
ABSTRACT
Background: Prior studies investigated regulatory actions that reflected a negative impact on drug risks. We aimed to evaluate occurrence of regulatory actions that reflected a negative or positive impact on benefits or risks, as well as relations between them.
Research design and methods: We followed EMA-approved innovative drugs from approval (2009–2010) until July 2020 or withdrawal to identify regulatory actions. We assessed these for impact on benefits or risks and relations between actions. Additionally, we scrutinized drug lifecycles for time-variant characteristics that may contribute to specific patterns of regulatory actions.
Results: We identified 14 letters and 361 label updates for 40 drugs. Of the label updates, 85 (24%) reflected a positive impact, mostly concerning indications, and 276 (76%) a negative impact, mostly adverse drug reactions. Many updates (54%) occurred simultaneously with other updates, also if these reflected a different impact. Furthermore, levels of patient exposure, innovativeness, needs for regulatory learning and unexpected risks may contribute to patterns of regulatory actions.
Conclusions: Almost a quarter of regulatory actions reflected a positive impact on benefits and risks. Also, simultaneous learning about benefits and risks suggests an important role for drug development in risk characterization. These findings may impact regulatory analyses and decision-making.
1. Introduction
Regulatory learning about drugs is an important process. At the time of initial drug approval, many uncertainties about its clinical aspects remain [Citation1]. Knowledge is often limited to efficacy and the most common adverse events when used to treat a specific disease in a specific patient population. While a well-designed clinical trial in a restricted patient population is paramount to establishing efficacy [Citation2], it limits generalizability of these findings to the broader patient population. At the same time, it limits the characterization of a drug’s safety profile in the broader population [Citation2]. Also, it often follows too few patients for a too short time period to identify rare adverse events and adverse events with a long latency [Citation3]. Thus, there is ample room for post-approval learning about benefits and risks of drugs. Indeed, efficacy in broader or completely other indications is studied years after initial approval [Citation4] and the methods to characterize the safety profile continue to be refined [Citation5].
Contemporary drug regulation reflects the idea that drug development is never finished. It aims to capture and stimulate continuous knowledge accrual throughout the entire drug lifecycle, rather than a one-off learning experience at initial approval decisions [Citation6,Citation7]. Regulators can stimulate this process in the post-approval phase through e.g. requests for additional studies and periodic reports of information available in company databases and scientific literature, among others. Consequently, new information comes available on a regular basis, which is then assessed in terms of their positive or negative impact on either the benefits or risks of drugs. The weighing of information on adverse effects and other potential risks against the desired, therapeutic benefits of a drug in a specific population is called benefit-risk assessment and is important to all regulatory decisions in any country [Citation8,Citation9]. It may lead to post-approval regulatory actions such as the approval or refusal of a new drug or indication, the broadening or restriction of an indication, the addition of a new warning or adverse drug reaction (ADR) to the drug label, or perhaps the removal of an existing one [Citation10–13]. In some cases, new information may question the available knowledge or suggest a critical risk not known or expected before. Then, a complete reassessment of the benefit-risk balance based on all available data may be considered, in Europe also known as a referral procedure [Citation14–16].
Post-approval regulatory decision-making thus universally considers both drug benefits and risks and whether new information has a positive or a negative impact on either. However, recent studies on the outcomes of regulatory decision-making mostly assessed safety-related post-approval regulatory actions, i.e. those that respond to new information with a negative impact on drug risks. This often concerns newly identified ADRs, and the respective regulatory actions include drug label updates [Citation17–23], healthcare professional letters or similar notifications [Citation24,Citation25], withdrawals [Citation14,Citation26–29], or a combination thereof [Citation15,Citation16,Citation30–40]. Of these studies, few also assessed benefit-related post-approval regulatory actions – such as amended indications – as outcomes [Citation15,Citation20,Citation21], let alone how they are associated with safety-related regulatory actions [Citation24]. The few studies that focused primarily on amended indications [Citation4] or posology changes [Citation41,Citation42] did not consider other regulatory actions. Most importantly, no study comprehensively assessed relations between various regulatory actions.
We aimed to build on this previous research by assessing the type and impact of European post-approval regulatory actions that occur during the drug lifecycle (i.e. whether reflecting new information with a positive or a negative impact on benefits or risks), any relations between them, and potential characteristics that seem to play a role in their occurrence. We therefore used publicly available European regulatory action data, including changes to the marketing authorization, Direct Healthcare Professional Communications (DHPC) and all newly available clinical information in the product label – the Summary of Product Characteristics (SmPC).
2. Patients and methods
2.1. Study design and cohort selection
We performed a retrospective cohort study of drugs approved by the European Medicines Agency (EMA) between 1 January 2009 and 31 December 2010 that contained a new active substance and followed these up until 1 July 2020 or withdrawal from the market if that occurred earlier. This allowed 10 years of follow-up that was considered a relevant time horizon to address the aim of this study. In case of duplicate applications, i.e. drugs approved under multiple product names, we included the one with the longest time on the market. We excluded vaccines approved for the prevention of seasonal disease (e.g. influenza) because these are often marketed only temporarily, rendering them incomparable to other drugs in terms of their lifecycle and consequently changes to their marketing authorization. For the remaining drugs, European Public Assessment Reports (EPAR) at the EMA websiteFootnote1 were consulted to extract baseline drug and regulatory characteristics. Confidential Periodic Safety Update Reports (PSUR) were accessed through the database of the Dutch Medicines Evaluation Board to extract information about cumulative patient exposure at end of follow-up.
