
ABSTRACT
Background
Interleukin (IL)-36 signaling has been shown to be increased in ulcerative colitis (UC). Spesolimab, a novel humanized monoclonal antibody, targets the IL-36 pathway.
Research Design and Methods
We report safety, immunogenicity, and efficacy data of intravenous (IV) spesolimab in UC. Study 1: phase II, randomized, placebo-controlled trial (300 mg single dose; 450 mg every 4 weeks [q4w]; or 1,200 mg q4w, three doses). Study 2: phase IIa, randomized, placebo-controlled trial (1,200 mg q4w). Study 3: phase IIa, open-label, single-arm trial (1,200 mg q4w). Studies lasted 12 weeks, with a 12-, 24-, and 16-week safety follow-up, respectively.
Results
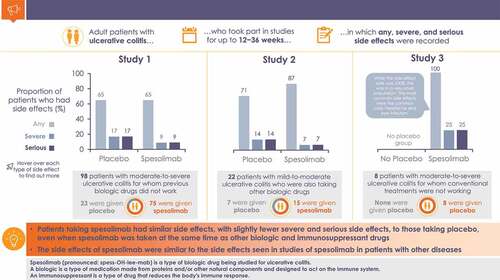
Adver+se event (AE) rates were similar for spesolimab and placebo in Studies 1 (N = 98; 64.9%; 65.2%) and 2 (N = 22; 86.7%; 71.4%); all patients in Study 3 (N = 8) experienced AEs. The most frequent investigator-assessed drug-related (spesolimab; placebo) AEs were skin rash (5.4%; 0%) and nasopharyngitis (4.1%; 0%) in Study 1; acne (13.3%; 0%) in Study 2; one patient reported skin rash, nasopharyngitis, headache, and acne in Study 3. Efficacy endpoints were not met.
Conclusions
Spesolimab was generally well tolerated, with no unexpected safety concerns. The safety data are consistent with studies in other inflammatory diseases.
Graphical abstract
1. Introduction
Ulcerative colitis (UC) is a chronic, relapsing-remitting inflammatory disease affecting the colon, leading to disabling, life-altering symptoms including bloody diarrhea, urgency, and abdominal pain [Citation1–3]. UC can be associated with considerable extraintestinal manifestations, and those with extensive disease have an increased risk of developing colorectal cancer [Citation1,Citation3,Citation4]. Colectomy may be required in up to 16% of patients to treat complications or medically refractory disease [Citation1–3,Citation5]. Overall, in addition to bowel symptoms, patients with active UC experience substantially reduced quality of life, increased levels of anxiety and depression, and detrimental effects on activities of daily living, social activities, and employment [Citation6,Citation7].
Various recommendations exist regarding treat-to-target goals in UC and the definition of clinical remission, which combines endoscopic remission (modified endoscopic subscore [mESS] of the Mayo Clinic Score [MCS] 0 or 1) and patient-reported outcomes such as resolved rectal bleeding and normalization of bowel habit [Citation2,Citation8]. Several treatment options, including conventional agents (e.g. aminosalicylates, corticosteroids, thiopurines), biologics (e.g. tumor necrosis factor alpha [TNF-α] inhibitors, vedolizumab, and ustekinumab), and small molecules (e.g. tofacitinib and ozanimod), are available and recommended to induce and maintain remission in patients with moderate-to-severe, active UC [Citation2,Citation3,Citation9–11]. However, many patients fail to achieve a clinical response or remission, experience loss of response after initial improvement, or experience use-limiting complications, and despite available treatments, around one-fifth will require surgery [Citation1,Citation2,Citation5,Citation9,Citation12,Citation13].
There is currently a clear unmet need for more effective, longer-lasting, and better-tolerated treatments, and for greater effectiveness particularly in patients previously treated with TNF-α inhibitors [Citation2,Citation14]. Such therapies are needed to achieve the disease modification goals outlined in the Selecting Endpoints for Disease-Modification Trials (SPIRIT) consensus [Citation15]. The need for safer treatments is exemplified by the fact that patients with moderate-to-severe UC experience adverse events (AEs) that lead to discontinuation of treatment [Citation16,Citation17]; a long-term safety study of vedolizumab reported a rate of 15% [Citation18].
Although the cause of UC remains unclear, a complex interplay of environmental and genetic factors is likely involved, along with changes in the colonic microbiome and immune system that result in pathological inflammatory responses [Citation1,Citation3,Citation19]. Increased interleukin (IL)-36 receptor (IL-36 R) signaling is a feature of chronic inflammatory conditions [Citation20–24], including Crohn’s disease, UC, palmoplantar pustulosis (PPP), and generalized pustular psoriasis (GPP). Studies have shown elevated levels of IL-36 R agonists in the colonic mucosa of patients with active UC [Citation21,Citation25]. Similarly, inhibition or knockout of the IL36R gene in murine models has shown reduced chronic colitis and intestinal fibrosis [Citation25,Citation26]. Subsequent IL-36 R signaling is thought to amplify the proliferation of gut cell populations that further promote recruitment and activate intestinal inflammation [Citation21]. Spesolimab, a novel, humanized monoclonal immunoglobulin G1 antibody that specifically targets and blocks IL-36 R signaling [Citation22,Citation27], has been shown to be an effective therapy for GPP and, in a post-hoc analysis, provide symptomatic improvement for patients with PPP. In these two indications, spesolimab has also been shown to be generally well tolerated, with no clinically relevant safety signals yet identified [Citation22,Citation24,Citation28].
