Abstract
A revisited synthesis of 2-aryl-6-methyl-1,2-dihydro-1H-pyridin-4-ones and their saturated analogues 2-aryl-6-methylpiperidin-4-ols is described. A five steps procedure, using the sulfinimine chemistry, to prepare a key intermediate β-(6-chloronicotinic)-β-amino ester is also reported. In vivo spontaneous working memory activity of these compounds has been investigated in the mouse. Results obtained with 2-(3-chlorophenyl)-6-methyl-1,2-dihydro-1H-pyridin-4-one 9b, the most effective derivative in this model, have been reported.
Introduction
The nicotinic acetylcholine receptors (nAChRs) belong to a superfamily of ligand-gated ion channels which includes muscle-type and neuronal-type nAChRs, 5-HT3, GABAA, GABAC and glycine receptors. Actually twelve neuronal nAChR subunits (α2–α10 and β2–β4) have been found. Several combinations of α and β subunits can be expressed in oocytes or other heterologous expression systems resulting in functional ion channels with distinct pharmacological properties. For a few decades neuronal nAChRs have hold considerable promise as therapeutic targets for the treatment of central nervous system disorders. Drugs aimed at nAChRs may have a potential for the treatment of neurodegenerative disorders such as Alzheimer's and Parkinson's diseases, Tourette's syndrome, schizophrenia, attention deficit, hyperactivity disorder, certain epilepsies, and nicotine addiction, as well Citation1-3.
Our laboratory has recently pointed out the interest of new 2-aryl-6-methyl-1,2-dihydro-1H-pyridin-4-one and 2-aryl-6-methylpiperidin-4-ol derivatives as memory enhancers in relation to their nicotinic acetylcholine receptor activity Citation4-6. Herein, we present a preliminary communication devoted to pharmacomodulations on our series and the first in vivo experiments concerning our lead compound.
Materials and methods
Chemistry
Instrumentation
Commercial reagents were used as received without additional purification. Melting points were determined on a Kofler melting point apparatus and are uncorrected. Infrared (IR) spectra were obtained in KBr pellets with a Genesis Series FTIR spectrometer. 1H NMR (400 MHz) and 13C NMR (100 MHz) were recorded on a JEOL Lambda 400 spectrometer. Chemical shifts are expressed in ppm downfield from the residual deuterated solvent or the internal standard tetramethylsilane. Thin layer chromatographies (TLC) were performed on 0.2 mm precoated plates of silica gel 60F-254 (Merck). Visualization was made with ultraviolet light (254 nm). Column chromatographies were carried out using silica gel 60 (0.063–0.2 mm) (Merck). Elemental analyses for new compounds were performed at the “Institut de Recherche en Chimie Organique Fine” (Rouen).
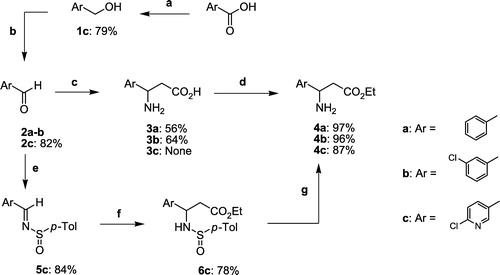
Synthesis method a: 6-chloropyridin-3-ylmethanol 1c
To a solution of 6-chloronicotinic acid (3.15 g, 20.0 mmol) in dry THF (60 mL), cooled to − 10°C, was added triethylamine (2.93 mL, 20.0 mmol). The reaction mixture was stirred for 30 min and ethyl chloroformate (2.10 mL, 22.0 mmol) was added dropwise. After stirring for 30 min the solution was filtered, H2O (30 mL) was added and the mixture was cooled to 0°C. NaBH4 (1.89 g, 50.0 mmol) was added and the reaction mixture was allowed to warm to RT under stirring for 2 h. The mixture was extracted with ethyl acetate. The organic phase was washed with a saturated aqueous NaHCO3 solution, dried (MgSO4) and concentrated. The residue was purified by chromatography on silica gel eluting with ethyl acetate/cyclohexane (1:1) to afford 1c (2.27 g) as a white solid. Yield: 79%. mp 44–45°C. IR (KBr) ν 3303, 2890, 1590, 1570, 1459, 1298, 1104, 1065, 1022, 821, 638 cm-1. 1H NMR (CDCl3) δ 8.31 (d, 4JHH = 2.0 Hz, 1H, H-2), 7.69 (dd, 3JHH = 8.2 Hz, 4JHH = 2.0 Hz, 1H, H-4), 7.31 (d, 3JHH = 8.2 Hz, 1H, H-5), 4.71 (s, 2H, CH2OH), 3.29 (bs, 1H, OH). 13C NMR (CDCl3) δ 150.4 (C-6), 148.1 (C-2), 137.8 (C-4), 135.5 (C-3), 124.2 (C-5), 61.6 (CH2OH).