2.2. Data collection
For the included drugs, we accessed their EPARs and extracted all regulatory procedures that occurred between drug approval and 1 July 2020 and led to regulatory actions (, inspired by work of Ebbers et al. [Citation43]). For each regulatory procedure, we extracted i) a high-level description of the type of information that prompted regulatory actions, ii) the decision date, iii) the type of regulatory action(s) (), and iv) the number of changes in case the regulatory action concerned an SmPC update. If any of iii–iv were unclear, we accessed the Union Register of medicinal products for human useFootnote2 to compare the SmPCs before and after the decision and extracted these data accordingly.
Figure 1. Potential regulatory actions, ordered according to the impact on benefits and risks
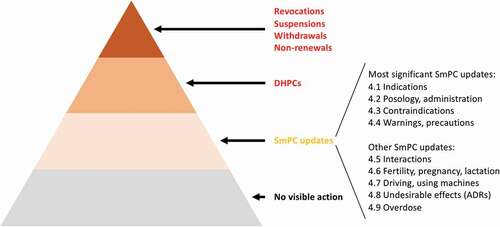
To limit our study to regulatory actions that reflected truly new information concerning benefits and risks, we only included SmPC updates that concerned topics that were not previously described in that specific section or if their description was substantially altered, e.g. updated warnings to note that fatal outcomes are possible. Also, we only included SmPC updates that reported in vitro or pharmacokinetic drug-drug interaction study results if these also noted implications or recommendations for clinical practice. We did not include SmPC updates that concerned (specifications of) clinical recommendations for topics already described in that specific section, confirmations of previously included information regarding expected or potential benefits and risks (e.g. concerning renally or hepatically impaired patients), study results, rewordings, clarifications, or cross-references to other SmPC sections.
Since EPARs do not provide information on DHPCs, we used European national regulatory authorities’ websites to identify relevant DHPCs for the included drugs.Footnote3 From each DHPC, we extracted i) the DHPC date, ii) a high-level description of the type of new information it addressed (less detailed than for SmPC updates), and iii) the number of key messages it communicated. Regulatory actions due to commercial reasons were excluded.
2.3. Categorization of regulatory actions according to the impact of new information on benefits and risks
While newly available information may have a positive or negative impact on knowledge of drug benefits and risks, detailed information is often not publicly available. Instead, regulatory actions are indicative of such information since they aim to ensure an optimal benefit-risk balance. Therefore, we reviewed the content of all changes to the marketing authorization, DHPCs and SmPC updates to understand what impact on benefits and risks these regulatory actions reflected. In addition to the impact being assessed as positive or negative, it was assessed as impact on benefits, defined as impact on the population eligible to use the drug and how to use it, or impact on risks, defined as safety aspects. This resulted in the following four categories: A) positive impact on benefits, e.g. a broadening of the indication; B) positive impact on risks, e.g. a decreased frequency of a known ADR; C) negative impact on benefits, e.g. a new contraindication; and D) negative impact on risks, e.g. a new ADR ().
Figure 2. Categories used to assess a positive or negative impact on benefits and risks reflected by regulatory actions

To consistently assess the category that each regulatory action belonged to, we created a list of subcategories (Table S1). This occurred in an explorative and iterative fashion, by creating an initial set of subcategories that were expected to be encountered based on input from LTB, MK, JH and AKMT. This already included the majority of the subcategories in Table S1. Subsequently, when these provided an insufficient basis for detailed and consistent assessment, existing subcategories were reworded or complemented by new subcategories by LTB and MK and agreed by the other authors. Examples of new subcategories were ‘Change in posology resulting in a reduced patient-burden’ and ‘Change of a contraindication into recommendation’. This process was repeated until each regulatory action was categorized and the subcategories were mutually exclusive.
In case an SmPC update comprised several changes to the same SmPC section that were considered to reflect an impact on eligible population and use characteristics as well as safety aspects (e.g. in case of multiple new interactions listed in the respective SmPC section), we categorized this SmPC section as the former (A or C). In case changes reflected both a positive and a negative impact (this only occurred in case of multiple updates to the SmPC section on ADRs, i.e. ‘Undesirable effects’), we categorized the updated SmPC section according to the most often occurring category. This way, each regulatory action was assessed by two researchers (LTB and MK). This ensured discussion about regulatory actions that were potentially difficult to categorize, which was predominantly the case for SmPC updates to the interactions section. There, we needed to establish the effect of the interaction on exposure to the drugs involved, and consequently their efficacy and/or safety (Table S1).
