Abstract
Anthracycline cardiotoxicity represents the most unfavorable side effect of these highly efficient anticancer drugs. Several biotransformation enzymes have been described to contribute to their cardiotoxicity. Besides the activities of CYP450 isoforms which lead to the generation of reactive oxygen species (ROS), the cytosolic reductases have attracted attention nowadays. The reductases known to metabolize anthracyclines to C13-hydroxyanthracyclines are carbonyl reductase (CR, 1.1.1.184) and the aldo-keto reductases (AKR1C2, 1.3.1.20; AKR1A1, 1.1.1.2). Their participation in the formation of the toxic C13-hydroxymetabolite has been investigated in rabbit using diagnostic inhibitors of CR and AKR1C2. The kinetics and the type of reductase inhibition exerted by the two inhibitors have been described and it was found that CR was the main daunorubicin reductase at both optimal and physiological pH with the kinetic parameters for daunorubicin reduction of and
The IC50 values for quercitrin and flufenamic acid were 5.45 ± 1.37 μM and 3.68 ± 1.58 μM, respectively. The inhibition was uncompetitive for both inhibitors and irreversible in the case of flufenamic acid.
1 Introduction
Anthracyclines are among the most efficient and widely used antineoplastic agents [Citation1]. It was about 50 years ago that daunorubicin (DAU) and doxorubicin were first isolated from the strain of bacteria Streptomyces and their antibiotic and antimitotic effects described [Citation2]. However, the anthracyclines were found to be too toxic for the therapy of human infectious diseases and they finally found their place in the chemotherapy of cancer. Many derivatives [Citation3,Citation4,Citation5] have been synthetised but even now none of them has actually replaced the first two parent compounds of the class—daunorubicin and doxorubicin.
Like many other cancer chemotherapeutics the anthracyclines possess a series of side effects which complicate therapy such as hair loss, nausea, vomiting etc. but the most inconvenient and potentially life-threatening effect of anthracyclines is their cardiotoxicity [Citation6]. The cardiotoxicity can be observed at different levels from acute to chronic and delayed toxicity [Citation7], the latter being especially serious for it can occur suddenly many years after chemotherapy has finished. It is well known that anthracycline-induced cardiomyopathy is most likely to develop upon administration of cumulative doses of anthracyclines of about 550 mg/m2 (the exact value depends on the particular anthracycline) [Citation8]. The cause of the cardiotoxicity has not yet been fully explained. There are many hypotheses about its development and several biotransformation enzymes seem to be involved. According to the popular freeradical theory, the cardiac injury results from the redox cycling of anthracyclines which leads to the generation of reactive oxygen species (ROS) and oxidative damage of the cell [Citation9,Citation10]. One-electron redox cycling of a quinone moiety of the molecule is catalyzed by CYP450 isoforms [Citation11] but some other enzymes have also been proposed to contribute to this proccess (e.g. nitric oxide synthase, NADH dehydrogenase) [Citation12]. However, this free-radical mechanism can hardly explain the development of the chronic toxicity. Moreover, it has been shown (in vitro) that the generation of ROS by anthracyclines can paradoxically protect the cardiac cell against iron-mediated toxicity [Citation13]. The role of iron in a freeradical-independent mechanism of cardiotoxicity has been studied intensively and it was suggested that anthracyclines disturb the homeostasis of iron by interferring with key iron regulatory proteins [Citation14,Citation15].
Some investigators have shown that the cardiotoxicity correlates with the reduction of anthracyclines to their C13-alcoholic metabolites [Citation16,Citation17,Citation18] (). This metabolic conversion is catalyzed by cytosolic reductases of the aldo–keto reductase class (AKR) and short chain dehydrogenase class (SDR) [Citation12]. The reducing capacity of the heart was shown to be much lower than that of the liver [Citation19], however, the cumulation of the metabolite in the heart has been described [Citation16] suggesting that the alcoholic metabolites might be transported and selectively trapped by cardiac cells. Three enzymes have actually been described to metabolize anthracyclines to C13-hydroxymetabolites [Citation20]: Carbonyl reductase (CR, 1.1.1.184) [Citation21,Citation22], AKR1C2 (1.3.1.20) [Citation23,Citation24] and AKR1A1 (1.1.1.2.) [Citation25]. (The latter two were previously referred to as dihydrodiol dehydrogenase DD2 and aldehyde reductase ALR1, respectively.) Unfortunately not much is known about the participation of these particular reductases on C13-alcoholic metabolite formation and it seems useful to know which enzyme is the most active regarding anthracycline reduction since this would allow the design of appropriate cardioprotective drugs on the basis of reductase inhibition. In this study we characterized the activities of anthracycline reductases in rabbit liver cytosol, the kinetics of daunorubicin reduction and the parameters of the reductase inhibition.
