Gen. 2004;75:65–74.
- Kishnani PS, Dickson PI, Muldowney L, et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab. 2016;117:66–83.
- Mehta A, Beck. M, Linhart A, et al. Chapter 42: monitoring and follow-up of patients. In: Fabry Disease: perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Accessed 2020 Nov 18. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11569/
- Byrne BJ, Geberhiwot T, Barshop BA, et al. A study on the safety and efficacy of reveglucosidase alfa in patients with late-onset Pompe disease. Orphanet J Rare Dis. 2017;12:144.
- Lenders M, Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with Fabry disease. J Am Soc Nephrol. 2018;29:2265–2278.
- Sasa H, Nagao M. Safety and effectiveness of enzyme replacement therapy with agalsidase alfa in patients with Fabry disease: post-marketing surveillance in Japan. Mol Genet Metab. 2019;126:448–459.
- Duro G, Zizzo C, Cammarata G, et al. Mutations in the GLA gene and lysoGb3: is it really Anderson-Fabry disease? Int J Mol Sci. 2018;19:3726.
- Sakuraba H, Tsukimura T, Togawa T, et al. Fabry disease in a Japanese population-molecular and biochemical characteristics. Mol Gen Metab Rep. 2018;17:73–79.
- MacDermot KD, Holmes A. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–775.
- Lin HY, Chong KW, Hsu JH, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2:450–456.
- Deegan PB, Bähner. F, Barba. M. Chapter 30: fabry disease in females: clinical characteristics and effects of enzyme replacement therapy. In: Mehta A, Beck M, Sunder-Plassmann G, editors Fabry Disease: perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Accessed 2020 Jul 17. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11591/
- Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112–128.
- Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52:353–358.
- Weidemann F, Niemann M, Störk S, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. 2013;274:331–341.
- Schellekens H, Casadevall N. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol. 2004;251(Suppl 2):i4–9.
- Bénichou B, Goyal S, Sung C, et al. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab. 2009;96:4–12.
- Rombach SM, Aerts JM, Poorthuis BJ, et al. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One. 2012;7:e47805.
- Lenders M, Stypmann J, Duning T, et al. Serum-mediated inhibition of enzyme replacement therapy in Fabry disease. J Am Soc Nephrol. 2016;27:256–264.
- Kanai T, Ito T, Odaka J, et al. Surges in proteinuria are associated with plasma GL-3 elevations in a young patient with classic Fabry disease. Eur J Pediatr. 2016;175:427–431.
- Goker-Alpan O, Gambello MJ, Maegawa GHB, et al. Reduction of plasma globotriaosylsphingosine levels after switching from agalsidase alfa to agalsidase beta as enzyme replacement therapy for Fabry disease. JIMD Rep. 2016;25:95–106.
- Liao H-C, Huang Y-H, Chen Y-J, et al. Plasma globotriaosylsphingosine (lysoGb3) could be a biomarker for Fabry disease with a Chinese hotspot late-onset mutation. Clin Chim Acta. 2013;426:114–120.
- Young E, Mills K, Morris P, et al. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr Suppl. 2005;94:51–54.
- Winchester B, Young E. Chapter 18: biochemical and genetic diagnosis of Fabry disease. In: Mehta A, Beck M, Sunder-Plassmann G, editors Fabry Disease: perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006. Accessed 2020 Jul 17. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11601/
- Maruyama H, Miyata K, Mikame M, et al. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet Med. 2019;21:44–52.
- Eto Y, Komuro I, Tsuji S, et al. Fabry Disease: Diagnosis and Management Handbook for Japanese, Revised 3rd Edition. In: Editorial Committee for Fabry's disease diagnosis and treatment handbook, editors. Tokyo: En Medix; 2018.