Here, we report safety, immunogenicity, and efficacy data from three phase II/IIa clinical trials of spesolimab in patients with moderate-to-severe UC.
2. Patients and methods
2.1. Study designs
The study designs for each of the three studies in UC presented in this paper are summarized in and in the following sections.
Table 1. Clinical trials of intravenous spesolimab as induction therapy in patients with ulcerative colitis.
Full criteria for the determination of sample sizes, inclusion and exclusion criteria, and methods for randomization and blinding in each of the three studies are listed in Supplemental Tables S1–S5. Each study assessed safety through AEs, serious AEs (SAEs), and treatment-emergent AEs (TEAEs; definitions are given in Supplemental Table S6); user-defined adverse event categories (UDAECs; definitions given in Supplemental Table S7), all safety analyses, laboratory test results, and immunogenicity analyses were descriptive, and no safety hypothesis testing was performed. In Study 2, TNF-α inhibitor levels were analyzed at baseline and at last treatment administration. In all three studies, the intensity of AEs and abnormal laboratory values was assessed using Rheumatology Common Toxicity Criteria (RCTC) version 2.0 [Citation32]; AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 22.1 or 23.0.
2.1.1. Study 1 (1368-0005; NCT03482635)
This was a multicenter, randomized, double-blind, placebo-controlled, parallel-group phase II trial comparing three dose groups of spesolimab with placebo over a 12-week treatment period (). The objectives were to evaluate the clinical activity of spesolimab as induction therapy in patients with moderate-to-severe UC who had failed previous biologics (TNF-α inhibitor and/or vedolizumab) and to identify an effective and safe spesolimab dose. Patients were randomized (1:1:1:1) to receive either one single intravenous (IV) infusion of spesolimab followed by placebo at Weeks 4 and 8, or three infusions of IV spesolimab 450 mg or 1,200 mg every 4 weeks (q4w) or placebo q4w (followed by a 12-week safety follow-up for patients not continuing into the open-label extension trial [1368–0017; NCT03648541]) [Citation33]. Owing to recruitment challenges, the study was prematurely discontinued.
2.1.2. Study 2 (1368-0010; NCT03123120)
This was a randomized, double-blind, placebo-controlled, parallel-group phase IIa trial with a 12-week treatment phase and subsequent 24-week follow-up (). The study objectives were to define the safety and efficacy of the induction of mucosal healing with spesolimab as add-on therapy in patients with mild-to-moderate UC and persistent endoscopic activity despite stable TNF-α inhibitor background therapy. Patients were randomized (2:1) to receive spesolimab IV 1,200 mg q4w or placebo, administered at Weeks 0, 4, and 8, on top of their background TNF-α inhibitor therapy. Anti-drug antibodies (ADAs) were assessed descriptively as part of the pharmacokinetic (PK) analyses. Owing to recruitment challenges, the study was prematurely discontinued.
2.1.3. Study 3 (1368-0004; NCT03100864)
This was an open-label, single-arm, phase IIa exploratory trial over a 12-week treatment period (). The main study objective was to understand the mode of action of spesolimab in patients with moderate-to-severe, active UC, naïve to advanced therapies or only exposed to and not failing TNF-α inhibitors; secondary study objectives were to explore the clinical effect, safety, and tolerability (including immunogenicity) of spesolimab induction treatment. Patients received spesolimab IV 1,200 mg q4w (administered at Weeks 0, 4, and 8) for 12 weeks, followed by a 16-week safety follow-up; patients who completed 12 weeks’ treatment could continue treatment in a long-term extension trial (1368–0017; NCT03648541).
2.2. Immunogenicity
Individual pre-dose spesolimab plasma samples were collected before the first dose and at subsequent treatment and follow-up visits. Samples were screened using a validated bridging electrochemiluminescent ADA assay, with sensitivity of 0.8 ng/mL, drug tolerance to 2,000 μg/mL, and precision coefficient of variance of 13.9% in UC. In a hierarchical approach, samples found to be putative positive were assessed using a confirmatory assay, and only those that were confirmed positive were then titrated. Full listings of all ADA-positive patients, on a study-by-study basis, are presented in Supplemental Tables S8–S10.
2.3. Efficacy
In Study 1, the primary efficacy endpoint was the proportion of patients in clinical remission at Week 12, which was defined as modified MCS ≤2 (with stool frequency score of 0 or 1 [if decrease ≥1 from baseline], rectal bleeding score 0, and mESS ≤1). In Study 2, the primary efficacy endpoint was endoscopic improvement (MCS mESS ≤1) at Week 12. The primary endpoint in Study 3 was the total number of deregulated genes between baseline and Week 12. A secondary endpoint was total clinical remission (defined as total MCS ≤2 points and all subscores ≤1 point) at Week 12.
2.4. Study ethics
All three studies were conducted in accordance with principles of the Declaration of Helsinki, with the International Conference on Harmonisation Good Clinical Practice, and in accordance with applicable regulatory requirements and standard operating procedures of the study sponsor and contract research organizations and with local legal requirements. All patients signed and dated an informed consent form before participation in any of the three studies. All trials were reviewed and approved by the Independent Ethics Committees and Competent Authorities of the respective investigational sites.
3. Results
3.1. Disposition of patients
Full details of patient flow through each of the three studies are provided in Supplemental Figures S1–S3. A total of 128 patients were included in the full analysis set (FAS; spesolimab, 97; placebo, 31) and 127 in the safety analysis set (SAS; spesolimab, 97; placebo, 30). In Study 2, one patient assigned to placebo was given spesolimab in error at Week 8 and was therefore analyzed in the placebo group in the FAS and in the spesolimab group in the SAS, in line with the population definitions (Supplemental Table S6). Accordingly, the FAS comprised 14 patients who received spesolimab and eight patients who received placebo and the SAS for analysis of exposure and safety comprised 15 patients who received spesolimab and seven patients who received placebo.