2.4. Data analysis
First, we used descriptive statistics to describe the cohort of drugs with regard to drug and regulatory characteristics as well as the regulatory actions that these drugs underwent during follow-up, and the type of information that prompted regulatory actions. Second, we categorized all regulatory actions according to the approach discussed above. Third, we described relations between regulatory actions, i.e. simultaneous updates within the same procedure. Last, we scrutinized individual drug lifecycles to identify time-variant characteristics that may typically play a role in specific patterns of regulatory actions, based on cohort characteristics, EPARs, previous research, and our own regulatory experience. For this analysis, we took into account the most significant regulatory actions, i.e. all but the ‘other SmPC updates’ listed in .
3. Results
3.1. Description of the cohort
We included 40 drugs that were approved by EMA in 2009 and 2010 (). Of these, five were later withdrawn by the company – all because of commercial reasons: autologous cartilage cells (brand name ChondroCelect), catumaxomab (Removab), collagenase Clostridium histolyticum (Xiapex), ofatumumab (Arzerra) and rilonacept (Rilonacept Regeneron). The remaining drugs were followed until 1 July 2020, resulting in a median follow-up of 10.5 years (interquartile range 9.8–10.8 years).
Table 1. Cohort characteristics
3.2. Occurrence of regulatory actions and their impact on benefits and risks
During the study period, there were no revocations, suspensions, withdrawals (apart from the five because of commercial reasons discussed above) or non-renewals. However, we identified 14 DHPCs that had been distributed after new information with negative impact on benefits and risks had come available. Of these, one mainly had a negative impact on the eligible population and use characteristics, i.e. it communicated restrictions of the indication and new contraindications for dronedarone (Multaq) following a referral procedure. In addition, it communicated new warnings with regard to liver injury, lung toxicity and cardiovascular risk. Another DHPC also for dronedarone communicated that an increased cardiovascular risk had been observed in a study in a non-approved indication, which did not lead to an SmPC update but informed the start of the referral procedure. The remaining 12 DHPCs communicated (clinical recommendations for) one or two safety issues. Of these, another also concerned dronedarone. Six concerned denosumab (Prolia), ofatumumab and tolvaptan (Samsca) – two for each product. Five concerned epoetin theta (Eporatio), pazopanib (Votrient), regadenoson (Rapiscan), saxagliptin (Onglyza), and vernakalant (Brinavess) – one for each product. These 12 DHPCs often led to an update of SmPC sections concerning posology and administration, warnings and precautions and/or ADRs. However, these SmPC updates were not all included in our study because some concerned (specifications of) clinical recommendations for topics already described in that SmPC section.
We also identified 266 regulatory procedures during which 361 SmPC sections had been updated with new information concerning benefits and risks. Of these 361 updates, 276 were considered to reflect a negative impact on benefits and risks (76%) and 85 to reflect a positive impact (24%). The updates most frequently concerned the ADR section (155, 43%), followed by the warnings and precautions section (85, 24%). For these sections, the majority of updates was considered to reflect a negative impact because it concerned new ADRs or increased frequencies of previously known ADRs (152/155, 98%), or new warnings or precautions (75/85, 88%), respectively. Another frequently updated SmPC section was the indications section (50, 14%), for which the majority of updates was considered to reflect a positive impact because it concerned new or broadened indications (48/50, 96%). Furthermore, while most updates to SmPC sections implemented one change to that specific section (252, 70%), in 109 instances (30%) two or more changes were implemented. These latter mostly concerned the ADR (81/109, 74%) and the warnings and precautions sections (18/109, 17%). A complete overview of all regulatory actions (DHPCs and updated SmPC sections) and their categorization according to impact on benefits and risks is provided in . In addition, an overview of the type of information that prompted the regulatory actions is provided in Table S2, according to the impact on benefits and risks.
Table 2. Overview of regulatory actions that reflected a positive or negative impact on benefits and risks
3.3. Relations between SmPC updates
Looking closer into the occurrence of the 361 SmPC updates, we observed that during 85 of 266 regulatory procedures (32%) multiple SmPC sections were updated simultaneously. During the majority of these procedures (68, 80%), two SmPC sections were updated simultaneously, while during 17 procedures (20%), three or more SmPC sections were updated. In total, 194 of 361 updates to SmPC sections (54%) occurred simultaneously with at least one other SmPC section. Of these, 44 reflected a positive impact on benefits and risks and 150 reflected a negative impact, i.e. 52% and 54%, respectively, of all updated SmPC sections that reflected a positive or negative impact. illustrates for each SmPC section the number and proportion of updates that occurred simultaneously with updates to other sections, the impact reflected by these updates and the relations between the sections.
Figure 3. Overview of updates to SmPC sections during the same regulatory procedure (194/361)
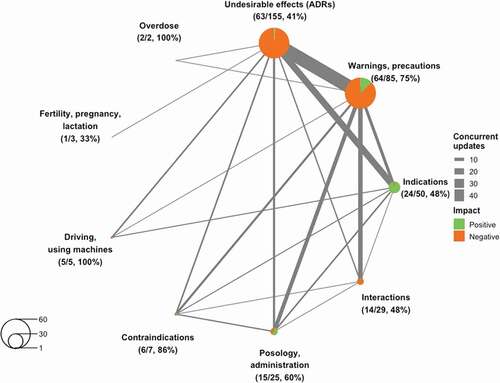
Of the 85 regulatory procedures that led to simultaneous updates to SmPC sections, 55 (65%) concerned updates that only reflected a negative impact. Of these, 30 procedures led to a simultaneous update to the warnings and precautions section and the ADR section – the most commonly observed combination. The procedure that led to the most SmPC updates was the referral for dronedarone discussed earlier, which led to restrictions of the indication, new contraindications, new warnings in the posology and administration as well as the warnings and precautions section, and new ADRs.