Figure 1 Scheme of metabolic conversion of daunorubicin to its C13-hydroxymetabolite daunorubicinol. The cytosolic reductases involved in this process are CR (EC 1.1.1.184), AKR1C2 (EC 1.3.1.20), and AKR1A1 (EC 1.1.1.2).
2 Materials and methods
2.1 Animals
Male rabbits (Oryctolagus cuniculus var.Chinchilla, about 3 kg weight) were obtained from Velaz, Prague, Czech Republic and kept on standard laboratory diet ad libitum. The animals were sacrificed by bleeding to death under anaesthesia and their livers removed and stored at − 80°C. All the procedures were permitted and supervised by the Ethics Committee of the Faculty of Pharmacy, Charles University in Prague.
2.2 Chemicals and reagents
Daunorubicin (Cerubidine® inj.) was obtained from Rhône-Poulenc, France. NADPH was obtained from ICN Biochemicals Inc., California, USA. Quercitrin, flufenamic acid and the protein assay reagents (BCA kit) were obtained from Sigma-Aldrich. All other chemicals and solvents used were of analytical grade and were obtained from local commercial sources.
2.3 Isolation of liver cytosolic fractions
Rabbit livers stored at − 80°C were thawed at room temperature (all the following procedures took place at 4°C). 20 g of liver tissue per rabbit were cut into small pieces and homogenised in 100 mL of sodium-phosphate buffer pH 7.4 using a Potter-Elvehjem's homogeniser. The homogenate was distributed in 8 cuvettes, buffer was added to a total volume of 50 ml per cuvette and the homogenate was centrifuged at 5,000 × g. The supernatant was collected and centrifuged at 20,000 × g. After this, the supernatant was ultracentrifuged at 100,000 × g for 1 h. The supernatant represented the cytosolic fraction and was stored at − 80°C for later use.
2.4 Reductase activity assay
Activities of selected cytosolic reductases were measured spectrophotometrically by monitoring the oxidation rate of NADPH at 340 nm [Citation26,Citation27]. Potassium-phosphate buffer 0.1 M, pH 6.0 was used for CR and AKR1C2 assay and TRIS-HCl buffer 0.2 M, pH 8.5 was used for AKR1A1 assay. The incubation mixture (1 mL) contained 930 μL of appropriate buffer, 50 μL of cytosolic fraction, 10 μL of 10 mM NADPH and 10 μL of 1 mM daunorubicin by which the reaction was started. Absorbance at 340 nm was continuously recorded for 4 min (linear) and enzymatic activity was calculated using and expressed in cat/mg protein.
2.5 Inhibition assays
Quercitrin [Citation21,Citation28], a specific inhibitor of CR and flufenamic acid [Citation23,Citation24] (NSAID), a specific inhibitor of AKR1C2 were used to separate the two enzymes' activities at pH 6.0. Inhibitors were dissolved in DMSO so that a series of stock solutions (1000-fold concentrated) was prepared to give final concentrations within the range of 0.5–50 μM. The concentration of DMSO in reaction mixture never exceeded 0.1% which did not affect enzyme activity. Dicoumarol was dissolved in 15 mM NaOH to give final concentrations of 1–50 μM. Typically, 50 μL of cytosolic fraction and 10 μL of 10 mM NADPH were put in 930 μL buffer, 1 μL of inhibitor stock solution was added and the reaction mixture was preincubated for 5 min. The reaction was then started by adding 10 μL of 1 mM DAU and the residual reductase activity was determined as described above. Sodium-phosphate buffer 0.1 M was used for measuring activity at pH 7.4. The fraction of the total reductase activity that was not inhibitable by quercitrin was considered to be due to AKR1C2 activity whereas the fraction of total activity that was not sensitive to flufenamic acid was considered as due to CR activity; these values allowed calculation of the CR/AKR1C2 ratios at pH 6.0 and 7.4.
