Abstract
Two series of variably N-substituted biperidines were synthesized by condensing various acid chlorides, alkyl halides and anhydrides with 1,4-bipiperidine. The new compounds were tested as tyrosinase inhibitors and a structure–activity relationship (SAR) study was carried out. Potent inhibition was observed in the case of the 4′-methylbenzyl substitution on this atom (IC50 = 1.72 μM) with this compound being a lead for future drug design. Additionally, calculations of the important QSAR molecular descriptors were done on the biperidine analogues after their 2 ps molecular dynamics (MD) simulations using molecular mechanics force field (MMFF) approaches. Using MD simulations potential and total energies were calculated for the energy minimized models of bipiperidine and the most active analogs 2, 3, 4, 6, 8 and 10.
1 Introduction
Tyrosinase (E.C. 1.14.18.1), also known as polyphenol oxidase (PPO), is a multifunctional copper-containing enzyme widely distributed in plants and animals [Citation1]. It catalyzes the o-hydroxylation of monophenols as well as the oxidation of o-diphenols to o-quinones. Tyrosinase is known to be a key enzyme for melanin biosynthesis in plants and animals. Tyrosinase inhibitors are clinically useful for the treatment of some dermatological disorders associated with melanin hyperpigmentation and also important in cosmetics for whitening and depigmentation after sunburn. Hyperpigmentation is associated with increased plasma melanocyte-stimulating hormone activity in peoples with insufficient production of glucocorticoids (Addison's disease). Recently, a number of inhibitors from natural products have been used in cosmetics [Citation1]. Tyrosinase plays a leading role in various biological and chemical processes [Citation2,Citation3]. So, there is a need to develop tyrosinase inhibitors to cure various skin diseases.
In continuation of our research work on bioactive molecules [Citation4,Citation5] we have synthesized a series of bipiperidine derivatives 2–12 and found them to be an active class of tyrosinase inhibitors which may provide lead compounds for future drug development.
2 Materials and methods
2.1 General
Melting points were determined on a Büchi 434 melting point apparatus and are uncorrected. NMR spectroscopy was performed on a Bruker AVANCE 400 MHz. Elemental analysis was carried out on a Carlo Erba Strumentazion-Mod-1106 Italy. UV and IR spectra were recorded on Perkin-Elmer Lambda-5 UV/VIS and JASCO IR-A-302 spectrophotometers, respectively. A FINNIGAN MAT-311A Germany spectrometer was used to record mass spectra. Thin layer chromatography (TLC) was performed on pre-coated silica gel glass plates (Kieselgel 60, 254, E. Merck, Germany) and chromatograms were visualized by UV light at 254 and 365 nm or iodine vapors.
2.2 General procedure for the preparation of compounds 2–12
Various anhydrides, alkyl halides, aromatic and aliphatic acid halides (1.1 mmol) dissolved in dry THF (10 mL) was added drop wise to 1,4-bipiperidine (0.5 mmol) in dry THF (25 mL) and the resultant reaction mixture was stirred for 48 h under argon at ambient temperature. The reaction was monitored by TLC analysis and upon completion; the reaction mixture was evaporated under reduced pressure on a rotary evaporator. The resultant product, if solid, was washed with excess of dry THF and recrystallized with an appropriate solvent or in the case of liquids, distilled under reduced pressure. The compounds were characterized using different spectroscopic techniques including UV, IR, NMR and mass and elemental analysis.
2.3 Tyrosinase inhibition assay
Tyrosinase inhibition assays were performed in a 96-well microplate format using SpectraMax® 340 (Molecular Devices, CA, USA) microplate reader according to the method developed earlier [Citation6].
Initially the compounds were screened for the o-diphenolase inhibitory activity of tyrosinase using l-DOPA as substrate. All the active inhibitors from the preliminary screening were subjected to IC50 studies. Briefly, the compounds were dissolved in DMSO to a concentration of 2.5%. Thirty Units mushroom tyrosinase (28 nM) was first preincubated with the compounds, in 50 nM Na-phosphate buffer (pH 6.8) for 10 min at 25°C. Then l-DOPA (0.5 mM) was added to the reaction mixture and the enzyme reaction was monitored by measuring the change in absorbance at 475 nm of the formation of the DOPAchrome for 10 min, at 37°C. The percent inhibition of the enzyme and IC50 values of the active compounds were calculated using a program developed with Java and Macro Excel® 2000 (Microsoft Corp., USA) for this purpose. The following equation was used:
Where ABSBlank and ABSSample are the absorbances for the blank and samples, respectively. All the studies were done at least in triplicates and the results here represents the Mean ± S.E.M. (standard error of the mean). All the reagents, enzyme, substrate and reference compounds, were purchased from Sigma Chem Co., MO, USA.