The remaining 30 procedures (35%) concerned simultaneous updates that either reflected only a positive impact on benefits and risks, or a combination of positive and negative impact. Apart from one procedure during which interactions and a warning about interactions were removed for ulipristal acetate (ellaOne), all these procedures were initiated by study results that supported a new or broadened indication or an update of posology and/or administration characteristics. While 2 procedures only concerned these aspects, 27 procedures also led to changes in ADRs, warnings and precautions and/or other risk-related SmPC sections, indicating an important role for further post-approval drug development in characterizing drug risk profiles. This does not always have to become more negative, e.g. a new indication for liraglutide (Victoza) also led to a less restrictive warning regarding patients with congestive heart failure, and the broadening of the indication of prucalopride (Resolor) to use in men led to removal of a warning that use in men was not recommended due to a lack of efficacy and safety data. Similarly, a new indication for golimumab (Simponi) led to multiple ADR frequencies being decreased based on the new study data. Lastly, such procedures may even positively impact contraindications, as illustrated by moderate hepatic impairment no longer being contraindicated based on study data supporting a new indication for ticagrelor (Brilique).
3.4. Identification and characterization of typical drug lifecycles
In , we plotted all 40 drug lifecycles according to the most significant regulatory actions () that reflected positive (vertical axis) versus negative (horizontal axis) impact on benefits and risks. We identified several typical drug lifecycles that seem to undergo specific patterns of regulatory actions. These are characterized by levels of post-approval patient exposure, innovativeness, need for further regulatory learning and unexpected risks. First, the level of post-approval patient exposure seems to play an important role in the occurrence of regulatory actions, thereby facilitating further development or learning about a drug. Of the 17 drugs that underwent up to two regulatory actions, eight (47%) had one or more specific characteristics that are often suggestive of low patient exposure, i.e. orphan designation (5/9 orphan drugs), market withdrawal (4/5 withdrawn drugs – all except ofatumumab, which was withdrawn to be remarketed in another disease area [Citation44,Citation45]), and approval under exceptional circumstances (2/3 exceptionally approved drugs). The latter approval pathway is applicable only when little evidence is available at approval and not expected to be supplemented post-approval. The patient exposure data from PSURs support these observations: of the 13 drugs (33%) with the lowest patient exposure, 11 underwent a maximum of two regulatory actions, including those discussed earlier.
Figure 4. Characterization of drug lifecycles according to most significant regulatory actions that reflected a positive versus negative impact on benefits and risks
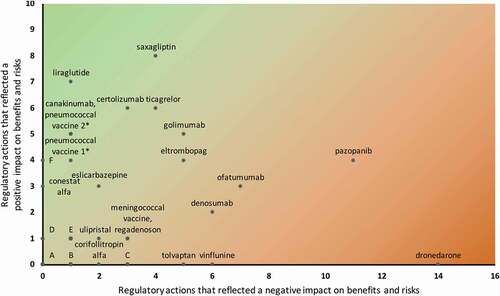
Second, the level of drug innovativeness also seems to play a role in the occurrence of regulatory actions, in various ways. For instance, five of the remaining six drugs that also underwent up to two regulatory actions but were exposed to substantially more patients, were of a drug class that had been available for many years. These are asenapine (Sycrest, an atypical antipsychotic), bazedoxifene (Conbriza, a selective estrogen receptor modulator), epoetin theta, indacaterol (Onbrez Breezhaler, a long-acting inhaled β2-agonist), and silodosin (Urorec, an α1-adrenoceptor antagonist). This reflects limited innovativeness and relevant knowledge about efficacy and safety may have already been available or expected and addressed in the SmPC at initial approval. In contrast, higher innovativeness may be reflected by further post-approval drug development, including conditional approval as a special case, and initiate many regulatory actions. These include drugs for which multiple truly new indications are approved after initial approval as well as drugs for which the initial indication is progressively broadened toward ‘blockbuster’ status. The first group includes for example the anti-inflammatory monoclonal antibodies canakinumab (Ilaris), certolizumab (Cimzia) and golimumab. Canakinumab’s initial approval under exceptional circumstances was – highly exceptional – later converted to a standard approval when comprehensive evidence had come available. This was due to evidence supporting three new indications in gouty arthritis, systemic juvenile idiopathic arthritis and periodic fever syndromes, as well as two extensions of existing indications. Similarly, for certolizumab and golimumab, also various new and extended indications were approved, four and five, respectively. The second group includes for example the antidiabetics liraglutide and saxagliptin. These both saw their initial indications progressively broadened to new combination regimens, lines of treatment and age groups. In line with our general findings discussed earlier, the further drug development of these five drugs also enabled further characterization of safety profiles, with several warnings added to their SmPCs – most after new or extended indications were approved. These drugs’ lifecycles confirm that relevant regulatory learning is conditional on sufficient patient exposure. However, the relatively limited number of regulatory actions reflecting a negative impact on benefits and risks suggests that baseline uncertainty at initial approval was quite low. This is different for the drugs that received conditional approval, i.e. ofatumumab and pazopanib, indicating that less comprehensive evidence was available at initial approval. While further post-approval drug development ultimately led to extensions of the indication and other SmPC updates that reflect a positive impact, it also led to various warnings as well as DHPCs. This supports the expectation that baseline uncertainty is much higher for drugs that received conditional instead of standard approval, and underscores the need for ‘regulator-induced learning’ through obligations to generate further evidence post-approval.