2.6 Reversibility assay (dialysis)
Reversibility of inhibition was tested by comparing the activities of inhibited reactions before and after dialysis. Briefly, after the reductase activity of inhibited sample was determined, the reaction mixture was placed in dialysis tubing (Visking®, MWCO 14000, Carl Roth GmbH) and dialysed against PBS for 12 h at 4°C with the buffer being changed every 2 h. After 12 h the contents of each dialysis tube was carefully moved to the cuvette and the reductase activity was measured again after adding 10 μL 10 mM NADPH and 10 μL 1 mM DAU. The non-inhibited reaction was dialysed and measured under the same conditions and served as a control to eliminate the influence of spontaneous loss of activity and/or minor changes in the enzyme concentration due to the sample manipulation during the experiment.
2.7 Protein determination
Protein was determined spectrophotometrically using bicinchoninic acid [Citation29], with bovine serum albumin as a standard.
2.8 Data analysis
The data were analysed using Graph Pad Prism v. 3.00. All values are given as means of at least 3 measurements ± SD.
3 Results
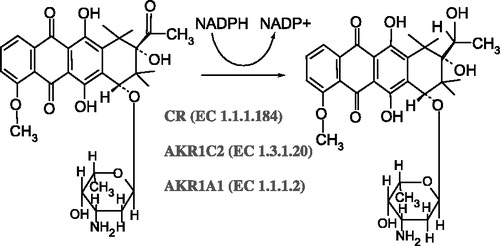
The reductase activities of CR, AKR1C2 and AKR1A1 were determined spectrophotometrically at their respective optimal pH (). However, the pH optimum for CR and AKR1C2 is the same (pH 6.0) so we were unable to distinguish the activities of the two enzymes initially and for this purpose used quercitrin and flufenamic acid, the diagnostic inhibitors of CR and AKR1C2, respectively. We measured the dependence of residual reductase activity on increasing inhibitor concentration and from the resulting inhibition curves () estimated the IC50 values as 5.45 ± 1.37 μM for quercitrin and 3.68 ± 1.58 μM for flufenamic acid. We also evaluated the contribution of CR and AKR1C2 to the total activity at pH 6.0 which was 3:1. The test of inhibitor sensitivity was repeated at pH 7.4 as well to get closer to physiological conditions and found that the difference between CR and AKR1C2 activity is then even more pronounced—in the ratio of 5:1. As the method for reductase activity determination is based on measuring the rate of NADPH oxidation instead of product determination (C13-OL) it could be regarded as relatively non-specific. We therefore investigated the possible interference of another cytosolic NADPH-dependent reductase which is known to metabolize daunorubicin, DT-diaphorase (EC 1.6.99.2). DT-diaphorase reduces the quinone moiety of the tetracyclic ring of daunorubicin and is not involved in C13-OL formation but theoretically it could interfere with our kinetic study. However, it was found that dicoumarol, an inhibitor of DT-diaphorase, had no effect on NADPH-dependent reduction of daunorubicin in our system indicating that the reduction the of quinone moiety of daunorubicin did not interfere with the determination of C13-carbonyl reduction (data not shown).
Figure 2 The activities of the three cytosolic reductases measured at their optimal pH. The first column represents the sum of activities of CR and AKR1C2 with the pH optimum at 6.0. The activity of AKR1A1 could be measured separately at pH 8.5.