3.2. Baseline demographics, clinical characteristics, and medical history
Baseline demographics and clinical characteristics in spesolimab-treated patients were generally similar across the three studies (); the proportion of female patients ranged from 28.6% to 37.5% across the three studies and the median ages were 44.0 years, 47.5 years, and 35.0 years in Studies 1, 2, and 3, respectively. Median time since first diagnosis of UC was substantially shorter in Study 3 than in the other two studies, as might be expected for patients naïve to advanced therapies. Extra-intestinal manifestations of UC were reported by similar proportions of patients in the spesolimab and placebo arms in Study 1 and included arthritis and erythema nodosum. In Study 2, extra-intestinal manifestations were only reported in the spesolimab group, and all were arthritis. As expected, because of the inclusion criteria, mean baseline total MCS was lower in Study 2 than Study 1, as were mean baseline levels of C-reactive protein and fecal calprotectin.
Table 2. Baseline demographics and clinical characteristics in clinical trials of intravenous spesolimab in patients with ulcerative colitis.
In Study 2, baseline TNF-α inhibitor trough levels of patients receiving infliximab were similar in both treatment groups (geometric mean [gMean] of 4.1 μg/mL with spesolimab and 4.8 μg/mL with placebo). Similar trough levels at baseline were observed for the two patients in the spesolimab group receiving adalimumab (gMean 4.8 μg/mL) and for the one patient receiving golimumab (3.8 μg/mL).
Historical and concomitant UC medications up to Week 12 are presented in and concomitant non-UC medications are given in Supplemental Table S11. Details of patient exposure to different concomitant medication categories are given in Supplemental Tables S12 and S13. Concomitant use of immunosuppressants was high across all studies. Use of concomitant medication with corticosteroids was recorded for 49.3% of spesolimab-treated patients in Study 1, 42.9% in Study 2, and 25.0% in Study 3; corresponding values for the placebo groups were 43.5% in Study 1 and 0% in Study 2. Relatively high rates of previous treatment with TNF-α inhibitors and vedolizumab for treatment of UC were reported in Study 1 (). In Study 2, patients received spesolimab or placebo as an add-on to existing TNF-α inhibitor background medication; the most commonly received TNF-α inhibitor was infliximab, accounting for 87.5% of patients in both arms.
Table 3. Concomitant and historical ulcerative colitis medications up to Week 12 in clinical trials of intravenous spesolimab in patients with ulcerative colitis.
3.3. Overall summary of adverse events
Overall, the incidences of any TEAE for spesolimab-treated patients were 64.9% (Study 1), 86.7% (Study 2), and 100% (Study 3); corresponding values for the placebo groups were 65.2% (Study 1) and 71.4% (Study 2), with no placebo comparator arm in Study 3 (). The most frequently reported TEAEs (absolute rates spesolimab; placebo), besides worsening of UC, were nasopharyngitis (6.8%; 0%), skin rash (6.8%; 0%), and headache (5.4%; 0%) in Study 1; and headache (26.7%; 14.3%), nasopharyngitis (20.0%; 14.3%), nausea (13.3%; 0%), acne (13.3%; 0%), and eczema (13.3%; 0%) in Study 2. The most common TEAEs in Study 3 were nasopharyngitis (50.0%) and headache (25.0%). The frequencies of infections and infestations in spesolimab-treated and placebo groups were 25.7% (spesolimab) and 8.7% (placebo) in Study 1; 26.7% and 28.6%, respectively, in Study 2 (note that all patients in the placebo group were receiving TNF-α inhibitor medication); and 87.5% in Study 3. Full listings of all TEAEs, on a study-by-study basis, are given in Supplemental Tables S14–S16.
Table 4. Overall summary of adverse events in clinical trials of intravenous spesolimab in patients with ulcerative colitis.
One patient each in Study 1 (receiving placebo, 4.3%; Clostridioides difficile colitis) and Study 3 (receiving spesolimab, 12.5%; diverticulitis) was reported to have AEs in the UDAECs under the severe (RCTC grade 3 or 4) infections and infestations category; none were reported in Study 2. The overall incidence of any UDAECs in spesolimab-treated patients in Study 1 was 13.5%, all of which came under the infusion or systemic hypersensitivity, including anaphylactic reactions category, with rash (6.8%) being the most common AE at the preferred term (PT) level; the corresponding value in the placebo group was one patient (4.3%) with infusion-related reaction at the PT level. In Study 2, similar proportions of patients in the spesolimab (13.3%) and placebo (14.3%) groups reported AEs under the infusion or systemic hypersensitivity, including anaphylactic reactions category, with eczema (13.3%) and skin rash (14.3%) being the most common at the PT level. Similarly, in Study 3, the most common UDAECs at the PT level were rash (12.5%) and acne (12.5%) (). Details of the MedDRA search criteria for UDAECs are described in Supplemental Table S7 and full listings of all UDAECs, on a study-by-study basis, are available in Supplemental Tables S17–S19.
In Studies 1 and 2, subgroup analyses of TEAEs by UDAECs were comparable in spesolimab-treated and placebo groups, with no differences between female and male patients (Supplemental Tables S20 and S21). In Study 3, equal proportions of female and male patients reported any AE (Supplemental Table S21). Similarly, subgroup analyses of TEAEs by age and treatment group were comparable across studies, with the majority of UDAECs reported under the infusion or systemic hypersensitivity, including anaphylactic reactions category (Supplemental Tables S22 and S23).