Last, the frequent occurrence of risks without clear factors that drive their occurrence may define another type of drug lifecycle. This constitutes extensive unexpected post-approval characterization of the drug risk-profile without significant drug development efforts, up to the point that the initial approval decision may be reconsidered. One may describe these as potential regulatory type I errors. For example, for dronedarone, three DHPCs were distributed (21% of all DHPCs sent for this cohort), the indication was restricted to last-line treatment and a cautionary note concerning use by the elderly, contraindications and new warnings were added. It was the only drug in our cohort that underwent a referral for safety reasons, and we did not identify any regulatory actions that reflected a positive impact on benefits and risks. Although the benefit-risk balance in the broader initial indication was thus considered negative, it is currently considered a valuable therapeutic option in a strictly limited setting – mainly because its safety profile is still better than that of the more efficacious alternative drug amiodarone [Citation46].
4. Discussion
We aimed to perform an in-depth evaluation of regulatory actions during the drug lifecycle, taking into account regulatory actions that reflected positive and negative impact on benefits and risks, and the relations between them. During more than ten years of follow-up of 40 innovative medicines, 14 DHPCs were distributed that reflected a negative impact on benefits and risks. Also, 361 SmPC sections were updated, of which 24% reflected a positive impact and 76% a negative impact on benefits and risks. Of these updates, 54% occurred simultaneously with at least one update to another SmPC section. Lastly, we found that levels of post-approval patient exposure, innovativeness, needs for further regulatory learning and unexpected risks may play a role in the occurrence of specific patterns of regulatory actions during a drug lifecycle.
Our findings that almost one-fourth of SmPC updates reflected a positive impact on benefits and risks and that more than half were updated simultaneously are important, for several reasons. First, the former highlights that safety-related regulatory actions may also reflect a positive impact on risks, as exemplified by 12 regulatory actions in our study such as removed warnings and ADRs. We encountered one other study that previously reported a similar finding [Citation19], while many others only reported safety-related regulatory actions that reflect a negative impact. Second, they highlight the relative importance of regulatory actions that reflect a positive impact on benefits. These 73 regulatory actions formed one-fifth of all regulatory actions and mostly concerned new or broadened indications or an update of posology and/or administration characteristics. These were especially important given their frequent role in the further characterization of risks, with 27 leading to changes in ADRs, warnings and precautions and other risk-related sections, including changes that reflected a positive impact on risks. One previous study reported a similar finding [Citation24].
These findings are of relevance to researchers and regulators, but also to healthcare professionals and patients. First, they may prompt researchers to investigate a broader range of regulatory outcomes than is often studied as well as relations between them, or discuss their findings in this broader context. As a consequence, such studies may better reflect regulatory practice and inform their public; other researchers, clinicians and regulators. Second, these findings may prompt regulators to stimulate simultaneous learning about benefits and risks. Although regulators typically have a greater influence on the generation of risk-related evidence, e.g. through requests for monitoring in PSURs and post-authorization safety studies (PASS) [Citation47], they may influence the generation of benefit-related evidence through post-authorization efficacy studies (PAES). These include so-called ‘specific obligations’ for e.g. conditionally [Citation48] and exceptionally [Citation49] approved drugs, but may also be requested for other drugs [Citation50]. Such PAESs also form an opportunity for further characterization of risks. Similarly, when companies request scientific advice on how to study a new or broadened indication, new pharmaceutical form or new method of administration, further characterization of risks can also be stimulated. Lastly, our findings confirm that by reporting their observations during clinical practice and daily use, healthcare providers and patients play an important role in the continuous regulatory learning process about drug risks, but also about benefits.
The drug lifecycle characteristics post-approval patient exposure [Citation24,Citation35], innovativeness [Citation24,Citation34,Citation39], and need for further regulatory learning [Citation19,Citation32,Citation40] have also previously been highlighted as factors that are associated with regulatory actions. They may help regulators to plan regulatory measures, including those discussed above. Currently, these characteristics are used to e.g. define the European PSUR submission frequency. By default, once a drug is marketed in Europe, PSURs are submitted every six months for two years, then every year for two years, and then every three years [Citation51]. However, regulators may deviate from this schedule using a risk-based approach that comprises, among others, the following criteria: ‘size of the safety database and exposure to the medicinal product,’ ‘new product for which there is limited safety information available,’ ‘significant changes to the product (e.g. new indication (…), new pharmaceutical form or route of administration (…)),’ ‘medicinal products subjected to additional monitoring’ [Citation52]. This last group of medicines also includes those that received conditional approval or approval under exceptional circumstances [Citation53]. Our findings support these criteria, which could potentially also be used to define the need for other regulatory tools, such as the electronic Reaction Monitoring Report (eRMR) used for signal detection [Citation54]. Moreover, where regulators play an active role in further learning about benefits through e.g. PAESs, similar criteria could help to define the need for simultaneous characterization of the safety profile.