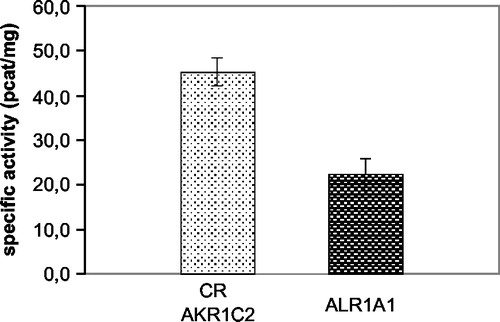
Figure 3 The inhibition curves of quercitrin (A) and flufenamic acid (B) at pH 6.0 and pH 7.4 for comparison. The participation of CR and AKR1C2 to the total reductase activity was calculated to be 3:1 at pH 6.0 (optimal) and 5:1 at pH 7.4 (physiological). The IC50 values at pH 6.0 were estimated to be 5.45 ± 1.37 μM for quercitrin and 3.68 ± 1.58 μM for flufenamic acid.
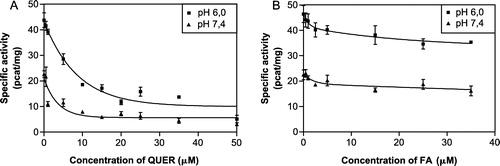
As the next step we were interested in discovering the mechanism of reductase inhibition exerted by quercitrin and flufenamic acid (this was done at optimal pH 6.0). Initially the kinetics of non-inhibited daunorubicin reduction () were studied and gave and
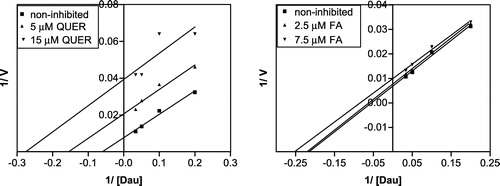
Four different concentrations of substrate (5–30 μM) and two concentrations of inhibitor (the lower concentration imitated the IC50 value and the other one was 3 fold higher) were used. The kinetics of the inhibited reactions were investigated (), the data were transformed according to Lineweaver and Burk and the Ki values were calculated (). An uncompetitive mechanism of inhibition was found for both inhibitors (). However, we are aware that attempting to estimate the kinetics of the minor component (AKR1C2) brought certain difficulties. The differences between the inhibitors concentrations were small and the error became relatively large so that the Ki value for flufenamic acid presented here did not come out as accurate as desired.
Figure 4 Michaelis-Menten kinetics of daunorubicin reduction at pH 6.0. The kinetic parameters calculated from the curve were:
Figure 5 Michaelis-Menten kinetics of daunorubicin reduction at pH 6.0 inhibited by quercitrin (A) and flufenamic acid (B) at two different concentrations by comparison with the non-inhibited reduction.
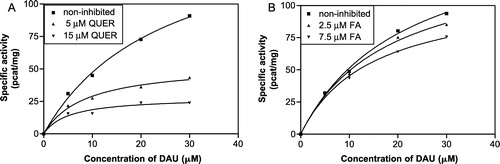
Table I. IC50 values, Ki constants and inhibition types of quercitrin and flufenamic acid for rabbit liver carbonyl reductase and aldo-keto reductase AKR1C2, respectively.
Figure 6 Linearization of the inhibition data according to Lineweaver and Burk for of daunorubicin reduction by quercitrin and flufenamic acid (at pH 6.0).
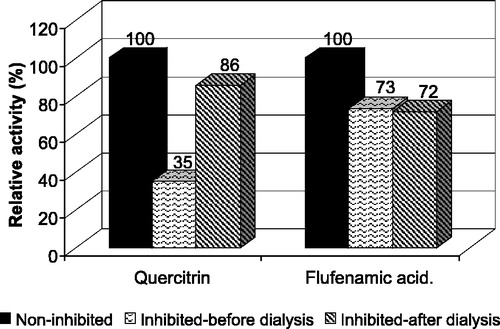
The reversibility of inhibition was tested by measuring the rate of inhibition before and after dialysis and comparing these values with those of a control (non-inhibited) sample to avoid misinterpretation of irreversibility of inhibition due to possible loss of activity during dialysis. The inhibition of CR by quercitrin was found to be reversible while flufenamic acid acted as an irreversible inhibitor of AKR1C2 ().
Figure 7 Inhibition reversibility assay. The reduction was inhibited by 10 μM quercitrin and 5 μM flufenamic acid to 35% and 73% of control, respectively. After 12 h of dialysis in PBS the inhibition by quercitrin was reversed and the activity reached 86% of control. In the case of flufenamic acid no significant changes in activity were observed due to dialysis suggesting reversible inhibition of CR by quercitrin and irreversible inhibition of AKR1C2 by flufenamic acid.
