
ABSTRACT
Background
MONONOFU, a multicenter, randomized, double-blind, placebo-controlled phase 2 study of Japanese patients with migraine, was pivotal for lasmiditan approval in Japan. However, treatment-emergent adverse events (TEAEs) were more common than in global studies. A detailed safety profile would assist patient management.
Research design and methods
Safety assessments in MONONOFU included specific terms reported, frequency, severity, time to onset, duration, TEAE management, common TEAE risk factors, and TEAE–efficacy associations.
Results
Of 846 participants, 691 were assessed for safety. The proportion of participants reporting ≥1 TEAE was 23.4% with placebo and 70.9% with lasmiditan; 87.3% of TEAEs with lasmiditan were mild. The most frequent TEAEs with lasmiditan, dizziness (39.4%) and somnolence (19.3%), started ≤1 hour postdose (median durations: 2.5 and 3.3 hours, respectively). Higher lasmiditan dose, but not patient factors including body size, was identified as a clinically meaningful predictor of dizziness and somnolence. There were no adverse consequences of neurological TEAEs, which did not appear to adversely affect lasmiditan efficacy.
Conclusions
In the MONONOFU study, TEAEs appeared typically mild, transient, and self-limiting. Lasmiditan may represent a useful and well-tolerated acute treatment option for smaller (body mass index <30 kg/m2) patients and Asian patients with migraine.
GRAPHICAL ABSTRACT

1. Introduction
Migraine is a severely disabling disease that is the second highest contributor to disease burden globally [Citation1] and is associated with high levels of unmet needs related to currently available treatments [Citation2–4]. In Japan, the prevalence of migraine is similar to other countries and is estimated to be 13.0% for women and 6.6% for men ≥15 years of age [Citation4]. As in other countries, the unmet needs and burden of migraine in Japan are high, with migraine contributing to approximately 2-fold higher absenteeism and work productivity losses and almost twice as many physician visits for individuals with migraine than for those without migraine [Citation3,Citation5].
Pharmacological management of migraine during an attack comprises acute treatments for the relief of pain or limiting the duration of the attack [Citation6]. A wide range of acute treatments is available worldwide, including analgesics, antiemetics, ergotamines, nonsteroidal anti-inflammatory drugs (NSAIDs), triptans (5-hydroxytryptamine [HT] receptor antagonists) [Citation6,Citation7], and the calcitonin gene-related peptide receptor antagonists and ditans that have become available recently in some countries [Citation8]. In clinical practice in Japan, the commonly prescribed acute treatments are triptans (e.g. sumatriptan, rizatriptan, and naratriptan) and NSAIDs (e.g. loxoprofen and diclofenac) [Citation9]. However, as many as 42% of patients in Japan may be unresponsive to acute treatment [Citation3], and triptans are contraindicated in patients with cardiovascular disease or who are at risk of cardiovascular disease [Citation10,Citation11]. In addition, because triptan side effects can include chest pain and tightness, concerns about this side effect and cardiovascular-related label language for triptans may affect patient adherence.
Lasmiditan is an acute treatment for migraine with a novel mechanism of action. As a centrally penetrant, high-affinity and highly selective 5-HT1F receptor agonist, lasmiditan acts on the trigeminal system to suppress hyperactivation of trigeminal neurons and inhibits neurotransmitter release without causing vasoconstriction [Citation12,Citation13]. Lasmiditan has been studied in four global phase 3 studies [Citation14–17] and is approved as an oral acute treatment for migraine in the United States. Consistent with these studies, findings from a randomized, double-blind, placebo-controlled phase 2 trial (MONONOFU) showed that, in general, lasmiditan is a well-tolerated and effective acute treatment for migraine in Japan [Citation18]. Although the types of treatment-emergent adverse events (TEAEs) reported in the MONONOFU study were similar to those in the global studies, the incidence of TEAEs in the 100-mg and 200-mg lasmiditan treatment groups in Japanese patients appeared to be higher than in non-Japanese patients enrolled in phase 3 studies [Citation17–19].
Given the novel mechanism of action of lasmiditan, the collection of detailed information on the safety of lasmiditan is required to inform its use in clinical practice. The population in the MONONOFU study had a smaller body size (mean body mass index [BMI] 22.6 kg/m2) compared with the phase 3 global study populations (BMI 30–31 kg/m2) [Citation14–16,Citation18] and therefore may be more vulnerable to adverse effects. In addition, the higher incidence of TEAEs in the MONONOFU study compared with the global studies presents an opportunity to describe and assess a greater variety of adverse events. The aim of this report is to provide a detailed assessment of the safety of lasmiditan for acute treatment of migraine from the MONONOFU study data, including any potential impact of TEAEs on the efficacy of lasmiditan.
