Abstract
We have previously reported the synthesis and evaluation of potent anti-human immunodeficiency virus compounds based on β-D-d4T analogues bearing a tether attached at the C-5 position and their β-L-counterparts. Initial study revealed a requirement for an alkyl side-chain with an optimal length of 12 carbons for a weak antiviral activity. As a continuation of that work, we have now prepared the corresponding phosphoramidate derivatives as possible membrane-permeable prodrugs. Phosphorochloridate chemistry gave the target phosphoramidates which were tested for anti-human immunodeficiency virus type 1 activity; unfortunately, they were devoid of anti-HIV activity.
Keywords::
Introduction
The nucleotide prodrug concept continues to be an area of intense interest since all antiviral nucleoside drugs require metabolic activation in their target cell to the bio-active triphosphorylated forms by kinase-mediated phosphorylation for the expression of their antiviral activity [Citation1]. Also, the principal objective of the pronucleotide approach is to confer activity upon nucleoside analogues that express poor activity due to the inefficient intracellular phosphorylation by host cell or virally-encoded nucleoside kinases leading to the production of the active triphosphorylated form.
In this way, the phosphoramidate approach is increasingly used to improve the pharmacological activity of modified nucleoside analogues requiring intracellular phosphorylation since these compounds would otherwise be inactive due to poor kinase-mediated phosphorylation. This prodrug methodology has been widely developed by McGuigan who has previously shown that the (aryloxy)phosphoramidate nucleoside prodrugs, that undergo facile P-N bond hydrolysis, can lead to markedly elevated levels of anti-HIV potency for a range of antiviral nucleoside analogues that successfully bypass dependency on nucleoside kinase Citation2-4. The phosphate group was masked with neutral lipohilic groups to obtain a lipophilic membrane-permeable derivative able to access intracellular target sites. The amino acid moiety has emerged as the key determinant of intracellular phosphate delivery and previous modifications have revealed L-alanine as the moiety for optimal antiviral activity [Citation3]. The masked-phosphate approach to the 2’,3’-didehydro-2’,3’-dideoxy thymidine (d4T, Stavudine) was particularly interesting since the initial monophosphorylation step appears to be rate-limiting. Previous studies investigating the activation of pronucleotides have proposed two possible pathways for the hydrolysis of the phosphate prodrugs to their corresponding free mononucleotides (Scheme ) [Citation5]. The alaninyl phosphoramidate derivatives were presumed to be cleaved by either an enzymatic (path A) or a chemical (path B) hydrolysis, path A being suggested as the major route. The first step in the enzymatic hydrolysis of the pronucleotides (path A) was presumed to be the cleavage of the methoxy group followed by spontaneous cyclization and displacement of the aryloxy group leading to the formation of the amino acyl metabolite; the last step is catalyzed by intracellular phosphoramidase [Citation5].
Scheme 1 Two possible pathways proposed for the degradation of the phosphate prodrugs to their corresponding free nucleotides [Citation5].
![Scheme 1 Two possible pathways proposed for the degradation of the phosphate prodrugs to their corresponding free nucleotides [Citation5].](/cms/asset/a72caa7a-9b89-447d-a66a-63005ecceb5d/ienz_a_122017_f0001_b.jpg)
In connection with our program on β-D-d4T analogues bearing an amino linker arm on the C-5 position, we were particularly interested in applying emerging phosphate prodrug approaches to this series of 5-(aminoalkyl)carbamoylmethyl-d4T analogues in order to increase the antiviral activity. Previously, we have reported that the β-D-d4T analogues bearing an (aminoalkyl)carbamoylmethyl tether containing twelve methylene units attached at the position C-5 exhibit micromolar anti-HIV-1 activity in the CEM-SS cells (IC50 2.3 μM) [Citation6]. Consequently, in this current work, we have investigated the conversion of this series of C-5 modified nucleosides to their corresponding prodrugs containing a phenyl group and the methyl ester of L-alanine linked to the phosphorus through a phosphoramidate bond with a primary amino moiety.