4 Discussion
The cardiac toxicity of anthracyclines is the main obstacle to their clinical use [Citation6]. This is quite unfortunate because anthracyclines are otherwise very efficient for the chemotherapy of a number of solid tumors as well as haematological malignancies [Citation1]. The mechanism of the cardiotoxicity development is not clear but it is believed that it is different from those toxic effects that anthracyclines exert against cancer cells [Citation36]. The generation of ROS during the redox-cycling of anthracyclines has been well described [Citation9,Citation10] and generally accepted as an important factor that can contribute to the cardiotoxicity but it is certainly not the only one. The role of cytosolic reductases have also been implied [Citation12,Citation19,Citation20] and the cardiodepressive effects of the secondary alcoholic metabolites have been described [Citation16,Citation17]. It has also been proved that the metabolites are accumulated in the heart and moreover that they are less potent cytostatics than the parent compounds [Citation20]. Three cytosolic reductases (CR, 1.1.1.184; AKR1C2, 1.3.1.20; AKR1A1, 1.1.1.2) have been described as participating in C13-carbonyl reduction of anthracyclines [Citation20], however, not much is known about the contribution of the particular enzymes to the formation of C13-OL. It was previously indicated that AKR1A1 which has a pH optimum at 8.5 was not sensitive to its specific inhibitor (phenobarbital) at pH 7.4 suggesting that it was not involved in the process of C13-OL formation from daunorubicin under physiological conditions [Citation19]. In that study Pröpper et al. also showed that carbonyl reductase was inhibited by its inhibitor at physiological pH and suggested that this single enzyme was responsible for C13-OL formation [Citation19]. However, the third enzyme, AKR1C2, was not investigated. The authors formulated their conclusions on the basis of a kinetic study at pH 7.4 which they indicated to be a single enzyme kinetics. The slight deviations from this model were explained as non-enzymatic daunorubicinol formation. Therefore it was our interest to find out whether it could be due to contribution of enzymatic reduction by AKR1C2.
This study was designed to estimate the contribution of CR and AKR1C2 to the total reductase activity at pH 6.0 (optimal) as well as at pH 7.4 (physiological). Using diagnostic inhibitors of the two enzymes it was found that the CR/AKR1C2 ratio was 3:1 at pH 6.0 and 5:1 at pH 7.4. These results indicate that CR is the major enzyme of daunorubicin reduction but there is still some residnal activity due to AKR1C2 even at physiological pH and this should not be neglected. On the other hand the Pröpper group's observation was confirmed that AKR1A1 was not active at pH 7.4 [Citation19]. This is due to the fact that the residual reductase activities in the presence of quercitrin or flufenamic acid together accounted for 100% of activity so that it is then very unlikely that any enzyme other than CR and AKR1C2 participates in daunorubicinol formation at pH 7.4.
Uncompetitive inhibition was found for both inhibitors of daunorubicin reduction in this study, unfortunately with low accuracy in the case of flufenamic acid which was unavoidable while working with the whole cytosolic fraction and not using purified enzymes. As for AKR1C2, it is evident that the members of AKR1C class have been much more studied in relation to the oxidation of the trans-dihydrodiols of aromatic hydrocarbons and their role in carcinogenesis [Citation30,Citation31]. The uncompetitive inhibition of AKR1C2 by flufenamic acid has been previously described with respect to NADP+ (i.e. in the direction of oxidation) [Citation24]. Enzymes, however, do not necessarily have to exert the same kinetic properties for the reactions of oxidation and reduction which is why we performed our own experiments using daunorubicin as the substrate of interest and examined its NADPH-dependent reduction. No information about the mechanism of inhibition of CR by quercitrin is available in the literature where quercitrin was only described as a noncompetitive inhibitor of lens aldose reductase (EC 1.1.1.21) [Citation32].