2. Patients and methods
2.1. Study design
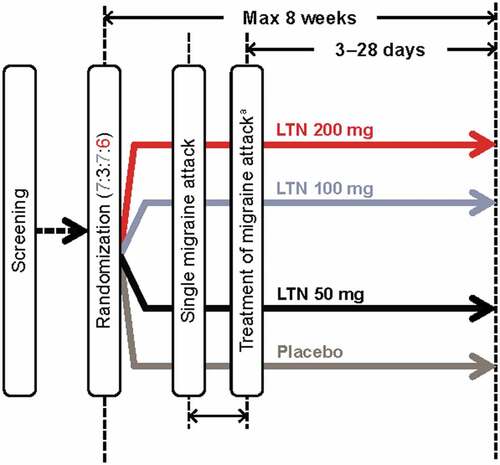
MONONOFU was a prospective, multicenter, randomized, double-blind, placebo-controlled phase 2 study conducted at 34 sites in Japan from 30 May 2019 to 8 June 2020 (). The primary objective was to assess the efficacy of lasmiditan 200 mg on migraine headache pain freedom compared with placebo in Japanese adults with migraine with or without aura as previously reported [Citation18]. The study was conducted in accordance with the Declaration of Helsinki, the relevant laws and regulations in Japan, and the Guidance on Ethnic Factors in the Acceptability of Foreign Clinical Data (ICH-E5). The protocol was approved by local institutional review boards for each site. All patients gave written informed consent to participate in the trial. The study was registered at https://clinicaltrials.gov/ct2/home (NCT03962738).
Figure 1. Study design. aHeadache severity must be at least moderate or severe and treated within 4 hours of pain onset. LTN lasmiditan, Max maximum.
2.2. Study population
Full details of the MONONOFU study have been previously described [Citation18]. Key inclusion criteria of the study were male or female patients ≥18 years of age who had migraine with or without aura fulfilling the International Headache Society (IHS) diagnostic criteria 1.1 and 1.2.1, a history of disabling migraine for ≥1 year, and a Migraine Disability Assessment score ≥11.
Patients were excluded if they had any of the following: a known hypersensitivity to lasmiditan or to any excipient of lasmiditan oral tablets; a history of chronic migraine or other forms of primary or secondary chronic headache disorder within the past 12 months; history or evidence of hemorrhagic stroke, epilepsy, or any other condition placing the patient at increased risk of seizures; or a history of recurrent dizziness and/or vertigo including benign paroxysmal positional vertigo, Ménière’s disease, vestibular migraine, other vestibular disorders, and orthostatic hypotension with syncope.
2.3. Randomization and treatment protocol
Eligible patients were randomized in a 7:3:7:6 ratio to placebo or lasmiditan 50 mg, 100 mg, or 200 mg using an interactive web-response system, stratified by current use of concomitant medications (yes/no). The study comprised a screening visit, a randomization visit, treatment for up to 8 weeks, and an end-of-study visit (3–28 days after treatment of the migraine attack) (). Participants were asked to treat a moderate-to-severe migraine attack within 4 hours of pain onset. A qualifying migraine attack was one that was not improving and for which no other acute migraine treatments had been taken within the previous 24 hours. Rescue medication (NSAIDs, acetaminophen, and/or antiemetics) could be taken after completion of the 2-hour postdose assessments if patients were not headache pain-free. Triptans, ergots, opioids, and barbiturates were not permitted within 24 hours postdose of study drug.
2.4. Outcome measures and study endpoints
2.4.1. Safety
All adverse events (AEs), concomitant medication use, nonstudy rescue/recurrence medication use, and menstrual cycle status were recorded by participants in a paper diary. A TEAE was defined as a new or worsening AE within 48 hours postdose of study drug and did not need to have a causal relationship with treatment. If multiple severities were reported for a given TEAE for a participant, the highest severity was counted. TEAEs were coded using the Medical Dictionary for Regulatory Activities Version 23.0 (MedDRA 23.0). Assessments of safety included the frequency and severity of TEAEs, treatment-relatedness of TEAEs (as determined by the investigators), serious AEs, discontinuations due to AEs, time from dosing to TEAE onset, and TEAE duration. Cardiovascular risk factors were defined in accordance with the Comprehensive Risk Management Chart for Cerebro-Cardiovascular Disease, 2019 [Citation20].