Moreover, in the last years, the L-nucleosides analogues have drawn considerable attention as potential antiviral drugs [Citation7]. The “unnatural” L-nucleoside derivatives have been described as being more efficient than the corresponding D enantiomers, owing to their powerful antiviral activity and favorable toxicity profile [Citation8]. In particular, the 2’,3’-dideoxy-3’-thiacytidine (3TC, lamivudine) which has the β-L(-) isomeric configuration was more potent and less toxic than its D-isomer and is now approved for the treatment of AIDS Citation9-10. However, 3TC needs to be converted to its 5’-triphosphate derivative by cellular enzymes to inhibit HIV reverse transcriptase. Moreover, it has been reported that the β-L-d4T had no anti-HIV activity whereas the 5’-β-L-d4T triphosphate was shown to be a potent inhibitor of HIV reverse transcriptase Citation11-12. In an attempt to circumvent the first intracellular step of our previously published β-L-d4T analogues bearing a linker at C-5 position and on the basis of enantiodifferentiation of cellular kinases, it was also of particular interest to convert these β-L-modified nucleosides into their corresponding phosphoramidate prodrugs.
As a continuation of our efforts to explore the requirements for optimal anti-HIV activity, we have consequently studied chemical modifications in the side-chain of the β-D-d4T analogues.
The detection of biomolecules at concentrations in the subnanogram range is now routinely achieved with labels based on enzymes and fluorophores. Techniques based on luminescent labels are replacing those with radioisotopes which have contributed greatly to the elucidation of biochemical mechanisms. Detection systems based on the quantitative measure of emitted or absorbed light generally consist of three components: [biomolecule]-[spacer arm]-[signal generator or label]. The spacer arm provides the crucial bridge between the biomolecule which is being detected and the signal molecule whose concentration can be quantified. Therefore, we wanted to prepare chain-terminating fluorescence-tagged d4T analogues. Although, the parent nucleosides were available for attaching a fluorescent dye. Fluorophores are a promising area of non-radioactive labels that can be detected directly. In the literature, dye-labeled terminators have been described [13–14]. Here we are reporting the covalent introduction of a fluorescent group (acridine moiety) to the β-D-d4T analogues bearing a spacer containing a terminal amino group. The spacer arm would provide a crucial bridge between the nucleoside which could be detected and the marker molecule.
Materials and methods
General
Tetrahydrofuran was distilled from sodium/benzophenone immediately prior to use. Anhydrous dichloromethane was prepared by using molecular sieves. Anhydrous N,N-dimethylformamide and pyridine were purchased from Carlo Erba and Aldrich, respectively. Reagent grade acetonitrile was refluxed and distilled from phosphorus pentoxide. Anhydrous ethanol was prepared by using magnesium turnings.
Unless otherwise stated, reactions were run under an atmosphere of argon and monitored by thin-layer chromatography (TLC) using precoated silica gel 60 F254 sheets (0.2 mm layer) purchased from Macherey-Nagel, and compounds were detected by UV absorption at 254 nm. Column chromatography was effected by using Merck silica gel 60 (0.063–0.200 mm) and silica gel Si-60 for flash chromatography (40–63 μm) was supplied by Merck. All samples were kept in a drying oven over P2O5 for at least 24 h prior to analysis.
Melting points were determined on a Kofler apparatus. IR spectra were recorded on a Fourier transform Mattson spectrometer Genesis DTGS using WinFIRST™ Macros and ApPro™: only noteworthy absorptions are listed. 1H and 13C-NMR spectra were obtained on a JEOL Lambda 400 using TMS as an internal standard. NH and OH signals appeared as broad singlets exchangeable with D2O (s = singlet, br = broad, d = doublet, t = triplet, q = quadruplet, m = multiplet) 31P NMR spectra were recorded on a Bruker DPX 400 spectrometer and chemical shifts are quoted in parts per million relative to an external phosphoric acid standard. Mass spectra were obtained from a JEOL JMS-GCmate spectrometer.