The frequencies of patients reported with infusion or systemic hypersensitivity reactions concomitantly using immunosuppressants and/or corticosteroids between treatment groups (spesolimab; placebo) were numerically higher in Study 1 (12.1%; 4.5%), and comparable in Study 2 (13.3%; 14.3%); in Study 3, 25.0% of such patients reported infusion or systemic hypersensitivity reactions (Supplemental Tables S24 and S25). Furthermore, one spesolimab-treated patient (12.5%) in Study 3, and none in Studies 1 and 2, reported severe, serious, or opportunistic infections; one patient (4.5%) in the placebo group in Study 1 reported a severe infection.
3.4. Serious adverse events
In the placebo groups, four patients (17.4%) in Study 1 and one (14.3%) in Study 2 had SAEs. In the spesolimab group, seven patients (9.5%) in Study 1, one patient (6.7%) in Study 2, and two patients (25.0%) in Study 3 had SAEs (). The most frequently reported SAE was worsening of underlying UC: Study 1: spesolimab, 6.8%; placebo, 4.3%; Study 2: spesolimab, 0%; placebo, 14.3%; Study 3: spesolimab, 12.5%. Overall, none of the infection TEAEs in spesolimab-treated patients were considered an SAE in Studies 1 and 2; one patient (12.5%) in Study 3 had an SAE of diverticulitis. In Study 1, one patient (4.3%) in the placebo group reported an SAE of C. difficile colitis; in Study 2, one patient in the placebo group (14.3%) reported an SAE of rectal abscess ().
Table 5. Serious adverse events in clinical trials of intravenous spesolimab in patients with ulcerative colitis.
In Study 1, an infusion-related reaction, assessed as drug-related by the investigator, was reported for one patient (4.3%) in the placebo group and one (1.4%) in the spesolimab group. Of note, the patient randomized to the spesolimab group received the planned 300 mg IV single dose on Day 1, with no AE reported during that dose or before the next administration. As planned, the second administration on Day 33 was placebo, reflecting the randomized treatment with single-dose spesolimab. The infusion-related reaction started approximately 25 minutes after the start of the placebo infusion. The reaction was reported by the investigator as being life-threatening: the patient experienced swollen lips, chest tightness with difficulty breathing, a decrease in systolic blood pressure (from 110 to 74 mmHg), and blurred vision. No other symptoms were reported. The infusion was stopped, and the patient was treated with an infusion of saline and diphenhydramine. The event resolved within 14 minutes after its onset and 5 minutes after start of the H1 antagonist. Immunoglobulin E levels were within normal range. The study medication was discontinued.
3.5. Drug-related adverse events
The most frequent AEs assessed by the investigator as drug related (absolute rates spesolimab; placebo) were skin rash (5.4%; 0%) and nasopharyngitis (4.1%; 0%) in Study 1, and acne (13.3%; 0%) in Study 2; in Study 3, one patient (12.5%) each experienced skin rash, nasopharyngitis, headache, and acne (Supplemental Table S26). Full listings of all drug-related AEs, on a study-by-study basis, are available in Supplemental Tables S27–S29.
3.6. Immunogenicity
In Study 1, the proportions of spesolimab-treated patients with treatment-emergent ADAs were 12.5% (300 mg single-dose group), 0% (450 mg q4w group), and 19.2% (1,200 mg q4w group). Overall, 8/73 patients (11.0%) were ADA positive after IV administration of spesolimab; however, a potential impact of ADAs on spesolimab PK was observed for only one patient (in the 1,200 mg q4w group), who developed ADAs at Week 4, and for whom spesolimab plasma levels were below the limit of quantification for the PK analytical assay at Weeks 8, 12, and 24 (Supplemental Table S8).
In Study 2, a positive ADA result was observed in only two patients (14.3%) during the follow-up period. ADA positivity persisted in only one case; the other had a positive result at Week 16 only that was characterized as induced and transient, and ADA formation did not alter spesolimab PK (Supplemental Table S9).
In Study 3, two patients (25.0%) had a positive ADA result: one had a positive result (induced, potentially persistent) at Week 28 only, whereas the other patient had positive results at Weeks 12, 18, and 28; the latter result was characterized as induced, persistent, although the ADA titer declined from 720 ng/mL at Week 12 to 180 ng/mL at Week 28. Overall, ADA formation did not alter spesolimab PK (Supplemental Table S10).
Further analysis concluded that it was highly unlikely that the hypersensitivity events were related to ADA development; in Study 1, hypersensitivity AE rates were similar before and after ADA development, and in Studies 2 and 3 all AEs occurred before ADA development.
3.7. Laboratory parameters
In Study 1, the numbers of patients with laboratory parameter values within the normal range at baseline but outside the normal range at any time during the 12-week treatment period and 16-week residual-effect period were generally low (Supplemental Table S30); moreover, from baseline to the end of treatment, no marked changes were observed in mean values for any laboratory parameter. Specifically, the most frequent potentially clinically significant laboratory abnormalities (PCSAs) up to Week 12 in spesolimab-treated patients were neutrophils >10 × 109/L (23.5% of patients), hematocrit <32% (12.3% of patients), and triglycerides >2.3 mmol/L (8.3% of patients) (Supplemental Table S30). For the placebo group, the most frequent PCSAs were hematocrit <32% (30.0% of patients), platelets >700 × 109/L (13.6% of patients), and neutrophils >10 × 109/L (11.1% of patients). In addition, one patient in the spesolimab 300 mg group and one in the placebo group had an increase in alanine aminotransferase and/or aspartate aminotransferase level to ≥3 times the upper limit of normal; however, this increase was not associated with concomitant liver-related AEs or eosinophilia >5%.