Other safety measures included cardiovascular safety; accidents/injury secondary to neurological TEAEs; hepatic safety; suicidal ideation and behavior and nonsuicidal self-injury; hypersensitivity events; vital signs; and body weight, electrocardiograms (ECGs), and laboratory data. TEAEs that could potentially be related to serotonin syndrome were medically reviewed to determine if the Hunter [Citation21] or Sternbach [Citation22] criteria were met. Because of the potential for misunderstanding between terms related to dizziness and vertigo [Citation23,Citation24], a descriptive analysis of all events associated with vertigo (consolidated term) was conducted. The preferred terms included in this analysis were vertigo, cervicogenic vertigo, vertigo central nervous system origin, vertigo positional, and vertigo labyrinthine. A stepwise regression analysis of the most influential baseline predictors of the most common TEAEs (dizziness, somnolence, and malaise) was also conducted (see Statistical analysis section for details).
Clinical questions were issued for AEs if the start date of the AE was the day of or the day after the study intervention was taken, the severity was moderate or severe, and if the AE was considered related to the study intervention. The clinical data questions included questions about activities that worsened or improved the AE (‘What activity made this AE worse?,’ ‘What activity made this AE better?’).
2.4.2. Efficacy
Efficacy responses were recorded in an electronic diary. There were four efficacy endpoints included in this safety analysis: (1) the proportion of participants in each group at 2 hours postdose who were headache pain-free (moderate or severe headache pain at predose becoming no pain), where headache severity was defined in accordance with the IHS 4-point headache severity rating scale from 0 (no pain) to 3 (severe pain); (2) the proportion of participants in each group at 2 hours postdose who were free from the most bothersome symptom associated with migraine (identified by participants from associated symptoms at predose, including nausea, phonophobia, or photophobia); (3) the proportion of participants in each group at 2 hours postdose who experienced no disability, where disability was assessed via the question ‘How much is your migraine interfering with your normal activities?’ (with four response options: not at all, mild interference, marked interference, and need complete bed rest); and (4) the proportion of participants in each group at 2 hours postdose who were classified as responders (very much better or much better) in accordance with the Patient Global Impression of Change (PGI-C) question ‘How do you feel after taking study medication?’ (response was measured on a 7-point scale from ‘very much better’ to ‘very much worse’).
2.5. Statistical analysis
Details of the calculation of sample size have been previously published [Citation18]. Safety analyses were conducted using the safety population, which included all randomized participants who received ≥1 dose of study drug. Efficacy analyses were conducted using the modified intent-to-treat (mITT) population, in which participants were treated with ≥1 dose of study drug, had ≥1 postdose assessment, and were treated for a qualifying migraine attack within 4 hours.
A subgroup safety analysis was conducted to determine the frequency of TEAEs in participants who had and had not used a triptan within 3 months of informed consent. Categorical safety measures are reported using descriptive statistics. Associations between the presence or absence of the five most common TEAEs (affecting ≥5% of all participants who received lasmiditan) and efficacy endpoints (pain-free, PGI-C, free of most bothersome symptom, and disability) at 2 hours postdose in participants treated with lasmiditan were calculated. These calculations are reported as fractional values in ‘yes’ or ‘no’ groupings, where yes/no refers to the presence/absence of TEAEs within 2 hours postdose in participants treated with lasmiditan.
To identify the most influential baseline predictors of dizziness, somnolence, and malaise, a forward stepwise logistic regression was used with a cutoff P-value = 0.05, following the method of Tepper and colleagues [Citation24]. Dizziness, somnolence, and malaise were selected as the TEAEs of interest because they affected ≥10% of all participants who received lasmiditan in the safety population. The analysis included 25 variables arranged into six variable groups based on their characteristics. Data types for each variable were specified as continuous, binary, or three categories (in the case of lasmiditan dose). Variables, variable groups, data types, and category values are summarized in Supplementary Table 1. The response variable in the forward stepwise logistic regression procedure was presence/absence of each of the TEAEs of interest (dizziness, somnolence, or malaise). In the first step, the group 1 variables were the predictors. In the second step, only the group 1 variables that were significant (below the cutoff P-value) were retained, and the group 2 variables were added as predictors. This process was repeated up to group 6 to identify the most influential predictors for each TEAE. These predictors were then included in a multiple logistic regression model. For each predictor in the final model, estimated odds ratios (ORs), P-values, and 95% confidence intervals (CIs), and the P-value for the overall effect of that predictor, were calculated. For continuous variables, ORs were based on 1-unit change of the variable.