Method b: 6-chloropyridine-3-carbaldehyde 2c
To a mixture of pyridinium chlorochromate (4.85 g, 22.5 mmol) and Celite® (5.0 g) in dry CH2Cl2 (100 mL), cooled to 0°C, was added a solution of 6-chloropyridin-3-ylmethanol 1c (2.15 g, 15.0 mmol) in CH2Cl2 (25 mL). After stirring for 2 h the mixture was filtered through Celite® and the filter cake was washed with diethyl ether. The organic layer was concentrated and the residue was purified by chromatography on silica gel eluting with CH2Cl2 to afford 2c (1.74 g) as a white solid. Yield: 82%. mp 79-80°C. IR (KBr) ν 3095, 2875, 1702, 1588, 1560, 1458, 1353, 1110, 1020, 832, 734, 538, 489 cm-1. 1H NMR (CDCl3) δ 10.07 (s, 1H, CHO), 8.84 (d, 4JHH = 1.9 Hz, 1H, H-2), 8.12 (dd, 3JHH = 8.2 Hz, 4JHH = 1.9 Hz, 1H, H-4), 7.49 (d, 3JHH = 8.2 Hz, 1H, H-5). 13C NMR (CDCl3) δ 189.2 (CHO), 156.9 (C-6), 152.4 (C-2), 138.0 (C-4), 130.4 (C-3), 125.2 (C-5).
Method e: N-(6-Chloropyridin-3-ylmethylene)-p-toluenesulfinamide 5c
A mixture of 6-chloropyridine-3-carbaldehyde 2c (991 mg, 7.0 mmol), p-toluenesulfinamide (1.14 g, 7.35 mmol) and titanium(IV) ethoxide (8.0 g, 35.0 mmol) in dry CH2Cl2 (100 mL) was refluxed for 12 h. The reaction mixture was quenched at 0°C by addition of H2O. The turbid solution was filtered through Celite® and the filter cake was washed with CH2Cl2. The organic layer was separated, dried (CaCl2) and concentrated. The residue was purified by chromatography on silica gel eluting with CH2Cl2 to afford 5c (1.64 g) as a white solid. Yield: 84%. mp 142–143°C. IR (KBr) ν 3086, 3056, 2923, 1594, 1583, 1556, 1454, 1373, 1333, 1100, 805, 673, 552 cm− 1. 1H NMR (CDCl3) δ 8.77 (s, 1H, HC = N), 8.76 (d, 4JHH = 2.2 Hz, 1H, H-2′), 8.13 (dd, 3JHH = 8.3, 4JHH = 2.2 Hz, 1H, H-4′), 7.6 (d, 3JHH = 8.2 Hz, 2H, H-2 and H-6), 7.41 (d, 3JHH = 8.3 Hz, 1H, H-5′), 7.32 (d, 3JHH = 8.2 Hz, 2H, H-3 and H-5), 2.40 (s, 3H, CH3). 13C NMR (CDCl3) δ 156.7 (HC = N), 154.9 (C-6′), 151.1 (C-2′), 142.0 (C-4), 140.8 (C-1), 138.0 (C-4′), 129.8 (C-3 and C-5), 128.4 (C-3′), 124.7 (C-5′), 124.5 (C-2 and C-6), 21.3 (CH3). Anal. Calcd. For C13H11N2OClS: C, 56.01; H, 3.98; N, 10.05. Found: C, 56.22; H, 4.24; N, 9.68%.