In conclusion, we describe here how CR and AKR1C2 participated in C13-OL formation and that CR was the more important enzyme in this process and, moreover, characterised the mechanism of inhibition of these enzymes by their specific inhibitors. This information can be especially valuable for designing new effective cardioprotective agents since it is clear that the C13-OL metabolites are at least partially responsible for the anthracycline-induced cardiotoxicity. It is of interest that quercitrin which belongs to a class of flavonoids is a potent inhibitor of C13-OL formation from daunorubicin. Flavonoids have been extensively studied as potential cardioprotectors since they act as antioxidants and iron-chelating agents [Citation33]. With regard to the freeradical theory and the involvement of iron in the development of cardiotoxicity it seems that the flavonoids could offer good multi-factorial protection. However, the group of flavonoids (either natural or semi-synthetic) is huge and while the relations between their structure and their antioxidant, and iron-chelating properties have been at least partially described in several studies [Citation34,Citation35], very little is known about the correlation with their inhibitory/induction effects which could perhaps be crucial for their clinical efficacy in protecting the heart from anthracycline-induced injury.
| Abbreviations | ||
| ROS: | = | reactive oxygen species |
| DAU: | = | daunorubicin |
| QUER: | = | quercitrin |
| FA: | = | flufenamic acid |
| CR: | = | carbonyl reductase |
| AKR: | = | aldo-keto reductase |
| C13-OL: | = | C13-hydroxyanthracycline |
Acknowledgements
This work was supported by the Research Centre LN00B125 of Czech Ministry of Education and GA čR No. 305/03/1511
References
- Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs 1997; 54: 1–7
- Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C, Spalla C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol Bioeng 1969; 11(6)1101–1110
- Giannini G. Fluorinated anthracyclines: Synthesis and biological activity. Med Chem Rev—Online 2004; 1: 47–71, (URL: http://www.bentham-mps.org/1-1/mcro1-1/Giuseppe%20Giannini.pdf, 10.8.2004)
- Monneret C. Recent developments in the field of antitumour anthracyclines. Eur J Med Chem 2001; 36(6)483–493
- Cottin Y, Touzery C, Dalloz F, Coudert B, Toubeau M, Riedinger A, Louis P, Wolf JE, Brunotte F. Comparison of epirubicin and doxorubicin cardiotoxicity induced by low doses: Evolution of the diastolic and systolic parameters studied by radionuclide angiography. Clin Cardiol 1998; 21(9)665–670
- Shadle SE, Bammel BP, Cusack BJ, Knighton RA, Olson SJ, Mushlin PS, Olson RD. Daunorubicin cardiotoxicity: Evidence for the importance of the quinone moiety in a free-radical-independent mechanism. Biochem Pharmacol 2000; 60(10)1435–1444
- Ferrans VJ, Clark JR, Zhang J, Yu ZX, Herman EH. Pathogenesis and prevention of doxorubicin cardiomyopathy. Tsitologiia 1997; 39(10)928–937
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. New Eng J Med 1998; 339(13)900–905
- Hrdina R, Geršl V, Klimtová I, Šimůnek T, Macháčková J, Adamcová M. Anthracycline-induced cardiotoxicity. Acta Medica 2000; 43(3)75–82
- Keizer HG, Pinedo HM, Schuurhui GJ, Joenje H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharm Ther 1990; 47(2)219–231
- Goeptar AR, Te Koppele JM, Lamme EK, Pique JM, Vermeulen NP. Cytochrome P450 2B1-mediated one-electron reduction of adriamycin: A study with rat liver microsomes and purified enzymes. Mol Pharmacol 1993; 44(6)1267–1277
- Mordente A, Minotti G, Martorana GE, Silvestrini A, Giardina B, Meucci E. Anthracycline secondary alcohol metabolite formation in human or rabbit heart: Biochemical aspects and pharmacologic implications. Biochem Pharmacol 2003; 66(6)989–998
- Corna G, Santambrogio P, Minotti G, Cairo G. Doxorubicin paradoxically protects cardiomyocytes against iron-mediated toxicity: Role of reactive oxygen species and ferritin. J Biol Chem 2004; 279(14)13738–13745