All analyses were conducted using SAS 9.4 (SAS Institute Inc., Cary, NC, USA).
3. Results
3.1. Participant disposition and baseline characteristics
A total of 846 participants were randomized to the placebo and lasmiditan dose groups (placebo N = 258, lasmiditan 50 mg N = 109, 100 mg N = 261, and 200 mg N = 218). Of those, 691 (placebo N = 214, lasmiditan 50 mg N = 87, 100 mg N = 208, and 200 mg N = 182) were treated with ≥1 dose of any study drug and were included in the safety population (), and 682 (placebo N = 211, lasmiditan 50 mg N = 85, 100 mg N = 207, and 200 mg N = 179) met the criteria for the mITT population. Baseline demographics and characteristics of participants in each treatment group were similar (). Approximately 35% of participants were ≥50 years old. Around 78% of participants had a mean BMI below 25 kg/m2 and 9.3% had a BMI below 18.5 kg/m2.
Table 1. Summary of baseline demographics and characteristics (safety population)a.
3.2. Safety and tolerability measures
Most TEAEs were of mild severity (87.3% for all patients who received lasmiditan; ), including the most common TEAEs (dizziness, somnolence, malaise, asthenia, and hypoesthesia, nausea and muscular weakness; and Supplementary Table 2). Among patients who experienced moderate or severe TEAEs and were asked the clinical questions, the most common answer to the question ‘What activity made this AE worse?’ was any movement (e.g. getting up, walking, reading). The most common answers to ‘What activity made this AE better?’ were lying down, having a sleep, or waiting for time to pass.
Figure 2. Severity of common TEAEs (≥5% in any lasmiditan group). TEAEs shown are: (a) dizziness; (b) somnolence; (c) malaise; (d) asthenia; (e) hypoesthesia; (f) nausea; and (g) muscular weakness. TEAE treatment-emergent adverse event.
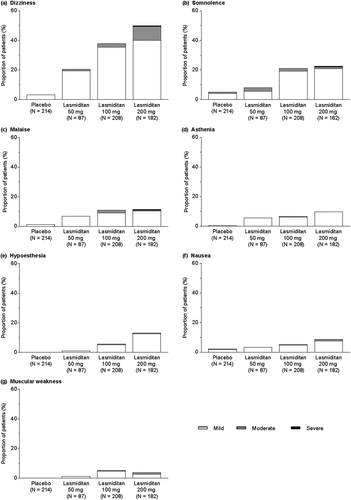
Table 2. Overview of AEs and TEAEs (safety population)a.
Dizziness was the most common TEAE among all participants treated with lasmiditan (39.4%; ). The MedDRA term ‘dizziness’ included several patient- and physician-reported terms, with the most common being ‘dizziness,’ ‘dizzy,’ ‘light-headed feeling,’ and ‘swaying feeling’; ‘somnolence’ included reported terms such as ‘sleepiness,’ ‘somnolence,’ and ‘drowsiness’ (Supplementary Table 3). The median time from dosing to onset of the most common TEAEs was ≤1 hour (). The median duration of dizziness among all patients who received lasmiditan was 2.5 hours and that of the other common TEAEs ranged from 1.1 hours (hypoesthesia) to 3.7 hours (muscular weakness).
Table 3. Common TEAEs (≥2.0% in any lasmiditan group)a.
Table 4. Time (dosing–onset) and duration of most common TEAEs (≥5% in any lasmiditan group).
Vertigo was reported in one (0.5%) participant in the placebo group and 16 (3.4%) participants in the all-lasmiditan group (all patients who received lasmiditan at any dose). Similar to the more common TEAEs, the severity of vertigo was mild in most participants (87.5%), and no TEAEs were rated as severe or serious. For the all-lasmiditan group, the median time to onset of vertigo was 0.6 hours postdose, and the median duration was 4.3 hours.
The incidence of TEAEs in participants who had and had not used triptans within 3 months of enrollment was 24% (47/196) and 16.7% (3/18), respectively, for the placebo group and was 70.0% (301/430) and 78.7% (37/47), respectively, for the all-lasmiditan group.