Method f: ethyl 3-(6-chloropyridin-3-yl)-3-[(p-tolylsulfinyl)amino]propanoate 6c
To a solution of dry THF (20 mL), cooled to − 78°C, was added NaHMDS 2M (4.40 mL, 8.8 mmol) followed by anhydrous ethyl acetate (806 μL, 8.25 mmol). The reaction mixture was stirred for 1 h and during this time a solution of N-(6-chloropyridin-3-ylmethylene)-p-toluenesulfinamide 5c (1.53 g, 5.5 mmol) in THF (5 mL) was added dropwise at − 78°C. After stirring for 6 h the reaction was quenched at − 78°C with a saturated aqueous NH4Cl solution. The organic phase was extracted with ethyl acetate, dried (MgSO4) and concentrated. The residue was purified by chromatography on silica gel eluting with ethyl acetate/petroleum ether (1:1) to afford 6c (1.57 g) as a yellow oil. Yield: 78%. Mixture of two diastereomers: 1H NMR (CDCl3) δ 8.42 and 8.12 (m, 1H, H-2″), 7.76 and 7.46 (m, 1H, H-4″), 7.57 and 7.45 (m, 2H, H-2′ and H-6′), 7.34 and 7.15 (m, 1H, H-5″), 7.32 and 7.18 (m, 2H, H-3′ and H-5′), 5.53 and 5.17 (m, 1H, NH), 4.85 and 4.75 (m, 1H, CH), 4.07 (q, 3JHH = 7.2 Hz, 2H, CH2O), 2.96 and 2.85 (m, 2H, CH2C = O), 2.43 and 2.36 (s, 3H, CH3), 1.18 (t, 3JHH = 7.2 Hz, 3H, CH3CH2O).
Method g: ethyl 3-amino-3-(6-chloropyridin-3-yl)propanoate 4c
To a stirred solution of ester 6c (1.47 g, 4.0 mmol) in MeOH (15 mL), cooled to 0°C, was added TFA (1.23 mL, 16.0 mmol). The reaction mixture was stirred at RT for 2 h and concentrated. The residue was dissolved in CH2Cl2 (30 mL) and the resulting solution was washed with a saturated aqueous NaHCO3 solution. The organic layer was dried (CaCl2) and concentrated. The residue was purified by chromatography on silica gel eluting with ethyl acetate/MeOH (9:1) to afford 4c (796 mg) as a yellow oil. Yield: 87%. IR (KBr) ν 3365, 3292, 2983, 2941, 1725, 1429, 1372, 1185, 1022, 711 cm-1. 1H NMR (CDCl3) δ 8.39 (d, 4JHH = 2.3 Hz, 1H, H-2′), 7.73 (dd, 3JHH = 8.2 Hz, 4JHH = 2.3 Hz, 1H, H-4′), 7.31 (d, 3JHH = 8.2 Hz, 1H, H-5′), 4.48 (dd, 3JHH = 5.5 and 8.1 Hz, 1H, CH), 4.14 (q, 3JHH = 7.1 Hz, 2H, CH2O), 2.68 and 2.64 (ABX, 2JAB = 16.1 Hz, 3JAX = 8.1 Hz, 3JBX = 5.5 Hz, 2H, CH2C = O), 1.97 (bs, 2H, NH2), 1.24 (t, 3JHH = 7.1 Hz, 3H, CH3CH2O). 13C NMR (CDCl3) δ 171.1 (C = O), 150.5 (C-6′), 148.2 (C-2′), 138.5 (C-3′), 137.0 (C-4′), 124.2 (C-5′), 60.8 (CH2O), 49.7 (CH), 43.5 (CH2C = O), 14.1 (CH3CH2O). Anal. Calcd. For C10H13N2O2Cl: C, 52.52; H, 5.73; N, 12.25. Found: C, 52.79; H, 5.97; N, 11.91%.