Chemistry
5′-O-Acetyl-5-(methoxycarbonylmethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (1) was prepared by the method of Delbederi et al [Citation6]
5-(Methoxycarbonylmethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (2). To a solution of the protected nucleoside 1 (400 mg, 1.23 mmol) in methanol (5 mL) was added crystals of NaCN (60 mg, 1 equiv.). The reaction mixture was stirred at room temperature [monitored by TLC dichloromethane:methanol (90/10)] until no starting material remained. The solvent was evaporated under reduced pressure and the oily residue was purified by silica-gel column chromatography eluted with methanol in chloroform (from 0%–10%) to yield the free nucleoside 2 as white crystals (285 mg); Yield 82%; m.p. 138°C; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.48; IR (KBr), cm− 1: 3527 (OH), 3369 (NH), 3159, 2961, 2833 (CH), 1737 (C = 0), 1687, (C = 0) 1444, 1105, 1030, 846, 801; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.27 (s, 1H, NH), 7.72 (s, 1H, H-6), 6.82 (s, 1H, H-1’), 6.40 (d, 1H, H-3’, 5.8), 5.92 (d, 1H, H-2’, 5.8), 5.00 (br s, 1H, OH), 4.76 (s, 1H, H-4’), 3.57 (s, 5H, OCH3 and H-5’), 3.23 (m, 2H, 5-CH2); 1H-NMR (CDCl3), δ, ppm, J, Hz: 7.77 (s, 1H, H-6), 7.05 (s, 1H, H-1’), 6.37 (d, 1H, H-2’, 5.8), 5.87 (d, 1H, H-3’, 5.8), 4.92 (s, 1H, H-4’), 3.87 (ddd, 2H, H-5’, 12.7, 2.6, 2.4), 3.70 (s, 1H, OCH3), 3.32 (dd, 2H, 5-CH2, 17.2); 13C-NMR (d6-DMSO), δ, ppm: 170.8 (C(O)OCH3), 163.1 (C-4), 150.6 (C-2), 138.9 (C-6), 135.1 (C-2’), 125.7 (C-3’), 107.5 (C-5), 89.2 (C-1’), 87.5 (C-4’), 62.5 (C-5’), 51.7 (OCH3), 31.6 (5-CH2); 13C-NMR (CDCl3), δ, ppm: 171.6 (C(O)OCH3, 162.8 (C-4), 150.4 (C-2), 139.7 (C-6), 135.1 (C-2’), 126.1 (C-3’), 107.7 (C-5), 90.0 (C-1’), 87.5 (C-4’), 63.2 (C-5’), 52.4 (OCH3), 31.0 (5-CH2).
5-{2-[6-(Trifluoroacetylamino)hexylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (3)
To a solution of compound 2 (400 mg, 1.42 mmol) in methanol (5 mL) was added 1,6-diaminohexane (1.65 g, 10 equiv.) and a catalytic amount of 4-dimethylaminopyridine (DMAP) and the reaction mixture was heated to reflux with stirring for 2 days. After cooling down to room temperature, the solvent was evaporated in vacuo. The oily residue was crystallized from hexane and the terminal amino group was protected by trifluoroacetyl group without further purification. Ethyl trifluoroacetate (0.5 mL, 4.26 mmol, 3 equiv.) was added dropwise to a solution of the crude product in methanol (10 mL) containing a catalytic amount of DMAP. The reaction mixture was stirred at 40°C for 2 h and concentrated to dryness in vacuo. The residue was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 3 as white crystals (410 mg); Yield 63%; m.p. 170°C; TLC Rf (CHCL3:CH3OH = 90:10) 0.47; IR (KBr), cm− 1: 3509 (OH), 3300 (NH), 3188 (NH), 3144, 3061, 2935, 2863, 1721 (C = O), 1703 (C = O), 1665 (C = O), 1630 (C = O), 1559, 1478, 1437, 1370, 1082; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.42 (s, 1H, NH), 9.46 (t, 1H, NHCOCF3, 6.4), 7.85 (t, 1H, CONH-linker, 5.5), 7.68 (s, 1H, H-6), 6.90 (d, 1H, H-1’, 1.5), 6.48 (d, 1H, H-3’, 6.0), 5.97 (d, 1H, H-2’, 6.0), 5.03 (t, 1H, OH, 5.4), 4.83 (s, 1H, H-4’), 3.64 (t, 2H, H-5’, 4.6), 3.41 (s, 2H, 5-CH2), 3.22 (q, 2H, CH2NHCOCF3, 13.2, 6.6), 3.05 (m, 2H, CONHCH2), 1.52 (t, 2H, CH2CH2NHCOCF3, 6.4), 1.42 (t, 2H, CONHCH2CH2, 6.4), 1.30 (m, 4H, CH2:linker); 13C-NMR (d6-DMSO), δ, ppm: 168.9 (CO), 163.3 (C-4), 156.1 (q, COCF3, 35.5), 150.7 (C-2), 138.6 (C-6), 135.1 (C-2’), 125.7 (C-3’), 116.0 (q, COCF3, 270.1), 108.6 (C-5), 89.2 (C-1’), 87.4 (C-4’), 62.6 (C-5’), 39.1-38.6-33.1-29.0-28.2-25.9-25.9 (5-CH2 and CH2:linker).