In Study 2, there were no marked changes in mean laboratory values over time, and only a very few, sporadic group means were outside the respective reference ranges. No shifts from RCTC grade ≤2 to worst RCTC grade ≥3 were observed up to Week 12, and only 11 PCSAs were observed in total in the spesolimab group (Supplemental Table S31). Specifically, the most frequent PCSAs up to Week 12 in the spesolimab group were elevated triglycerides (30.8% of patients), low hematocrit (13.3%), and low leukocytes (<3.0 × 109/L; 6.7%) (Supplemental Table S31). Three PCSAs were observed in the placebo group: elevated triglycerides (14.3%), low leukocytes (<3.0 × 109/L; 14.3%), and high creatinine (>150 µmol/L; 14.3%).
In Study 3, four patients (50.0%) developed PCSAs during treatment that were not present at baseline. The most frequently reported PCSA was low hemoglobin (28.6% of patients) (Supplemental Table S32).
3.8. Efficacy
In Study 1, only 1/24 (4.2%), 2/23 (8.7%), and 2/28 (7.1%) patients in the spesolimab 300 mg single dose, 450 mg q4w, and 1,200 mg q4w groups, respectively, achieved the primary endpoint (clinical remission at Week 12), and differences from placebo (0/23 patients; 0%) were not statistically significant. In Study 2, only two spesolimab-treated patients (14.3%) achieved the primary endpoint (endoscopic improvement at Week 12), and the unadjusted risk difference relative to placebo was –0.232 (95% confidence interval: –0.568 to 0.118). In Study 3, limited changes in gene expression were observed, with very few genes deregulated (primary endpoint). No patients (0/8; 0%) achieved the secondary efficacy endpoint (total clinical remission at Week 12).
4. Discussion
Studies highlighting the link between IL-36-driven inflammation and epithelial inflammation have led to the hypothesis that IL-36 signaling may play an important role in inflammatory bowel diseases such as UC [Citation20,Citation21,Citation25]. We report safety, immunogenicity, and efficacy data for three phase II proof-of-concept trials for the treatment of a first-in-class IL-36 R inhibitor, spesolimab, in patients with UC. Although efficacy endpoints were not met, valuable safety information on spesolimab was collected during these studies and provides insights into key safety considerations for this patient population.
Across the three studies, the AEs were of mild or moderate intensity for most patients, and no deaths occurred. The overall frequency of patients with AEs was generally similar between the spesolimab and placebo groups in patients who received concomitant TNF-α inhibitor treatment (Study 2) and those who did not receive concomitant TNF-α inhibitor treatment (Study 1).
With respect to safety, it is important to note that the data presented here across all three studies originate from patient populations who were co-treated with other medications with likely immunomodulatory and/or immunosuppressive activity. For instance, almost all patients in Study 1 used at least one background medication, of which approximately half were corticosteroids. In the placebo-controlled Studies 1 (patients with moderate-to-severe UC who had failed previous biologics) and 2 (patients with mild-to-moderate UC receiving TNF-α inhibitor therapy), all doses of spesolimab were well tolerated, and safety profiles were generally similar to placebo, with the most frequently reported AEs mild or moderate in intensity. There were low rates of SAEs, most of which reflected worsening of UC. One patient in the single-dose spesolimab group in Study 1 reported no AEs during or after spesolimab 300 mg IV on Day 1, but experienced a life-threatening infusion reaction (as classified by the investigator), including hypotension and lip swelling, during infusion of the first placebo dose on Day 33. The infusion was stopped and the event resolved. There were lower rates of drug-related SAEs in spesolimab arms than placebo arms. Similarly, for AEs by PT level, apart from worsening of UC, overall reported rates were low.
With respect to UDAECs, there was limited evidence for severe, serious, or opportunistic infections, or hypersensitivity in the spesolimab group compared with the placebo group. The most frequently reported UDAECs in Study 1 and Study 2 were rash and eczema, respectively. There were relatively low rates of treatment discontinuation, although AEs and lack of efficacy were among the most frequent reasons for discontinuation. In the open-label Study 3 (patients with moderate-to-severe UC who were biologically naïve or TNF-α inhibitor experienced but had not failed treatment), relatively higher rates of AEs were reported compared with Studies 1 and 2, including two SAEs that required or prolonged hospitalization. Overall, no deaths were reported in any of the three studies, and no unexpected safety observations were made.
Subgroup analyses of the data revealed some differences: overall, a higher proportion of female patients and patients under 50 years of age in the spesolimab arms experienced AEs compared with male patients and patients aged 50 years or over; however, interpretation is limited by the small subgroup sizes in Studies 2 and 3. In terms of concomitant medications, a higher percentage of patients receiving immunosuppressants alone appeared to experience AEs than patients receiving other combinations of treatment, although, as noted previously, sample sizes were small in Studies 2 and 3. However, the data suggest that co-administration of spesolimab with other medications, including other biologics, does not negatively impact its safety profile. Overall, infection-related TEAEs in patients treated with spesolimab did not translate into serious, severe, or opportunistic infections. Furthermore, rates of serious infections were low in patients who received concomitant immunosuppressants and/or corticosteroids. These data suggest that spesolimab does not lead to a risk of serious infection, even in immunosuppressed patients with UC.