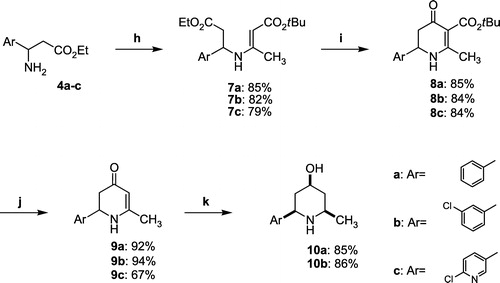
Method h: tert-Butyl 3-[1-(6-chloropyridin-3-yl)-3-ethoxy-3-oxopropylamino]but-2-enoate 7c
To a stirred solution of amino ester 4c (572 mg, 2.5 mmol), in dry benzene (15 mL), was added AcOH (430 μL, 7.5 mmol), and tert-butylacetoacetate (497 μL, 3.0 mmol). The mixture was heated under reflux for 4 h and the water formed was removed azeotropically using a Dean-Stark apparatus. After cooling to RT, CHCl3 (15 mL) was added and the solution was washed with a saturated aqueous NaHCO3 solution. The organic layer was dried (CaCl2), the solvents were evaporated and the residue was purified by chromatography on silica gel eluting with diethyl ether/petroleum ether (6:4) to afford 7c (729 mg) as a colorless oil. Yield: 79%. IR (KBr) ν 3269, 3221, 2972, 2937, 1732, 1651, 1610, 1448, 1422, 1365, 1270, 1152, 1026, 788, 715 cm-1. 1H NMR (CDCl3) δ 9.03 (d, 3JHH = 9.3 Hz, 1H, NH), 8.34 (d, 4JHH = 2.7 Hz, 1H, H-2′), 7.62 (dd, 3JHH = 8.3 Hz, 4JHH = 2.7 Hz, 1H, H-4′), 7.32 (d, 3JHH = 8.3 Hz, 1H, H-5′), 5.03 (m, 1H, CH), 4.48 (s, 1H, C = CH), 4.14 and 4.10 (ABX3, 2JAB = 10.7 Hz, 3JAX = 7.2 Hz, 3JBX = 7.2 Hz, 2H, CH2O), 2.85 and 2.78 (A′B′X′, 2JA′B′ = 15.6 Hz, 3JA′X′ = 7.8 Hz, 3JB′X′ = 6.4 Hz, 2H, CH2C = O), 1.80 (s, 3H, CH3), 1.47 (s, 9H, CH3tBu), 1.22 (t, 3JHH = 7.2 Hz, 3H, CH3CH2O). 13C NMR (CDCl3) δ 170.4 (C = O), 169.8 (C = O), 159.0 (C = CH), 150.7 (C-6′), 147.9 (C-2′), 136.7 (C-3′), 136.6 (C-4′), 124.5 (C-5′), 87.6 (C = CH), 78.4 (CtBu), 61.2 (CH2O), 51.1 (CH), 42.9 (CH2C = O), 28.5 (CH3tBu), 19.4 (CH3), 14.0 (CH3CH2O).