Our study was the first to comprehensively assess all regulatory actions that occurred during a long period of follow-up of EMA-approved innovative drugs, and assess relations between them. The findings can improve the methodology and interpretation of future studies that evaluate regulatory decision-making, and regulatory actions specifically. While manual extraction and categorization of the data were highly resource intensive, future studies could employ data science methods to perform these tasks. In addition, our findings may help regulators to plan and implement regulatory measures. However, our study also had several limitations. First, we assessed the occurrence of regulatory actions for a relatively small sample of 40 innovative drugs that were approved relatively long ago. Although the results provide important insights in regulatory decision-making that are relevant for researchers and regulators in general, our specific findings may not be generalizable to every cohort of drugs. Future studies may thus use and perhaps expand our categorization and analyses for other drugs. Second, we performed an assessment of European post-approval regulatory actions for innovative drugs. We do not expect that these are substantially different in other jurisdictions such as the United States. However, regulatory actions in one jurisdiction may play a role in the occurrence of regulatory actions in other jurisdictions. Future studies may thus employ a multi-jurisdiction perspective to evaluate these relations. Third, our unit of analysis comprised the number of regulatory actions that were encountered during each regulatory procedure, including DHPCs, rather than the precise number of issues addressed during each regulatory procedure. This mainly affected the ADR section that underwent most updates involving two or more changes. However, we considered that counting these changes as separate updates would skew our analysis of simultaneously occurring SmPC updates, which we found most important to assess in detail. Fourth, EPARs often provided limited to no details about specific information that led to regulatory actions, which required us to interpret reflected impact on benefits or risks. Specifically, we may have categorized regulatory actions as reflecting negative impact on benefits since they concerned the eligible population and use characteristics, while it would have been more correct to categorize these as reflecting negative impact on risks since they formally occurred due to safety issues. However, these concern only 15 regulatory actions that definitely reflected a negative impact, whether on benefits or risks, and thus did not impact any other results.
4.1. Conclusions
In conclusion, we identified 375 regulatory actions – 14 DHPCs and 361 SmPC updates – that occurred for 40 EMA-approved innovative drugs during more than ten years of follow-up. Of the SmPC updates, 24% reflected a positive and 76% a negative impact on benefits and risks. Moreover, simultaneous learning about benefits and risks suggests an important role for drug development in characterization of risks. Lastly, we found that the drug lifecycle characteristics post-approval patient exposure, innovativeness, need for further regulatory learning and unexpected risks play a role in the occurrence of specific patterns of regulatory actions. These findings may support the methodology and interpretation of future comprehensive regulatory analyses, and impact regulatory decision-making by stimulating simultaneous regulatory learning about benefits and risks. Also, they may help to define the need for further evidence generation.
Article highlights
As opposed to previous studies that mainly evaluated post-approval regulatory actions that reflected a negative impact on drug risks, we provide the first comprehensive evaluation of all impactful post-approval regulatory actions that occurred during drug lifecycles, for a sample of 40 innovative drugs and over ten years of follow-up.
We identified five withdrawals for commercial reasons, 14 letters to healthcare professionals and 361 label updates.
While 276 (76%) label updates reflected a negative impact and were mainly risk-related, we also identified 85 (24%) that reflected a positive impact and were mainly benefit-related, for example new or broadened indications, but also included removal of risk-related information.
Of the label updates, 194 (54%) occurred simultaneously with other label updates during 85 regulatory procedures. Of these procedures, 27 highlighted that evidence generated for post-approval drug development such as new indications simultaneously helped to further characterize drug risk profiles.
Specific patterns in post-approval regulatory actions that reflect a positive or negative impact seem to be influenced by levels of patient exposure, innovativeness, needs for regulatory learning and unexpected risks.
These findings may support future comprehensive regulatory analyses, stimulate simultaneous regulatory learning about benefits and risks, and help identify regulatory needs for evidence generation.
Declaration of interest
HGM Leufkens reported that he is a member of the Lygature Leadership Team. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
A reviewer on this manuscript has disclosed that they receive personal fees from MNES Inc., outside the submitted work. All other peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Disclaimer
The views expressed in this article are the personal views of the authors and must not be understood or quoted as being made on behalf of the Dutch Medicines Evaluation Board or any of its committees.
Supplemental Material
Download PDF (180.8 KB)Acknowledgments
The authors would like to thank colleagues of the Copenhagen Centre for Regulatory Science at the University of Copenhagen, Denmark for their kind assistance in and contributions to the identification of Dear Healthcare Professional Communications (DHPC).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14740338.2021.1952981.
Additional information
Funding
Notes
3. See for an overview of national regulatory authorities’ web pages where information on DHPCs is published
References
- Eichler HG, Pignatti F, Flamion B, et al. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat Rev Drug Discov. 2008;7(10):818–826.