There were no injuries or accidents secondary to neurological TEAEs, no TEAEs related to hepatic injury and function, and no AEs related to suicidal ideation and behavior or nonsuicidal self-injury. In addition, there were no clinically relevant changes in vital signs, laboratory tests, or ECG results collected and interpreted by investigators at the last study visit as part of AE follow-up. Anaphylactic reaction, hypersensitivity, and angioedema were not reported in lasmiditan- or placebo-treated participants. No participants met the Hunter or Sternbach criteria for serotonin syndrome based on medical review. The incidence of likely cardiovascular TEAEs was low (all-lasmiditan 3.8% [18/477], placebo 1.4% [3/214]); the most common likely cardiovascular TEAE was palpitations (all-lasmiditan 3.6% [17/477], placebo 1.4% [3/214]). One patient in the lasmiditan group reported mild ‘faintness’ 2.3 hours postdose that resolved immediately; this was coded as the preferred term ‘syncope’ under MedDRA 23.0. The patient had no cardiovascular-related medical history and the event resolved with no action. All cardiovascular events were nonserious, and all participants recovered without requiring intervention. There were no ischemic cardiovascular TEAEs reported. One participant in the lasmiditan 200-mg group experienced treatment-emergent palpitations of moderate severity; all other cardiovascular TEAEs were considered to be mild in severity.
3.2.1. Relationship between TEAEs and efficacy endpoints
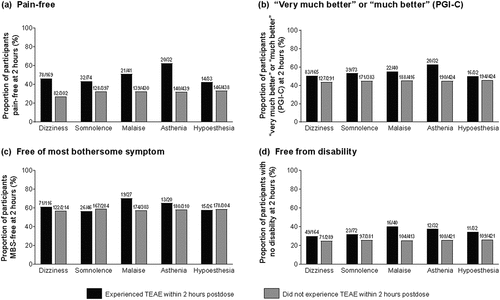
We assessed whether the occurrence of the five most common TEAEs (dizziness, somnolence, malaise, asthenia, and hypoesthesia) affected the efficacy of lasmiditan and found no negative relationship between TEAEs and efficacy endpoints (). In fact, the proportion of patients at 2 hours postdose who were free from pain, free from their most bothersome symptoms, felt very much better or much better (PGI-C), or experienced no disability was generally numerically higher in those who experienced the top five most common TEAEs in the 2 hours postdose compared with those who did not ().
Figure 3. Association between top five most common TEAEs and efficacy endpoints in patients treated with lasmiditan. Presence or absence within 2 hours postdose of TEAEs (≥5%) among: (a) pain-free participants; (b) participants who felt ‘very much better’ or ‘much better’ (PGI-C); (c) participants who were free of their most bothersome symptom; and (d) participants who experienced no disability. MBS most bothersome symptom, PGI-C Patient Global Impression of Change, TEAE treatment-emergent adverse event.
3.2.2. Predictors of dizziness, somnolence, and malaise
Dizziness, somnolence, and malaise affected ≥10% of all participants who received lasmiditan in the safety population and were selected for predictor analysis to identify the most influential risk factors for each TEAE. Among patients who received lasmiditan and experienced dizziness, the majority (~90%) received 100-mg or 200-mg doses (). Higher lasmiditan dose was identified as a statistically significant risk factor for dizziness (P < 0.001; ), with the highest ORs seen for lasmiditan doses of 200 mg versus 50 mg and 100 mg versus 50 mg (4.05 [95% CIs: 1.94–8.46] and 2.39 [1.16–4.95], respectively). Differences were also observed in mean age, body weight, and BMI between patients who did and did not experience dizziness (), but these differences were small and not clinically relevant. The only significant risk factor identified for somnolence was a higher lasmiditan dose (P < 0.01) (; ). No significant risk factors were identified for malaise (data not shown).
Figure 4. Forest plots of statistically significant predictors of (a) dizziness and (b) somnolence in patients receiving lasmiditan. P-values are shown for each OR. For the continuous variables of age and BMI, ORs were based on 1-unit change of the variable. The overall effect P-values for lasmiditan dose were P < 0.0001 and P = 0.0184 for dizziness and somnolence, respectively. BMI body mass index, CI confidence interval, OR odds ratio.
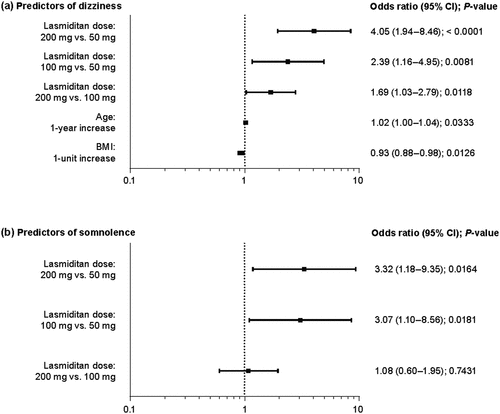
Table 5. Risk factors associated with dizziness and somnolence among patients who received lasmiditan.