Method i: tert-Butyle 6-(6-chloropyridin-3-yl)-2-methyl-4-oxo-1,4,5,6-tetrahydropyridine-3-carboxylate 8c
To a stirred solution of enamine 7c (738 mg, 2.0 mmol), in dry t-BuOH (10 mL), was added t-BuOK (269 mg, 2.4 mmol), and the mixture was heated at 50°C. After completion (4 h monitored by TLC analysis), the reaction mixture was quenched with an aqueous HCl 0.5 M solution and extracted with CHCl3. The organic layer was dried (CaCl2) and concentrated. The crude product was purified by chromatography on silica gel eluting with ethyl acetate/MeOH (8:2) to give 8c (542 mg) as a yellow solid. Yield: 84%. mp 180–181°C. IR (KBr) ν 3279, 3081, 2975, 2931, 1696, 1619, 1558, 1529, 1430, 1366, 1163, 1096, 715 cm-1. 1H NMR (CDCl3) δ 8.35 (d, 4JHH = 2.3 Hz, 1H, H-2′), 7.69 (dd, 4JHH = 2.3 Hz, 3JHH = 8.3 Hz, 1H, H-4′), 7.33 (d, 3JHH = 8.3 Hz, 1H, H-5′), 6.52 (bs, 1H, NH), 4.77 (dd, 3JHH = 5.9 and 11.4 Hz, 1H, CH), 2.61 and 2.49 (ABX, 2JAB = 15.8 Hz, 3JAX = 11.4 Hz, 3JBX = 5.9 Hz, 2H, CH2C = O), 2.31 (s, 3H, CH3), 1.50 (s, 9H, CH3tBu). 13C NMR (CDCl3) δ 186.7 (C = O), 166.4 (CO2tBu), 165.5 (HN-C = C), 151.7 (C-6′), 148.3 (C-2′), 137.3 (C-3′), 134.0 (C-4′), 124.7 (C-5′), 100.4 (HN-C = C), 80.8 (CtBu), 51.5 (CH), 42.8 (CH2C = O), 28.4 (CH3tBu), 22.6 (CH3). Anal. Calcd. For C16H19N2O3Cl: C, 59.54; H, 5.93; N, 8.68. Found: C, 59.19; H, 5.98; N, 8.91%.
Method j: 2-(6-Chloropyridin-3-yl)-6-methyl-2,3-dihydro-1H-pyridin-4-one 9c
To a stirred solution of tetrahydropyridinone 8c (484 mg, 1.5 mmol) in CH2Cl2 (5 mL) was added TFA (1.16 mL, 15.0 mmol) and the reaction mixture was heated at 50°C. After completion (4 h monitored by TLC analysis), the reaction mixture was neutralized with a saturated aqueous K2CO3 solution and extracted with CH2Cl2. The organic layer was dried (CaCl2) and concentrated. The residue was purified by chromatography on silica gel eluting with ethyl acetate/MeOH (7:3) to afford 9c (224 mg) as a yellow solid. Yield: 67%. mp 155-156°C. IR (KBr) ν 3235, 3059, 2963, 2936, 1616, 1581, 1536, 1435, 1358, 1269, 1105, 810 cm-1. 1H NMR (CDCl3) δ 8.36 (d, 4JHH = 1.9 Hz, 1H, H-2′), 7.68 (d, 4JHH = 1.9 Hz, 3JHH = 8.1 Hz, 1H, H-4′), 7.35 (d, 3JHH = 8.1 Hz, 1H, H-5′), 6.23 (bs, 1H, NH), 5.02 (s, 1H, C = CH), 4.72 (dd, 3JHH = 5.9 and 12.6 Hz, 1H, CH), 2.58 and 2.47 (ABX, 2JAB = 15.8 Hz, 3JAX = 12.6 Hz, 3JBX = 5.9 Hz, 2H, CH2C = O), 2.03 (s, 3H, CH3). 13C NMR (CDCl3) δ 190.0 (C = O), 161.1 (C = CH), 149.2 (C-6′), 151.1 (C-2′), 136.2 (C-3′), 134.0 (C-4′), 124.7 (C-5′), 99.0 (C = CH), 54.3 (CH), 42.8 (CH2C = O), 21.6 (CH3). Anal. Calcd. For C11H11N2OCl: C, 59.33; H, 4.98; N, 12.58. Found: C, 59.49; H, 5.17; N, 12.64%.