The described studies had some limitations: comparison of the safety results across all three studies with spesolimab-treated patients are descriptive in nature, and no formal analyses between studies were performed, mainly due to differences in patient populations and study designs. Studies 1 and 2 were terminated prematurely due to recruitment challenges, resulting in a smaller sample size than initially planned. The small sample size for Study 3 (n = 8) and its lack of a placebo control arm may limit the interpretation of its results.
The safety profiles reported here – that spesolimab is well tolerated with limited evidence for severe, serious, or opportunistic infections or hypersensitivity – are consistent with the overall safety profile of spesolimab reported in previous studies investigating spesolimab in healthy volunteers and in patients with other chronic inflammatory diseases, such as GPP [Citation28] and PPP [Citation22]. In a study of patients with GPP, a single dose of spesolimab (IV 10 mg/kg of body weight) reduced disease severity, with no SAEs reported [Citation24]. A more recent study in patients with GPP reported drug reactions with eosinophilia and systemic symptoms (DRESS) in two patients (4%) who received a 900 mg single dose of spesolimab up to Week 12. However, the scoring by RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) was noted as RegiSCAR score 1 (‘no DRESS’) in one patient, and RegiSCAR score 3 (‘possible DRESS’) in the other; both patients had received concomitant medications that could potentially be associated with DRESS. Moreover, the rapid occurrence of symptoms post spesolimab administration in one case makes a causal relationship with spesolimab implausible; in the other case, cutaneous symptoms after positive rechallenge with spiramycin suggest this antibiotic as an alternative explanation [Citation28]. In patients with PPP, spesolimab treatment (300 or 900 mg IV q4w) and placebo had similar safety profiles; most AEs were graded mild to moderate, with two patients in each arm reporting SAEs [Citation22].
Patients treated with spesolimab in these three studies of UC had a higher exposure to spesolimab and duration of treatment than patients in studies in other indications. The fact that the safety profile remained consistent with that observed in studies of single-dose spesolimab lends further support to the overall conclusion that spesolimab can be well tolerated by patients with inflammatory diseases. Furthermore, the low rates of treatment-related infections in patients receiving spesolimab vs placebo, in a population that was receiving concomitant immunomodulatory or immunosuppressive therapy, are encouraging.
5. Conclusions
Spesolimab was demonstrated to be generally well tolerated in patients with UC; efficacy endpoints were not met. There was no evidence for clinically relevant hypersensitivity or severe, serious, and opportunistic infections related to spesolimab, even when used in combination with other immunosuppressants or TNF-α inhibitors. Overall, AEs were mostly mild or moderate in intensity, with most of the few SAEs representing aggravations or complications of the underlying disease.
Declaration of interest
M Ferrante has received research grants from AbbVie, Amgen, Biogen, Janssen, Pfizer, Takeda, and Viatris; consultancy fees from AbbVie, Boehringer Ingelheim, Celgene, Celltrion, Eli Lilly, Janssen, Medtronic, MSD, Pfizer, Regeneron, Samsung Bioepis, Sandoz, Takeda, and Thermo Fisher; and speaker fees from AbbVie, Amgen, Biogen, Boehringer Ingelheim, Dr Falk Pharma, Ferring, Janssen, Lamepro, MSD, Mylan, Pfizer, Sandoz, Takeda, and Truvion Healthcare. PM Irving has received honoraria for speaking on behalf of AbbVie, BMS, Celgene, Celltrion, Dr Falk Pharma, Ferring, Galapagos, Gilead, MSD, Janssen, Eli Lilly, Pfizer, Takeda, Tillotts, Sapphire Medical, Sandoz, Shire, and Warner Chilcott, and for acting in an advisory capacity to AbbVie, Arena, Boehringer Ingelheim, BMS, Celgene, Celltrion, Genentech, Gilead, Hospira, Janssen, Eli Lilly, MSD, Pfizer, Pharmacosmos, Prometheus, Roche, Sandoz, Samsung Bioepis, Takeda, TopiVert, VH2, Vifor Pharma, and Warner Chilcott, and has received research grants from Celltrion, Galapagos, MSD, Pfizer, and Takeda. CP Selinger has received unrestricted research grants from Warner Chilcott, Janssen, and AbbVie; has provided consultancy to Warner Chilcott, Dr Falk Pharma, AbbVie, Takeda, Fresenius Kabi, Galapagos, Arena, and Janssen; and had speaker arrangements with Warner Chilcott, Dr Falk Pharma, AbbVie, MSD, Pfizer, Celltrion, and Takeda. G D’Haens has received consulting and/or lecture fees from AbbVie, ActoGeniX, AIM, Boehringer Ingelheim, Centocor, Chemo Centryx, Cosmo Technologies, Elan Pharmaceuticals, enGene, Dr Falk Pharma, Ferring, Galapagos, Giuliani, Given Imaging, GlaxoSmithKline, Janssen Biologics, MSD, Neovacs, Novo Nordisk, Otsuka, PDL BioPharma, Pfizer, Receptos, Salix, Schering-Plough, SetPoint Medical, Shire, Takeda, Tillotts Pharma, UCB Pharma, Versant, and Vifor Pharma; research grants from AbbVie, Dr Falk Pharma, Given Imaging, Janssen, MSD, and PhotoPill; and speaking honoraria from