4. Discussion
This is the first study to provide a detailed assessment of the safety profile of lasmiditan for acute treatment of migraine with and without aura in Asian patients, who typically have a smaller body size than patients from global populations. Although the incidence of TEAEs was relatively high, most TEAEs were mild in severity, occurred within 1 hour of treatment, and were transient and self-limiting. The most common TEAEs (affecting ≥10% of patients) were dizziness, somnolence, and malaise; the most influential risk factor for dizziness and somnolence was a higher lasmiditan dose; no significant risk factors were identified for malaise. Body size was not considered a clinically meaningful risk factor for any of these TEAEs. Moreover, the occurrence of TEAEs did not appear to negatively affect the efficacy of lasmiditan. In fact, relief from migraine symptoms appeared to be associated with the most common TEAEs.
Consistent with studies conducted in non-Asian patients [Citation19] and the pharmacokinetic profile of lasmiditan in healthy Japanese individuals [Citation25], the proportion of patients reporting any TEAE increased with increasing lasmiditan dose. As previously reported [Citation18], the incidence of TEAEs in the MONONOFU study was higher than that reported for patients in the global clinical trials [Citation14–17]. However, the types of TEAEs reported in the MONONOFU study were consistent with those in the global clinical trials [Citation14–17]. The TEAEs that occurred most commonly (≥5% of patients in the all-lasmiditan group) in the MONONOFU study were dizziness (including the reported terms light-headed feeling and swaying feeling), somnolence (including sleepiness and drowsiness), malaise, asthenia (including feeling of weakness and general weakness), and hypoesthesia. Similarly, the most common TEAEs in a pooled analysis of the global phase 3 studies of lasmiditan were dizziness, paresthesia, somnolence, fatigue, nausea, muscular weakness, and hypoesthesia [Citation19]. In general, these adverse effects are in line with the specific effects of 5-HT1F receptor agonist activity on the central nervous system and do not include chest pain that is associated with the use of triptans [Citation11]. The adverse effects were typically transient and self-limiting, and patients commonly reported that lying down or having a sleep were sufficient to resolve the symptoms of a moderate-to-severe adverse event. The transient nature of the central nervous system effects is supported by a recent placebo-controlled study demonstrating that although simulated driving ability was impaired at 1.5 hours after lasmiditan, there was no impairment at or after 8 hours [Citation26]. Therefore, the warning statement was included in the lasmiditan label.
The most influential risk factor for dizziness with lasmiditan in the current analysis was higher lasmiditan dose. Older age and lower BMI were not considered to be clinically meaningful risk factors, given the ORs were close to 1 and there were only small differences in mean age and BMI between patients who did and did not experience dizziness. These results are somewhat consistent with a pooled safety analysis of data from global clinical trials of lasmiditan, which showed that higher lasmiditan dose and mild or moderate severity of a migraine attack were clinically relevant risk factors for dizziness [Citation24]. We also identified that higher lasmiditan dose was a risk factor for somnolence; no significant risk factors were identified for malaise. Risk factors for somnolence and malaise were not determined in the global safety analysis. These predictor analyses demonstrate that the most clinically relevant risk factor for TEAEs with lasmiditan is drug dose rather than patient factors.
The presence or absence of common TEAEs did not appear to negatively affect the efficacy of lasmiditan, consistent with the pooled analysis of data from the global clinical trials [Citation19]. On the contrary, the proportion of patients who were pain-free appeared to be numerically higher in those reporting the five most common TEAEs, although no statistical analysis was conducted. This pattern was seen to a lesser extent in patients who were free from their most bothersome symptoms, felt very much better or much better (PGI-C), and experienced no interference with their daily activities at 2 hours postdose. This is consistent with the finding in patients from the global clinical trials that dizziness appeared to be associated with numerically greater freedom from pain and freedom from their most bothersome symptoms [Citation24]. One implication of this result, as previously suggested for dizziness [Citation24], is that dizziness and other common TEAEs may be linked with efficacy through a common mechanism of action, a possibility that appears likely given that the central nervous system is the primary location of 5-HT1F receptors. Alternatively, it is possible that patients who experience more freedom from pain and/or relief from other migraine symptoms may be more aware of and able to report side effects [Citation24], although this seems less likely given that the TEAEs in the current analysis were recorded within 2 hours postdose.