Method k: 2-(3-Chlorophenyl)-6-methylpiperidin-4-ol 10b
To a stirred solution of pyridinone 9b (332 mg, 1.5 mmol) in EtOH (10 mL), cooled to 0°C, was added NaBH4 (454 mg, 12.0 mmol) and the reaction mixture was stirred for 12 h at RT. The reaction mixture was made alkaline with a saturated aqueous K2CO3 solution and extracted with CH2Cl2. The organic layer was dried (CaCl2) and concentrated. The residue was purified by chromatography on silica gel eluting with ethyl acetate/MeOH (8:2) to afford 10b (291 mg) as a white solid. Yield: 86%. mp 94-95°C. IR (KBr) ν 3278, 3248, 3031, 2965, 2928, 2835, 1605, 1452, 1374, 1307, 1088, 1031, 847, 755, 699 cm-1. 1H NMR (CDCl3) δ 7.39 (s, 1H, H-2′), 7.26-7.24 (m, 3H, H-4′, H-5′ and H-6′), 3.79 (m, 1H, H-4), 3.67 (dd, 3JHH = 2.1 and 11.5 Hz, 1H, H-2), 2.84 (ddq, 3JHH = 2.2, 6.2 and 11.0 Hz, 1H, H-6), 2.07 (m, 1H, H-3eq.), 1.99 (m, 1H, H-5eq.), 1.85 (bs, 2H, NH and OH), 1.37 (m, 1H, H-3ax.), 1.17 (d, 3JHH = 6.2 Hz, 3H, CH3), 1.16 (m, 1H, H-5ax.). 13C NMR (CDCl3) δ 145.9 (C-1′), 134.3 (C-3′), 129.7 (C-5′), 127.4 (C-4′), 126.9 (C-2′), 124.9 (C-6′), 69.5 (C-4), 59.2 (C-2), 50.9 (C-6), 43.3 (C-3), 43.2 (C-5), 22.4 (CH3). Anal. Calcd. For C12H16NOCl: C, 63.86; H, 7.14; N, 6.21. Found: C, 63.99; H, 7.21; N, 6.06%.
Pharmacology
Animals
Experiments were performed on male NMRI mice (Centre d'Elevage René Janvier, Le Genest, France) weighing 25–30 g. Mice were housed in standard polycarbonate cages containing ten animals and maintained in a regulated environment (22 ± 1°C) under 12h–12 h light/dark cycle (light on between 20:00 and 8:00) with food and water freely available in the home cage. Behavioral tests were conducted during the dark phase of the cycle from 14:00 to 20:00.
Apparatus
Immediate spatial working memory performance was assessed by recording spontaneous alternation behavior in a single session in a Y-maze made of black painted wood. The maze consisted of three equally spaced arms (22 cm long and 6.5 cm wide with walls 10 cm high). An inclined mirror was suspended over the maze to allow recording of the behavior at distance (1.5 m). Temperature in the test room was kept at 22 ± 1°C.
Testing procedure [Citation7,Citation8]
Each mouse, naive to the maze, was placed at the end of one arm and allowed to freely explore the maze during a 5-min session. The number and the sequence of arms entries and the number of rears were recorded by the observer. An arm entry was scored when all four feet crossed into the arm. Alternation behavior, defined as entries into all three arms on consecutive occasions, was expressed in percent of total arm entries. Percentage alternation was calculated as the ratio of actual to possible alternation (defined as the total number of arm entries minus two), multiplied by 100 as shown in the following equation: % Alternation = {(Number of alternations)/(Total arm entries-2)} × 100. The sequence of arm entered (for example ACBCBACAB…) provides a measure of spontaneous alternation behavior and thus immediate working memory (spontaneous alternation required recalling of precedent visited arm). Each experience was completed for ten mice per group.
Drug administration
Compound 9b was tested at 0.1, 0.3 and 1 mg/kg. For each dose, four groups were constituted: control (saline+saline), scopolamine (saline+scopolamine), tested compound (compound+saline) and association (compound+scopolamine). Scopolamine (scopolamine hydrobromide, Sigma) was administered at 1 mg/kg. Arecoline (arecoline hydrobromide, Sigma) used as pharmacological anti-amnesic drug was tested at 1 mg/kg. All drugs, dissolved in saline as the vehicle, were administered intraperitoneally (i.p.) in a volume of 10 mL kg− 1, 30 min before testing.
Statistical analyses
Percentage alternation scores were statistically analyzed by a one-way analysis of variance (ANOVA) with “alternation” as interdependent factors and “pharmacological treatment” as an independent factor. In the case of significant differences in the variances, post-hoc multiple comparisons (PLSD of Fisher) were undertaken to locate a principal effect. p-Values less than 0.05 were considered to be significant. Results are expressed as means ± SEM.