5-{2-[8-(Trifluoroacetylamino)octylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (4)
Compound 2 (300 mg, 0.83 mmol) was converted to 4 using 1,8-diaminoctane (1.20 g, 10 equiv.) by the same procedure as described for 3. The residue was purified by silica-gel chromatography using dichloromethane:methanol (98/2) as eluent to yield 4 as white crystals (470 mg); Yield 68%; m.p. 140°C; TLC Rf (CHCL3:CH3OH = 90:10) 0.50; IR (KBr), cm− 1: 3510 (OH), 3312 (NH), 3191 (NH), 3111, 3062, 2928, 2860, 1720 (C = O), 1704 (C = O), 1663 (C = O), 1633 (C = O), 1562, 1478, 1409, 1373, 1182, 1083; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.26 (s, 1H, NH), 9.34 (s, 1H, NHCOCF3), 7.72 (br s, 1H, CONH-linker), 7.63 (s, 1H, H-6), 6.89 (s, 1H, H-1’), 6.47 (d, 1H, H-3’, 5.7), 5.96 (d, 1H, H-2’, 5.7), 4.92 (br s, 1H, OH), 4.83 (s, 1H, H-4’), 3.66 (m, 2H, H-5’), 3.32 (s, 2H, 5-CH2), 3.20 (m, 2H, CH2NHCOCF3), 3.06 (m, 2H, CONHCH2), 1.52 (m, 2H, CH2CH2NHCOCF3), 1.42 (m, 2H, CONHCH2CH2), 1.30 (m, 8H, CH2:linker).
5-{2-[10-(Trifluoroacetylamino)decylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (5)
Compound 2 (300 mg, 0.83 mmol) was converted to 5 using 1,10-diaminodecane (1.43 g, 10 equiv.) by the same procedure as described for 3. The residue was purified by silica-gel chromatography using dichloromethane:methanol (98/2) as eluent to yield 5 as white crystals (430 mg); Yield 58%; TLC Rf (CHCl3:CH3OH = 90:10) 0.57; IR (KBr), cm− 1: 3514 (OH), 3312 (NH), 3191 (NH), 3111, 3062, 2928, 2860, 1720 (C = O), 1704 (C = O), 1663 (C = O), 1633 (C = O), 1562, 1478, 1409, 1373, 1182, 1083; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.26 (s, 1H, NH), 9.34 (s, 1H, NHCOCF3), 7.72 (br s, 1H, CONH-linker), 7.63 (s, 1H, H-6), 6.89 (s, 1H, H-1’), 6.47 (d, 1H, H-3’, 5.7), 5.96 (d, 1H, H-2’, 5.7), 4.92 (br s, 1H, OH), 4.83 (s, 1H, H-4’), 3.66 (m, 2H, H-5’), 3.32 (s, 2H, 5-CH2), 3.20 (m, 2H, CH2NHCOCF3), 3.06 (m, 2H, CONHCH2), 1.52 (m, 2H, CH2CH2NHCOCF3), 1.42 (m, 2H, CONHCH2CH2), 1.30 (m, 12H, CH2:linker).
5-{2-[12-(Trifluoroacetylamino)dodecylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (6)
Compound 2 (300 mg, 0.83 mmol) was converted to 6 using 1,12-diaminodecane (1.66 g, 10 equiv.) by the same procedure as described for 3. The residue was purified by silica-gel chromatography using dichloromethane:methanol (98/2) as eluent to yield 6 as white crystals (520 mg); Yield 67%; m.p. 136–138°C; TLC Rf (CHCl3:CH3OH = 90:10) 0.60; IR (KBr), cm− 1: 3510 (OH), 3310 (NH), 3189 (NH), 3111, 2925, 2853, 1719 (C = O), 1704 (C = O), 1662 (C = O), 1632 (C = O), 1557, 1472, 1408, 1374, 1180, 1084; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.34 (s, 1H, NH), 9.38 (s, 1H, NHCOCF3), 7.77 (t, 1H, CONH-linker, 5.3), 7.60 (s, 1H, H-6), 6.82 (s, 1H, H-1’), 6.40 (d, 1H, H-3’, 5.8), 5.96 (d, 1H, H-2’, 5.8), 4.96 (br s, 1H, OH), 4.75 (s, 1H, H-4’), 3.56 (m, 2H, H-5’), 3.40 (s, 2H, 5-CH2), 3.15 (m, 2H, CH2NHCOCF3), 2.96 (m, 2H, CONHCH2), 1.44 (m, 2H, CH2CH2NHCOCF3), 1.34 (m, 2H, CONHCH2CH2), 1.22 (m, 16H, CH2:linker).