AbbVie, Ferring, MSD, Norgine, Shire, Tillotts, Tramedico, and UCB Pharma. T Kuehbacher has lectured and/or been on advisory boards for AbbVie, Almirall, Amgen, Arena, BMS, Celgene, Celltrion, Ferring, Dr Falk Pharma, Gilead, Galapagos, Janssen, MSD, Mundipharma, Pfizer, Shire, Takeda, Tillotts, and Vifor Pharma. U Seidler has received research grants from AbbVie, Janssen, Eli Lilly, Celgene, Gilead, Galapagos, Abivax, and Takeda, and speaker fees from Galapagos and AbbVie. S Gropper,T Haeufel, and S Forgia are employees of Boehringer Ingelheim. S Danese declares consultancy for AbbVie, Alimentiv, Allergan, Amgen, AstraZeneca, Athos Therapeutics, Biogen, Boehringer Ingelheim, BMS, Celene, Celltrion, Dr Falk Pharma, Eli Lilly, Enthera, Ferring Pharmaceuticals, Gilead, Hospira, Inotrem, Janssen, Johnson & Johnson, MSD Mundipharma, Mylan, Pfizer, Roche, Sandoz, Sublimity Therapeutics, Takeda TiGenix, UCB Pharma and Vifor Pharma, and has received honoraria for speaking on behalf of AbbVie, Amgen, Ferring Pharmaceuticals, Gilead, Janssen, Mylan, Pfizer, and Takeda. J Klaus declares no conflicts of interest. BG Feagan declares consultancy for AbbVie, AdMIRx, AgomAB Therapeutics, Akebia, Alivio Therapeutics, Allakos, Amgen, Applied Molecular Transport, Arena Pharma, AVIR, Azora Therapeutics, Boehringer Ingelheim, Boston Pharma, Celgene/BMS, Connect BioPharma, Cytoki Pharma, Disc Medicine, Everest Clinical Research Corp., Eli Lilly, Equillium, Ferring, Galapagos, Galen Atlantica, Genentech/Roche, Gilead, Glenmark, Gossamer Pharma, GlaxoSmithKline, Hoffmann-LaRoche, Hot Spot Therapeutics, Index Pharma, ImmuNext, Imhotex, Intact Therapeutics, Janssen, Japan Tobacco, Kaleido Biosciences, Leadiant Biosciences, Millennium, MiroBio, Morphic Therapeutics, Mylan, OM Pharma, Origo BioPharma, Otsuka, Pandion Therapeutics, Pfizer, Prometheus Therapeutics and Diagnostics, Progenity, PTM Therapeutics, Q32 Bio, Rebiotix, RedHill, Biopharma, Redx, Sandoz, Sanofi, Seres Therapeutics, Surrozen, Takeda, Teva Pharmaceuticals, Thelium Therapeutics, Theravance, TiGenix, Tillotts, UCB Pharma, VHsquared, Viatris, Ysios, and Zealand Pharma; speakers fees from AbbVie, Janssen, and Takeda; attendance of advisory boards for AbbVie, Amgen, Boehringer Ingelheim, Celgene/BMS, Genentech/Roche, Janssen, Novartis, Origo BioPharma, Pfizer, Prometheus, Takeda, Tillotts Pharma, Teva Pharmaceuticals, Progenity, Index, EcoR1 Capital, Morphic, and GlaxoSmithKline; stocks with Gossamer Pharma; employment by Western University, Alimentiv. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from Expert Opinion on Drug Safety for their review work but have no other relevant financial relationships to disclose.
Author contributions
All authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE) and made the decision to submit the manuscript for publication. The authors did not receive payment related to the development of the manuscript. Agreements between Boehringer Ingelheim and the authors included the confidentiality of the study data. All authors participated in the interpretation of study results, drafting, critical revision, and approval of the manuscript.
Data availability
To ensure independent interpretation of clinical study results and enable authors to fulfill their role and obligations under the ICMJE criteria, Boehringer Ingelheim grants all external authors access to relevant clinical study data. In adherence with the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data, scientific and medical researchers can request access to clinical study data after publication of the primary manuscript in a peer-reviewed journal, regulatory activities are complete and other criteria are met. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/msw/datasharing for further information.
Supplemental Material
Download MS Word (170.7 KB)SOM_Fig_3_Patient_disposition_Study_3__1_.jpg
Download JPEG Image (841.8 KB){kind=link}
SOM_Fig_2_Patient_disposition_Study_2__1_.jpg
Download JPEG Image (1.1 MB){kind=link}
SOM_Fig_1_Patient_disposition_Study_1__1_.jpg
Download JPEG Image (1.9 MB){kind=link}
Acknowledgments
We thank all the patients, study investigators, and investigational center teams who participated in the three clinical studies described here. The authors also thank for their contributions and advice Dr. Wulf O Böcher (Uniklinik Mainz, Mainz, Germany) and the following individuals from Boehringer Ingelheim: Slawomir Godyński (Biostatistics & Data Science), Carla Haefner (Clinical Development & Operations), Oliver Koersgen (Clinical Development & Operations), Jens Laass (Clinical Development & Operations), Sutirtha Mukhopadhyay (Patient Safety and Pharmacovigilance), Grace Richmond (Global Scientific Communications), Michael Shear (Biostatistics & Data Sciences), and Nikolaos Sitaridis (Biostatistics & Data Science).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14740338.2022.2103536.
Additional information
Funding
Related Research Data
References
- Feuerstein JD, Moss AC, Farraye FA. Ulcerative colitis. Mayo Clin Proc. 2019;94(7):1357–1373.
- Rubin DT, Ananthakrishnan AN, Siegel CA, et al. ACG clinical guideline: ulcerative colitis in adults. Am J Gastroenterol. 2019;114(3):384–413.