The strengths of the current study included the fact that there was no upper age limit (oldest patient was 73 years of age) and that those with comorbid cardiovascular disease and/or cardiovascular risk factors [Citation18], taking preventive medications, or with prior triptan experience were not excluded. The population had at least moderate migraine disability and is representative of the general Asian population expected to seek improved treatment for their migraine. However, the safety analyses were not powered for statistical significance. There were also fewer patients in the lasmiditan 50-mg dose group than in the other dose groups, which limits the interpretation of findings from this subgroup. Despite this, the current study provides clinically relevant information on the use of lasmiditan in a study population that is of smaller body size compared with the phase 3 global study populations.
5. Conclusions
Findings from this study of lasmiditan for acute treatment of migraine in Japanese patients have shown that despite the high TEAE frequency reported, most TEAEs were mild in severity, transient, self-limiting, and typically resolved using interventions such as rest or sleep. Patients in this study were of a smaller body size than in the global clinical trials and thus provide a useful proxy for Asian populations in general. Lasmiditan has not been associated with any major safety issues such as ischemic cardiovascular events, accidents, or injuries. Lasmiditan dose, rather than any patient factor, was the only clinically relevant predictor of common TEAEs. The occurrence of TEAEs did not appear to have an adverse effect on the efficacy of lasmiditan. Lasmiditan may represent a useful acute treatment option for smaller patients and Asian patients with migraine.
Declaration of interest
K Hirata received research funding from the Japanese Ministry of Health, Labour and Welfare and the Japan Agency for Medical Research and Development and has received personal fees from Amgen Astellas BioPharma K.K., Daiichi Sankyo Company, Limited, Eisai Co., Ltd., Eli Lilly Japan K.K., MSD K.K., Otsuka Pharmaceutical Co., Ltd., and Pfizer Japan Inc. Y Matsumori received personal consultancy fees from Amgen Astellas BioPharma K.K., Daiichi Sankyo Company, Limited, Eli Lilly Japan K.K., and Otsuka Pharmaceutical Co., Ltd. during the conduct of the study. Y Tanji, A Ozeki, and M Komori are employees of Eli Lilly Japan K.K. and have minor shareholdings in Eli Lilly and Company. R Khanna is an employee of Eli Lilly and Company, Limited – UK and has minor shareholdings in Eli Lilly and Company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from Expert Opinion on Drug Safety for their review work but have no other relevant financial relationships to disclose.
Ethics approval and consent to participate
The MONONOFU study was registered at https://clinicaltrials.gov/ct2/home (NCT03962738). The protocol was reviewed and approved by a central ethical review board and the appropriate institutional review boards for each site, which also reviewed any amendments to the protocol (Supplementary Table 4). The study was conducted in accordance with the Declaration of Helsinki 1964 and its later amendments, the relevant laws and regulations in Japan, the Guidance on Ethnic Factors in the Acceptability of Foreign Clinical Data (ICH-E5), and the Council for International Organizations of Medical Sciences International Ethical Guidelines. All participants provided written informed consent to participate in the study.
Author contributions
K Hirata and Y Matsumori were investigators in the study. Y Tanji was involved in data curation and validation. A Ozeki conducted the statistical analysis and was involved in data curation and validation. M Komori was involved in study conceptualization, data curation, and validation. All authors participated in the interpretation of study results, and in the drafting, critical revision, and approval of the final version of the manuscript. All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole, agree to be accountable for all aspects of the work, and have given their approval for this version to be published.
Supplemental Material
Download MS Word (28.1 KB)Acknowledgments
We thank the patients for their participation, and the investigators and their support staff in this study. The authors thank John Krege of Eli Lilly and Company for his critical input for the study design and interpretation of the data, Koichi Yamaguchi and Gosuke Homma of Eli Lilly Japan K.K. for their contributions to the statistical analyses, and Yasuchika Yamamoto of Eli Lilly Japan K.K. for his contributions to the case review of the study report.
Data availability statement
Eli Lilly and Company provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the United States and the European Union and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14740338.2022.2087630
Additional information
Funding
References
- Steiner TJ, Stovner LJ, Jensen R, et al. Migraine remains second among the world’s causes of disability, and first among young women: findings from GBD2019. J Headache Pain. 2020;21(1):137.
- Lipton RB, Buse DC, Serrano D, et al. Examination of unmet treatment needs among persons with episodic migraine: results of the American Migraine Prevalence and Prevention (AMPP) study. Headache. 2013;53(8):1300–1311.