- Minotti G, Ronchi R, Salvatorelli E, Menna P, Cairo G. Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: Evidence for distinct metabolic pathways and implications for iron-mediated cardiotoxicity of antitumor therapy. Cancer Res 2001; 61(23)8422–8428
- Cairo G, Recalcati S, Pietrangelo A, Minotti G. The iron regulatory proteins: Targets and modulators of free radical reactions and oxidative damage. Free Radic Biol Med 2002; 32(12)1237–1243
- Cusack BJ, Mushlin PS, Voulelis LD, Li X, Boucek RJ, Olson RD. Daunorubicin-induced cardiac injury in the rabbit: A role for daunorubicinol?. Toxicol Appl Pharmacol 1993; 118(2)177–185
- Olson RD, Mushlin PS. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J 1990; 4(13)3076–3086
- Boucek RJ, Olson RD, Brenner DE, Ogunbunmi EM, Inui M, Fleischer S. The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J Biol Chem 1987; 262(33)15851–15856
- Pröpper D, Maser E. Carbonyl reduction of daunorubicin in rabbit liver and heart. Pharmacol Toxicol 1997; 80(5)240–245
- Ax W, Soldan M, Koch L, Maser E. Development of daunorubicin resistance in tumour cells by induction of carbonyl reduction. Biochem Pharmacol 2000; 59(3)293–300
- Forrest GL, Gonzales B. Carbonyl reductase. Chem Biol Interact 2000; 129(1–2)21–40
- Imamura Y, Higuchi T, Nozaki Y, Sugino E, Hibino S, Otagiri M. Purification and properties of carbonyl reductase from rabbit kidney. Arch Biochem Biophys 1993; 300(2)570–576
- Miyabe Y, Amano T, Deyashiki Y, Hara A, Tsukada F. Kinetic studies of the inhibition of a human liver 3 alpha-hydroxysteroid/dihydrodiol dehydrogenase isozyme by bile acids and anti-inflammatory drugs. Biol Pharm Bull 1995; 18(1)9–12
- Hara A, Matsuura K, Tamada Y, Sato K, Miyabe Y, Deyashiki Y, Ishida N. Relationship of human liver dihydrodiol dehydrogenases to hepatic bile-acid-binding protein and an oxidoreductase of human colon cells. Biochem J 1996; 313: 373–376
- Flynn TG, Green NC. The aldo-keto reductases: An overview. Exp Med Biol 1993; 328: 251–257
- Felsted RL, Bachur NR. Mammalian carbonyl reductases. Drug Metab Rev 1980; 11(1)1–60
- Penning TM, Mukharji I, Barrows S, Talalay P. Purification and properties of a 3 alpha-hydroxysteroid dehydrogenase of rat liver cytosol and its inhibition by anti-inflammatory drugs. Biochem J 1984; 222(3)601–611
- Atalla A, Maser E. Characterization of enzymes participating in carbonyl reduction of 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) in human placenta. Chem Biol Interact 2001; 130–132(1–3)737–748
- Brown RE, Jarvis KL, Hyland KJ. Protein measurement using bicinchoninic acid: Elimination of interfering substances. Anal Biochem 1989; 180(1)136–139
- Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J Biol Chem 2002; 277(27)24799–24808
- Burczynski ME, Lin HK, Penning TM. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: Implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res 1999; 59(3)607–614
- Varma SD, Mikuni I, Kinoshita JH. Flavonoids as inhibitors of lens aldose reductase. Science 1975; 188(4194)1215–1216
- Korkina LG, Afanas'ev IB. Antioxidant and chelating properties of flavonoids. Adv Pharmacol 1997; 38: 151–163
- Van Acker SABE, Van den Berg DJ, Tromp MNJL, Griffionen DH, Van Bennekom WP, Van der Vijgh WJF, Bast A. Structural aspects of antioxidant activity of flavonoids. Free Radic Biol Med 1996; 20(3)331–342
- Van Acker SABE, Van Balen GJ, Van der Berg DJ, Bast A, Van der Vijgh WJF. Influence of iron chelation on the antioxidant activity of flavonoids. Biochem Pharmacol 1998; 56(8)935–943
- Kluza J, Marchetti P, Gallego MA, Lancel S, Fournier C, Loyens A, Beauvillain JC, Bailly C. Mitochondrial proliferation during apoptosis induced by anticancer agents: Effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene 2004; 23(42)7018–7030