2.4 Molecular modeling studies
2.4.1 Energy minimization
The 2D structures of the molecules were drawn with CS ChemDraw® Pro version 6.0 (CambridgeSoft Corporation), which were further changed into 3D forms and hydrogen atoms were added at the 3D structures with the help of HyperChem® Professional version 7.1 (HyperCube Inc., Gainesville, FL, USA).
Energy minimization experiments were carried out with HyperChem® using the Block-Diagonal Newton-Raphson algorithm at the RMS (root mean square) gradients of 0.1 Kcal/(Å mol) for different cycles in vacuo depending on the specific molecule. For rendering and refinement of the 3D structures of the molecules the Persistence of Vision (tm) Ray-Tracer (Pov-Ray®) MS windows XP® version was used [Citation7,Citation8].
2.4.2 Molecular dynamics (MD) simulations
The molecular dynamics (MM+, molecular mechanics force field, MMFF and Amber 94) simulation experiments were done using HyperChem® Professional version 7.1 (HyperCube Inc., Gainesville, FL, USA) on a Pentium™ IV single processor computer using OS MS Windows XP® service pack 2 [Citation4,Citation5]. Electrostatic bond dipoles were taken for the option of MM+ simulations. In these experiments total run time was fixed for 1.0, heating and cooling times were 0.5 and step size was 0.001 pico second, respectively. The simulations were run in vacuo at temperatures of 300°K. Initial and final temperature was 0°K and the temperature step was 20°K. For the force field analysis, bond angle, torsion, non-bonded, electrostatic, and hydrogen bonded aspects were taken as the components for the MM+ simulations Citation7-9. For each of the molecule the approximate CPU time for 2 ps MD simulation was about 4 h.
2.5 Calculation of molecular descriptors
Some molecular descriptors were calculated such as, net charge (not shown in , as the values for all the compounds were calculated to be ‘0’), approximate and grid surface area, volume, hydration energy, log P, polarizability, refractivity and molecular mass using the QSAR module integrated with the program HyperChem® Professional version 7.1 (HyperCube Inc., Gainesville, FL, USA) on a Pentium™ IV single processor computer using OS MS Windows XP® service pack 2 Citation4Citation5Citation7-9.
3 Results and discussion
Tyrosinase inhibition studies were conducted on eleven N-substituted synthetic bipiperidine compounds 2–12 () were conducted and it was found that inhibitory activity changed with the substituent. The unsubstituted parent molecule 1 exhibited weak inhibition against the enzyme tyrosinase (IC50 = 110.79 μM), whereas, N-acetylation enhanced the inhibition (IC50 = 29.94 μM) as in compound 2, while the 2-naphthoyl derivative 3 also exhibited inhibitory properties (IC50 = 6.64 μM). 4′-Methylbenzylation at the nitrogen as in compound 4 greatly increased activity (IC50 = 1.72 μM) and made this molecule the most active compound in the series. The trifluoroacetylated derivative 6 exhibited moderate inhibition (IC50 = 18.08 μM), while, 4′-nitrobenzenesulfonylation and 3-phenoxypropylation at the N-atom (compounds 8 and 10) led to moderate activity with IC50 = 8.76 μM, IC50 = 19.52 μM, respectively, whilst, benzoyl, myristoyl, 3′,4′,5′-trimethoxybenzoyl, methyl-3-propanoate and nicotinoyl substitution at the same position, as in compounds 5, 7, 9, 11 and 12, respectively, led to a remarkable decline in activity with completely inactivity. We have selected only eleven compounds to include in this communication for comparison purposes; other screened compounds were found to be inactive.
Table I. Physical and tyrosinase inhibitory data for compounds 1–12. .
.
The tyrosinase inhibitory activities of these compounds were tested in reactions using l-DOPA as a substrate with reference to Kojic acid (IC50 = 16.67 μM) and l-mimosine (IC50 = 3.68 μM) (). The Present study suggests that compound 4 might be a lead compound for further drug development for the management of several skin disorders.