Results and discussion
Chemistry
Key intermediates in the synthesis of the target compounds described here are the corresponding β-aryl-β-amino esters 4a–c. To obtain these derivatives we applied the Rodionow-Johnson reaction Citation9-11. This synthesis involves a single step in which corresponding arylcarboxaldehydes were refluxed with malonic acid and ammonium acetate. Contrary to previously described [Citation12], in the case of the 6-chloro-3-pyridine carboxaldehyde reagent, prepared from 6-chloronicotinic acid via a reduction-oxidation sequence, this method failed. Alternatively, transformation of the aldehyde 2c to N-sulfinyl aldimine 5c was achieved by refluxing with racemic para-toluenesulfinamide in the presence of titanium(IV) ethoxide [Citation13]. According to the Davis's methodology [Citation14,Citation15], treatment of derivative 5c with the sodium enolate of ethyl acetate, generated from NaHMDS in THF at − 78°C, afforded 6c. The sulfinamide 6c was then hydrolyzed by treatment with trifluoroacetic acid, affording after alkalinization, the aimed β-amino ester 4c (Scheme ).
Scheme 1 Synthesis of β-aryl-β-amino esters 4. (a) i) Et3N, EtOCOCl, THF, 0°C, ii) NaBH4, THF/H2O, 0°C; (b) PCC, CH2Cl2, RT; (c) CH2(CO2H)2, AcONH4, EtOH, reflux; (d). i) SOCl2, EtOH, reflux,. ii) NH4OH, CH2Cl2, RT; (e) p-Toluenesulfinamide, Ti(OEt)4, CH2Cl2, reflux; (f) NaHMDS, AcOEt, THF, − 78°C; (g) i) TFA, MeOH, 0°C, ii) NaHCO3, CH2Cl2, RT.
Treatment of β-aryl-β-amino esters 4a–c with tert-butyl acetoacetate, in the presence of three equivalent amounts of acetic acid in refluxing benzene with azeotropic removal of water, generated the tert-butyl enaminoesters 7a–c. The latter were then converted to the cyclic derivative 8a–c using potassium tert-butoxide in tert-butanol. Removal of the tert-butyl group was finally performed by refluxing in dichloromethane in the presence of trifluoroacetic acid to give the target 2-aryl-6-methyl-2,3-dihydro-1H-pyridin-4-ones 9a–c [Citation16]. Access to the saturated 2-aryl-6-methylpiperidin-4-ols 10a and 10b was carried out using an excess of NaBH4. Under these conditions only the diastereomers with substituents in the cis orientation were detected (1H NMR and HPLC) and isolated (Scheme ).
Scheme 2 Synthesis of 2-aryl-6-methyl-2,3-dihydro-1H-pyridin-4-ones 9. (h) t-Butyl acetoacetate, AcOH, C6H6, reflux; (i) t-BuOK, t-BuOH, 50°C; (j) TFA, CH2Cl2, reflux, (k) NaBH4, EtOH, RT.
Pharmacology
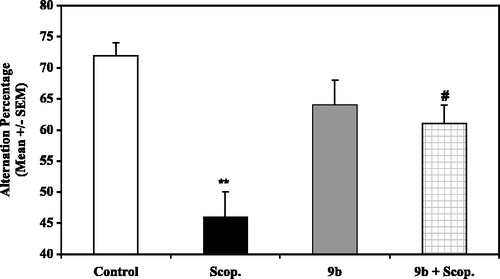
Administered alone, or in association with scopolamine, the ability of compound 9b to affect exploratory behavior in the Y-maze was investigated at doses of 0.1, 0.3 and 1 mg/kg. Statistical analysis revealed that, at the doses tested, compound 9b administered alone had no effect on the alternation behavior when compared to saline controls (see for 0.3 mg/kg in ). Scopolamine significantly reduced the percentage of alternation when compared to saline control (p < 0.01; PLSD of Fisher). However, in association with scopolamine experiments, 9b being ineffective at 0.1 mg/kg dosage, reversed, at the doses of 0.3 mg/kg () and 1 mg/kg (data not showns), the scopolamine-induced impairment of spontaneous alternation. This was illustrated for the 0.3 mg/kg dosage, both i) by a significant difference between the co-treatment and the scopolamine groups (p < 0.05; PLSD of Fischer), and ii) by the lack of significant difference between controls and co-treated animals. Under the same conditions, arecoline (1 mg/kg), used as a pharmacological reference, significantly reversed the scopolamine-induced deficit (49% for scopolamine group versus 67% for arecoline + scopolamine group, p < 0.0001 (PLSD of Fisher). The present findings demonstrate that compound 9b at 0.3 mg/kg is able to reverse alternation deficits induced by scopolamine in the Y-maze test.