- Segal JP, LeBlanc JF, Hart AL. Ulcerative colitis: an update. Clin Med (Lond). 2021;21(2):135–139.
- Magro F, Gionchetti P, and Eliakim R, et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11(6):649–670.
- Kühn F, and Klar E. Surgical principles in the treatment of ulcerative colitis. Viszeralmedizin. 2015;31(4):246–250.
- Yarlas A, Rubin DT, Panes J, et al. Burden of ulcerative colitis on functioning and well-being: a systematic literature review of the SF-36(R) health survey. J Crohns Colitis. 2018;12(5):600–609.
- Rapport F, Clement C, Seagrove AC, et al. Patient views about the impact of ulcerative colitis and its management with drug treatment and surgery: a nested qualitative study within the CONSTRUCT trial. BMC Gastroenterol. 2019;19(1):166.
- Peyrin-Biroulet L, Sandborn W, Sands BE, et al. Selecting therapeutic targets in inflammatory bowel disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol. 2015;110(9):1324–1338.
- Harbord M, Eliakim R, Bettenworth D, et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 2: current management. J Crohns Colitis. 2017;11(7):769–784.
- Feuerstein JD, Isaacs KL, Schneider Y, et al. AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology. 2020;158(5):1450–1461.
- Sandborn WJ, Feagan BG, D’Haens G, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2021;385(14):1280–1291.
- Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723–1736.
- Baker DM, Folan AM, Lee MJ, et al. A systematic review and meta-analysis of outcomes after elective surgery for ulcerative colitis. Colorectal Dis. 2021;23(1):18–33.
- O’Hagan P, Limdi J, Akbar A, et al. Ulcerative colitis: understanding the impact of ulcerative colitis on everyday life and exploring the unmet needs of patients. Curr Med Res Opin. 2021;37(11):1901–1911.
- Le Berre C, Peyrin-Biroulet L, SPIRIT -IOIBD study group. group S-Is. Selecting end points for disease-modification trials in inflammatory bowel disease: the SPIRIT Consensus from the IOIBD. Gastroenterology. 2021;160(5):1452–1460.
- European Medicines Agency. Guideline on the development of new medicinal products for the treatment of Crohn’s disease. CPMP/EWP/2284/99 Rev. 2; 2018 [cited 2022 May 30]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-new-medicinal-products-treatment-crohns-disease-revision-2_en.pdf.
- Herrlinger KR, and Stange EF. Twenty-five years of biologicals in IBD: what’s all the hype about? J Intern Med. 2021;290(4):806–825.
- Loftus EV Jr., Feagan BG, Panaccione R, et al. Long-term safety of vedolizumab for inflammatory bowel disease. Aliment Pharmacol Ther. 2020;52(8):1353–1365.
- Shimodaira Y, Watanabe K, Iijima K. Clinical course of ulcerative colitis associated with an age at diagnosis: a recent Japanese database survey. Tohoku J Exp Med. 2021;255(1):33–39.
- Nishida A, Hidaka K, Kanda T, et al. Increased expression of interleukin-36, a member of the interleukin-1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis. 2016;22(2):303–314.
- Neufert C, Neurath MF, and Atreya R. Rationale for IL-36 receptor antibodies in ulcerative colitis. Expert Opin Biol Ther. 2020;20(4):339–342.
- Mrowietz U, Burden AD, and Pinter A, et al. Spesolimab, an anti-interleukin-36 receptor antibody, in patients with palmoplantar pustulosis: results of a Phase IIa, multicenter, double-blind, randomized, placebo-controlled pilot study. Dermatol Ther (Heidelb). 2021;11(2):571–585.
- Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol. 2019;15(9):907–919.
- Bachelez H, Choon SE, and Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380(10):981–983.
- Russell SE, Horan RM, and Stefanska AM, et al., IL-36α expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016;9(5):1193–1204.
- Scheibe K, Kersten C, and Schmied A, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. 2019;156(4): 1082–97.e11.
- Ganesan R, Raymond EL, Mennerich D, et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs. 2017;9(7):1143–1154.
- Bachelez H, Choon SE, and Marrakchi S, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med. 2021;85(26):2431–2440.
- ClinicalTrials.gov. NCT03482635. BI655130 (spesolimab) induction treatment in patients with moderate-to-severe ulcerative colitis. [cited 2021 Dec 16] Available from: https://www.clinicaltrials.gov/ct2/show/NCT03482635.
- ClinicalTrials.gov. NCT03123120. A study in patients with mild or moderate ulcerative colitis who take a TNF inhibitor. The study investigates whether bowel inflammation improves when patients take BI 655130 in addition to their current therapy. [cited 2021 Dec 16] Available from: https://www.clinicaltrials.gov/ct2/show/NCT03123120.
- ClinicalTrials.gov. NCT03100864. This study tests how BI 655130 works in patients with active ulcerative colitis. The study also tests how well BI 655130 is tolerated and whether it helps the patients. [cited 2021 Dec 16] Available from: https://www.clinicaltrials.gov/ct2/show/NCT03100864.
- Woodworth T, Furst DE, Alten R, et al. Standardizing assessment and reporting of adverse effects in rheumatology clinical trials II: the Rheumatology Common Toxicity Criteria v.2.0. J Rheumatol. 2007;34(6):1401–1414.
- ClinicalTrials.gov. NCT03648541. BI 655130 long-term treatment in patients with moderate-to-severe ulcerative colitis. [cited 2022 May 30]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03648541.