Another purpose of this study was to identify the biologically active stable conformers of the bipiperidine analogues and to calculate the volume and several other very importent molecular descriptors such as net charge, approximate and grid surface area, hydration energy, log P, refractivity, polarizability and mass. Compound 4 exhibited excellent tyrosinase inhibitory activity (IC50 = 1.72 μM) when compared with the standard tyrosinase inhibitors kojic acid (IC50 = 16.67 μM) and l-mimosine (IC50 = 3.68 μM). In the energy minimized 3D model of the molecule, it showed 720 geometrical conformations at the RMS gradient of 0.1 kcal/(Å mol) and the final energy minimized structures are shown in . Its approximate surface area was calculated as 277.111 Å2, volume as 461.46 Å3, and the hydration energy and log P were − 0.051 kcal/mol and 1.695, respectively (). Here, log P indicates the lipid solubility of the molecule [Citation7]. From the molecular dynamic (MD) simulation experiments, kinetic, potential and total energy was calculated as presented in . The highest kinetic energy calculated at the 514th step after 0.512 ps of simulation time was 96.54 kcal/mol. The highest potential energy calculated at the 494th step after 0.472 ps was 147.39 kcal/mol and the highest total energy calculated at the 510th step after 0.508 ps was 182.08 kcal/mol. The whole MD simulations were done with 2001 steps at 2.0 ps.
Figure 1 Energy minimized 3D models of the bipiperidine and its most active analogs.
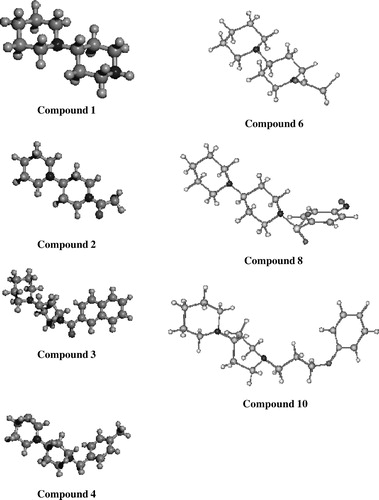
Table II. Calculations of the molecular descriptors of the bipiperidine molecules.
Figure 2 Energy [Potential (EPOT) and Total (ETOT)] calculations plots [time (x, in ps) vs energy (y, kcal/mol)] of Molecular Dynamics (MD) of compounds 1, 2, 3, 4, 6, 8 and 10, respectively.
![Figure 2 Energy [Potential (EPOT) and Total (ETOT)] calculations plots [time (x, in ps) vs energy (y, kcal/mol)] of Molecular Dynamics (MD) of compounds 1, 2, 3, 4, 6, 8 and 10, respectively.](/cms/asset/62697fd9-a994-41bc-9c3d-7da08159da72/ienz_a_117916_f0002_b.jpg)
Acknowledgements
The authors are very grateful to the HyperCube Inc., Gainesville, FL, USA, for providing the license of HyperChem® Professional version 7.1 by waiving the cost of the software. One of us M.T.H.K. also gratefully acknowledges travel support from Third World Academy of Sciences (TWAS), Italy, under the “South-South Fellowship” scheme.
Related Research Data
References
- Iida K, Hase K, Shimomura K, Sudo S, Kadota S, Namba T. Potent inhibitors of tyrosinase activity and melanin biosynthesis from Rheum officinale. Planta Med 1995; 61: 425–428
- Shiino M, Watanabe Y, Umezawa K. Synthesis of N-substituted N-nitrosohydroxylamines as inhibitors of Mushroom Tyrosinase. Bioorg Med Chem 2001; 9: 1233–1240
- Lee HS. Tyrosinase inhibitors of Pulsatilla cernua root-derived materials. J Agric Food Chem 2002; 50: 1400–1403
- Khan KM, Maharvi GM, Khan MTH, Parveen S, Choudhary MI, Atta-ur-Rahman. A facile and improved synthesis of sildenafil (Viagra®) analogues through solid support microwave irradiation possessing tyrosinase inhibitory potential, their conformational analysis and molecular dynamics simulation studies. Mol Divers 2005; 9: 15–26
- Khan KM, Maharvi GM, Perveen S, Khan MTH, Abdel-Jalil RJ, Shah STA, Choudhary MI, Atta-ur-Rahman, Voelter W. Synthesis of some methyl ether analogues of sildenafil (Viagra®) possessing tyrosinase inhibitory potential. Chem Biodiver 2005; 2: 470–476
- Hearing VJ. Methods Enzymol 1987; 142: 154–165
- McEachern MJ, Krauskopf A, Blackburn EH. Telomers and their control. Annu Rev Genet 2000; 34: 331–358
- Ren J, Qu X, Dattagupta N, Chaires JB. Molecular recognition of a RNA:DNA hybrid structure. J Am Chem Soc 2001; 123: 6742–6743
- Hong X, Hopfinger AJ. 3D-pharmacophores of flavonoid binding at the benzodiazepine GABAA receptor site using 4D-QSAR analysis. J Chem Inf Comput Sci 2003; 43: 324–336