Figure 1 Effects of compound 9b on spontaneous working memory in mouse. Mice (n = 10 in each group) received two injections 30 min before the test. Control = saline+saline; Scop. = saline+scopolamine (1 mg/kg); 9b = compound 9b (0.3 mg/kg) + saline; 9b+ Scop. = compound 9b (0.3 mg/kg) + scopolamine (1 mg/kg). **p < 0.01 versus control (saline), #p < 0,05 versus scopolamine treated mice (ANOVA + PLSD of Fisher).
Conclusion
The in vivo pharmacological findings indicated that these new nicotinic ligands (especially compound 9b) may provide a potential therapeutic approach to restore memory deficits in neurodegenerative diseases in which short-term working memory disorders were associated with cholinergic hypofunction. These promising results prompt us to continue the exploration of these new pyridinone and piperidinol series.
Acknowledgements
We appreciate the financial support provided by the “Ministère de la Recherche et de la Technologie” for a doctorate grant to N.L. (1999-2002) and the “Groupe de Recherche Servier”.
References
- Holladay MW, Dart MJ, Lynch JK. J Med Chem 1997; 40: 4169–4194
- Léna C, Changeux JP. J Physiology (Paris, Fr.) 1998; 92: 63–74
- Lloyd GK, Williams M. J Pharmacol Exp Ther 2000; 292: 461–467
- Rault S, Renault O, Guillon J, Dallemagne P, Pfeiffer B, Lestage P, Lebrun MC. Eur Pat Appl 2000, EP1050530, Chem. Abstr. 133, 335170 (2000)
- Leflemme N, Dallemagne P, Rault S. Tetrahedron 2004; 60: 4861–4865
- Rault S, Leflemme N, Dallemagne P, Lestage P, Lockhart B, Danober L, Pfeiffer B, Renard P. Chem Abstr 2004, FR2846654140, 375077 (2004)
- Sarter M, Bodewitz G, Stephens DN. Psychopharmacology (Berlin, Ger.) 1988; 94: 491–495
- Lelong V, Lhonneur L, Dauphin F, Boulouard M. Naunym-Schmiedeberg's Arch Pharmacol 2003; 367: 621–628
- Rodionow VM, Postovskaja EA. J Am Chem Soc 1929; 51: 841–847
- Johnson TB, Livak JE. J Am Chem Soc 1936; 58: 299–303
- Tan CYK, Weaver DF. Tetrahedron 2002; 58: 7449–7461
- Hoekstra WJ, Maryanoff BE, Damiano BP, Andrade-Gordon P, Cohen JH, Costanzo MJ, Haertlein BJ, Hecker LR, Hulshizer BL, Kauffman JA, Keane P, McComsey DF, Mitchell JA, Scott L, Shah RD, Yabut SC. J Med Chem 1999; 42: 5254–5265
- Davis FA, Zhang Y, Andemichael Y, Fang T, Fanelli DL, Zhang H. J Org Chem 1999; 64: 1403–1406
- Davis FA, Reddy RE, Szewczyk JM. J Org Chem 1995; 60: 7037–7039
- Davis FA, Szewczyk JM, Reddy RE. J Org Chem 1996; 61: 2222–2225
- Leflemme N, Dallemagne P, Rault S. Synthesis 2002; 1740–1746