Phenyl (methoxyalaninyl)phosphorochloridate (7) [Citation22]
A solution of triethylamine (4 mL, 28.7 mmol) in dichloromethane (40 mL) was added dropwise with vigorous stirring to a solution of L-alanine methyl ester hydrochloride (2 g, 14.32 mmol) and phenyl phosphorodichloridate (2.16 mL, 14.6 mmol) in dichloromethane (40 mL) at − 78°C. The reaction mixture was then allowed to come to room temperature with stirring over 6 h and concentrated to dryness in vacuo. The residue was treated with diethyl ether (2 × 50 mL), the mixture filtered and the filtrate evaporated to yield 7 as a colourless oil (3.76 g); Yield 94%; 1H-NMR (CDCl3), δ, ppm, J, Hz: 7.21–7.37 (m, 5H, H-arom), 4.90 (m, 1H, NH), 4.18 (m, 1H, CH), 3.78* (s, 3H, OCH3), 1.51* (d, 3H, CH3, 6.9); 13C-NMR (CDCl3), δ, ppm: 173.0 (CO-Ala), 149.5 (ipso-Ph), 130.6* (meta-Ph), 121.3* (para-Ph), 121.1* (ortho-Ph), 53.3* (CH), 51.0 (OCH3), 20.8 (CH3); 31P-NMR (CDCl3), δ, ppm: 8.67, 8.50.
5-{2-[6-(Trifluoroacetylamino)hexylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine 5′-(phenyl methoxyalaninyl phosphate) (8)
Phenyl(methoxyalaninyl)phosphorochloridate 7 (450 mg, 1.62mmol, 3 equiv.) was added to a stirred solution of 3 (250 mg, 0.54 mmol) and N-methylimidazole (266 mg, 3.25 mmol, 6 equiv.) in anhydrous tetrahydrofuran (7 mL) at room temperature under an argon atmosphere. After 4 h, the solvent was removed under reduced pressure. The gum was dissolved in chloroform (20 mL) and washed with 1M HCl, saturated sodium bicarbonate solution (20 mL), and water (20 mL). The organic layer was dried (MgSO4), filtered and the solvent was removed in vacuo. The residue was purified by silica-gel column chromatography (silica pre-equilibrated with 1% Et3N) using dichloromethane:methanol (97/3) as eluent. Pooling and evaporation of appropriate fractions gave the product 8 (50 mg) as white crystals; Yield 13%; TLC Rf (CHCl3:CH3OH = 95:5) 0.46; IR (KBr), cm− 1: 3306 (NH), 3079, 2939, 1712 (C = O), 1686 (C = O), 1592, 1553, 1491, 1465, 1376, 1334, 1259, 1210, 1153, 1109, 1089, 1036, 936; 1H-NMR (CDCl3), δ, ppm, J, Hz: 7.77* (s, 1H, H-6), 7.27-7.07 (m, 6H, Ph and NHCOCF3), 6.95* (s, 1H, H-1’), 6.69 (br s, 1H, CONH-linker), 6.28* (d, 1H, H-3’, 5.8), 5.86* (d, 1H, H-2’, 5.8), 5.21 (m, 1H, NH-Ala), 4.98 (m, 1H, H-4’), 4.41-4.20 (m, 2H, H-5’), 3.98 (m, 1H, CH-Ala), 3.62* (s, 3H, OCH3-Ala), 3.22-3.07 (m, 6H, 5-CH2 and CH2NHCOCF3 and CONHCH2), 1.46-1.12 (m, 11H, CH2:linker and CH3-Ala); 13C-NMR (CDCl3), δ, ppm: 174.4* (CO-Ala), 170.2 (5-CH2CONH), 164.2* (C-4), 157.3 (q, COCF3, 36.2), 150.4 (C-2), 150.3* (Ph-ipso), 139.1* (C-6), 133.5* (C-2’), 129.6 (Ph-meta), 127.0* (C-3’), 125.0* (Ph-para), 125.1* (Ph-ortho), 115.9 (q, COCF3, 286.2), 109.2* (C-5), 89.9*(C-1’), 85.4* (C-4’), 66.7* (C-5’), 52.3 (O CH3-Ala), 50.5* (CH-Ala), 39.6*-39.2*-35.0*-28.9*-28.5-25.9*-25.8* (5-CH2 and CH2:linker), 20.6* (CH3-Ala); 31P-NMR, (CDCl3) δ, ppm: 5.84, 5.27.