- Hirata K, Ueda K, Ye W, et al. Factors associated with insufficient response to acute treatment of migraine in Japan: analysis of real-world data from the Adelphi migraine disease specific programme. BMC Neurol. 2020;20(1):274.
- Sakai F, Igarashi H. Prevalence of migraine in Japan: a nationwide survey. Cephalalgia. 1997;17(1):15–22.
- Igarashi H, Ueda K, Jung S, et al. Social burden of people with the migraine diagnosis in Japan: evidence from a population-based cross-sectional survey. BMJ Open. 2020;10(11):e038987.
- Japanese Society of Neurology and the Japanese Headache Society. Clinical practice guideline for chronic headache 2013 [cited 2021 Mar 30]. Available from: https://www.neurology-jp.org/guidelinem/ch/documents/preface.pdf
- Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American headache society evidence assessment of migraine pharmacotherapies. Headache. 2015;55(1):3–20.
- American Headache Society. The American headache society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59(1):1–18.
- Ueda K, Ye W, Lombard L, et al. Real-world treatment patterns and patient-reported outcomes in episodic and chronic migraine in Japan: analysis of data from the Adelphi migraine disease specific programme. J Headache Pain. 2019;20(1):68.
- Dodick D, Lipton RB, Martin V, et al. Consensus statement: cardiovascular safety profile of triptans (5-HT agonists) in the acute treatment of migraine. Headache. 2004;44(5):414–425.
- Gonzalez-Hernandez A, Marichal-Cancino BA, MaassenVanDenBrink A, et al. Side effects associated with current and prospective antimigraine pharmacotherapies. Expert Opin Drug Metab Toxicol. 2018;14(1):25–41.
- Nelson DL, Phebus LA, Johnson KW, et al. Preclinical pharmacological profile of the selective 5-HT1F receptor agonist lasmiditan. Cephalalgia. 2010;30(10):1159–1169.
- Clemow DB, Johnson KW, Hochstetler HM, et al. Lasmiditan mechanism of action - review of a selective 5-HT(1F) agonist. J Headache Pain. 2020;21(1):71.
- Kuca B, Silberstein SD, Wietecha L, et al. Lasmiditan is an effective acute treatment for migraine: a phase 3 randomized study. Neurology. 2018;91(24):e2222–e2232.
- Goadsby PJ, Wietecha LA, Dennehy EB, et al. Phase 3 randomized, placebo-controlled, double-blind study of lasmiditan for acute treatment of migraine. Brain. 2019;142(7):1894–1904.
- Brandes JL, Klise S, Krege JH, et al. Interim results of a prospective, randomized, open-label, phase 3 study of the long-term safety and efficacy of lasmiditan for acute treatment of migraine (the GLADIATOR study). Cephalalgia. 2019;39(11):1343–1357.
- Ashina M, Reuter U, Smith T, et al. Randomized, controlled trial of lasmiditan over four migraine attacks: findings from the CENTURION study. Cephalalgia. 2021;41(3):294–304.
- Sakai F, Takeshima T, Homma G, et al. Phase 2 randomized placebo-controlled study of lasmiditan for the acute treatment of migraine in Japanese patients. Headache. 2021;61(5):755–765.
- Krege JH, Rizzoli PB, Liffick E, et al. Safety findings from phase 3 lasmiditan studies for acute treatment of migraine: results from SAMURAI and SPARTAN. Cephalalgia. 2019;39(8):957–966.
- The Japanese Council on Cerebro-Cardiovascular Disease. Comprehensive risk management chart for cerebro-cardiovascular disease 2019. Nihon Naika Gakkai Zasshi. 2019;108(5):1024–1074.
- Dunkley EJC, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635–642.
- Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):705–713.
- Kudrow D, Krege JH, Hundemer HP, et al. Issues impacting adverse event frequency and severity: differences between randomized phase 2 and phase 3 clinical trials for lasmiditan. Headache. 2020;60(3):576–588.
- Tepper SJ, Krege JH, Lombard L, et al. Characterization of dizziness after lasmiditan usage: findings from the SAMURAI and SPARTAN acute migraine treatment randomized trials. Headache. 2019;59(7):1052–1062.
- Komori M, Mimura H, Tsai M, et al. Safety, tolerability, and pharmacokinetics of lasmiditan in healthy Japanese and caucasian subjects. Rinsho Yakuri. 2020;51(3):119–127.
- Pearlman EM, Wilbraham D, Dennehy EB, et al. Effects of lasmiditan on simulated driving performance: results of two randomized, blinded, crossover studies with placebo and active controls. Hum Psychopharmacol. 2020;35(5):e2732.