- Vandenbroucke JP. Observational research, randomised trials, and two views of medical science. PLoS Med. 2008;5(3):e67.
- Duijnhoven RG, Straus SM, Raine JM, et al. Number of patients studied prior to approval of new medicines: a database analysis. PLoS Med. 2013;10(3):e1001407.
- Langedijk J, Whitehead CJ, Slijkerman DS, et al. Extensions of indication throughout the drug product lifecycle: a quantitative analysis. Drug Discov Today. 2016;21(2):348–355.
- Liu F, Jagannatha A, Yu H. Towards drug safety surveillance and pharmacovigilance: current progress in detecting medication and adverse drug events from electronic health records. Drug Saf. 2019;42(1):95–97.
- Santoro A, Genov G, Spooner A, et al. Promoting and protecting public health: how the european union pharmacovigilance system works. Drug Saf. 2017;40(10):855–869.
- Moseley J, Vamvakas S, Berntgen M, et al. Regulatory and health technology assessment advice on postlicensing and postlaunch evidence generation is a foundation for lifecycle data collection for medicines. Br J Clin Pharmacol. 2020;86(6):1034–1051. .
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). M4E(R2) guideline: the common technical document – efficacy. 2016.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). E2C(R2) Guideline: periodic Benefit-Risk Report (PBRER). 2012.
- Califf RM. Benefit-Risk assessments at the us food and drug administration: finding the balance. JAMA. 2017;317(7):693–694.
- European Medicines Agency. Day 180 joint response assessment report - Overview template rev. July.18. 2018.
- Arlett P, Portier G, De Lisa R, et al. Proactively managing the risk of marketed drugs: experience with the EMA Pharmacovigilance risk assessment committee. Nat Rev Drug Discov. 2014;13(5):395–397. .
- Potts J, Genov G, Segec A, et al. Improving the safety of medicines in the european union: from signals to action. Clin Pharmacol Ther. 2020;107(3):521–529.
- Bouvy JC, Huinink L, De Bruin ML. Benefit-risk reassessment of medicines: a retrospective analysis of all safety-related referral procedures in Europe during 2001-2012. Pharmacoepidemiol Drug Saf. 2016;25(9):1004–1014.
- Brown JP, Wing K, Evans SJ, et al. Use of real-world evidence in postmarketing medicines regulation in the European Union: a systematic assessment of European Medicines Agency referrals 2013-2017. BMJ Open. 2019;9(10):e028133.
- Farcas A, Balcescu T, Anghel L, et al. A description of medicines-related safety issues evaluated through a referral procedure at the EU level after 2012. Expert Opin Drug Saf. 2020;19(6):755–762.
- Sekine S, Pinnow EE, Wu E, et al. Assessment of the impact of scheduled postmarketing safety summary analyses on regulatory actions. Clin Pharmacol Ther. 2016;100(1):102–108.
- Ishiguro C, Misu T, Iwasa E, et al. Analysis of safety-related regulatory actions by Japan’s pharmaceutical regulatory agency. Pharmacoepidemiol Drug Saf. 2017;26(11):1314–1320.
- Mostaghim SR, Gagne JJ, Kesselheim AS. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study. BMJ. 2017;358:j3837.
- Insani WN, Pacurariu AC, Mantel-Teeuwisse AK, et al. Characteristics of drugs safety signals that predict safety related product information update. Pharmacoepidemiol Drug Saf. 2018;27(7):789–796.
- Shepshelovich D, Tibau A, Goldvaser H, et al. Postmarketing modifications of drug labels for cancer drugs approved by the US food and drug administration between 2006 and 2016 with and without supporting randomized controlled trials. J Clin Oncol. 2018;36(18):1798–1804.
- Watanabe K, Murakami M, Masuyama K, et al. The association between concerns toward adverse reactions during pre-approval drug reviews and the post-approval addition of clinically significant adverse reactions to package inserts: a retrospective analysis of pre-approval drug review reports and safety updates. Pharmacoepidemiol Drug Saf. 2018;27(11):1265–1276.
- Shepshelovich D, Tibau A, Goldvaser H, et al. Postmarketing Safety-Related modifications of drugs approved by the US Food and Drug Administration between 1999 and 2014 without randomized controlled trials. Mayo Clin Proc. 2019;94(1):74–83.
- Fujikawa M, Ono S. Analysis of Safety-Related regulatory actions for new drugs in Japan by nature of identified risks. Pharmaceut Med. 2017;31(5):317–327.
- Nakayama H, Matsumaru N, Tsukamoto K. Safety-Related regulatory actions and risk factors for anticancer drugs in Japan. Pharmaceut Med. 2019;33(1):45–52.
- Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med. 2016;14(1):10.
- Saluja S, Woolhandler S, Himmelstein DU, et al. Unsafe drugs were prescribed more than one hundred million times in the United States before being recalled. Int J Health Serv. 2016;46(3):523–530.
- Lane S, Lynn E, Shakir S. Investigation assessing the publicly available evidence supporting postmarketing withdrawals, revocations and suspensions of marketing authorisations in the EU since 2012. BMJ Open. 2018;8(1):e019759.
- Craveiro NS, Lopes BS, Tomas L, et al. Drug withdrawal due to safety: a review of the data supporting withdrawal decision. Curr Drug Saf. 2020;15(1):4–12.
- Kwon CS, Seoane-Vazquez E, Rodriguez-Monguio R. FDA safety actions for antidiabetic drugs marketed in the US, 1980-2015. Int J Risk Saf Med. 2016;28(4):197–211.
- Zeitoun JD, Lefevre JH, Downing NS, et al. Regulatory anticipation of postmarket safety problems for novel medicines approved by the EMA between 2001 and 2010: a cross-sectional study. Pharmacoepidemiol Drug Saf. 2016;25(6):687–694.
- Downing NS, Shah ND, Aminawung JA, et al. Postmarket safety events among novel therapeutics approved by the us food and drug administration between 2001 and 2010. JAMA. 2017;317(18):1854–1863. .
- Schick A, Miller KL, Lanthier M, et al. Evaluation of Pre-marketing factors to predict post-marketing boxed warnings and safety withdrawals. Drug Saf. 2017;40(6):497–503.
- Ikeda J, Kaneko M, Narukawa M. Analysis of factors related to the occurrence of important drug-specific postmarketing safety-related regulatory actions: a cohort study focused on first-in-class drugs. Pharmacoepidemiol Drug Saf. 2018;27(12):1393–1401.
- Pacurariu AC, Hoeve CE, Arlett P, et al. Is patient exposure preapproval and postapproval a determinant of the timing and frequency of occurrence of safety issues? Pharmacoepidemiol Drug Saf. 2018;27(2):168–173. .
- Pinnow E, Amr S, Bentzen SM, et al. Postmarket safety outcomes for New Molecular Entity (NME) drugs approved by the food and drug administration between 2002 and 2014. Clin Pharmacol Ther. 2018;104(2):390–400. .
- Bulatao I, Pinnow E, Day B, et al., Postmarketing Safety-Related regulatory actions for new therapeutic biologics approved in the United States 2002–2014: similarities and differences with new molecular entities. Clin Pharmacol Ther. 108(6): 1243–1253. 2020. .
- Gafter-Gvili A, Tibau A, Raanani P, et al. Safety-Related postmarketing modifications of drugs for hematological malignancies. Acta Haematol. 2020;143(1):73–77.
- Ikeda J, Kaneko M, Narukawa M. Post-marketing safety-related regulatory actions on first-in-class drugs: a double-cohort study. J Clin Pharm Ther. 2020;45(3):496–502.
- Kim J, Nair A, Keegan P, et al. Evaluation of serious postmarket safety signals within 2 years of FDA approval for new cancer drugs. Oncologist. 2020;25(4):348–354. .
- Ehmann F, Papaluca M, Di Giuseppe F, et al. Changes and determination of dosing recommendations for medicinal products recently authorised in the European Union. Expert Opin Pharmacother. 2015;16(6):903–911.
- Minnema LA, Giezen TJ, Gardarsdottir H, et al. Post-marketing dosing changes in the label of biologicals. Br J Clin Pharmacol. 2019;85(4):715–721.
- Ebbers HC, Mantel-Teeuwisse AK, Moors EHM, et al. A cohort study exploring determinants of safety-related regulatory actions for biopharmaceuticals. Drug Saf. 2012;35(5):417–427.
- European Medicines Agency. Arzerra – withdrawal of the marketing authorisation in the European Union (EMA/149805/2019). 28 February 2019.
- European Medicines Agency. Kesimpta: pending EC decision. https://www.ema.europa.eu/en/medicines/human/summaries-opinion/kesimpta. Accessed 19 March 2021.
- Adlan AM, Lip GY. Benefit-risk assessment of dronedarone in the treatment of atrial fibrillation. Drug Saf. 2013;36(2):93–110.
- European Commission. Commission Implementing Regulation (EU) No 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council. Off J Eur Union. 2012;L 159/5–25.
- European Medicines Agency. CHMP Guideline on the scientific application and the practical arrangements necessary to implement Commission Regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004 (EMA/CHMP/509951/2006, Rev. 1). 25 February 2016.
- European Medicines Agency. CHMP Guideline on procedures for the granting of a marketing authorisation under exceptional circumstances, pursuant to article 14 (8) of Regulation No 726/2004 (EMEA/357981/2005). 15 December 2005.
- European Medicines Agency. PDCO/CAT/CMDh/PRAC/CHMP Scientific guidance on post-authorisation efficacy studies (EMA/PDCO/CAT/CMDh/PRAC/CHMP/261500/2015). 12 October 2016.
- European Commission. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. Off J Eur Communities. 2001;L 311/67–128.
- European Medicines Agency, Heads of Medicines Agencies. Guideline on good pharmacovigilance practices (GVP) Module VII – Periodic safety update report (Rev 1) (EMA/816292/2011 Rev 1). 9 December 2013.
- European Medicines Agency. List of medicines under additional monitoring. https://www.ema.europa.eu/en/human-regulatory/post-authorisation/pharmacovigilance/medicines-under-additional-monitoring/list-medicines-under-additional-monitoring. Accessed 19 March 2021.
- European Medicines Agency. Screening for adverse reactions in EudraVigilance (EMA/849944/2016). 19 December 2016.