5-{2-[8-(Trifluoroacetylamino)octylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine 5′-(phenyl methoxyalaninyl phosphate) (9)
Compound 4 (400 mg, 0.81 mmol) was converted to 9 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (97/3) as eluent to yield 9 as white crystals (60 mg); Yield 10%; m.p. decomposed at 130°C; TLC Rf (CHCl3:CH3OH = 95:5) 0.50; IR (KBr), cm-1: 3306 (NH), 3079, 2939, 1712 (C = O), 1686 (C = O), 1592, 1553, 1491, 1465, 1376, 1334, 1259, 1210, 1153, 1109, 1089, 1036, 936; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.86 (br s, 1H, NH), 7.77* (s, 1H, H-6), 7.26-7.08 (m, 6H, Ph and NHCOCF3), 6.95* (s, 1H, H-1’), 6.75 (br s, 1H, CONH-linker), 6.29* (d, 1H, H-3’, 5.6), 5.85* (d, 1H, H-2’, 5.6), 5.27 (m, 1H, NH-Ala), 4.97 (m, 1H, H-4’), 4.41-4.22 (m, 2H, H-5’), 3.97 (m, 1H, CH-Ala), 3.62* (s, 3H, OCH2-Ala), 3.22-3.08 (m, 6H, 5-CH2 and CH2NHCOCF3 and CONHCH2), 1.46-1.12 (m, 15H, CH2:linker and CH3-Ala); 13C-NMR (CDCl3), δ, ppm: 174.4* (CO-Ala), 170.1 (5-CH2CONH), 164.3* (C-4), 157.3 (q, COCF3, 36.2), 150.6* (C-2), 1505* (Ph-ipso), 139.0* (C-6), 133.5* (C-2’), 129.6 (Ph-meta), 126.9* (C-3’), 125.0* (Ph-para), 120.5* (Ph-ortho), 115.9 (q, COCF3, 286.2), 109.2* (C-5), 89.9*(C-1’), 85.4* (C-4’), 66.7* (C-5’), 52.2 (OCH3-Ala), 50.5* (CH-Ala), 39.8*-34.8*-29.0*-28.8-28.7*-26.5* (5-CH2 and CH2:linker), 20.5* (CH3-Ala); 31P-NMR (CDCl3), δ, ppm: 6.40, 5.89.
5-{2-[10-(Trifluoroacetylamino)decylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine 5′-(phenyl methoxyalaninyl phosphate) (10)
Compound 5 (400 mg, 0.77 mmol) was converted to 10 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (98/2) as eluent to yield 10 as white crystals (150 mg); Yield 25%; m.p. 135°C; TLC Rf (CHCl3:CH3OH = 95:5) 0.46; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.80 (br s, 1H, NH), 7.78* (s, 1H, H-6), 7.25-7.08 (m, 5H, Ph), 7.01 (br s, 1H, NHCOCF3), 6.95* (s, 1H, H-1’), 6.74 (br s, 1H, CONH-linker), 6.28* (d, 1H, H-3’, 5.8), 5.85* (d, 1H, H-2’, 5.8), 5.34 (m, 1H, NH-Ala), 4.97 (m, 1H, H-4’), 4.38-4.24 (m, 2H, H-5’), 3.98 (m, 1H, CH-Ala), 3.23* (s, 3H, OCH3-Ala), 3.23-3.07 (m, 6H, 5-CH2 and CH2NHCOCF3 and CONHCH2), 1.47-1.16 (m, 19H, CH2:linker and CH3-Ala); 31P-NMR (CDCl3), δ, ppm: 7.00, 6.47.
5-{2-[12-(Trifluoroacetylamino)decylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine 5′-(phenyl methoxyalaninyl phosphate) (11)
Compound 6 (400 mg, 0.73 mmol) was converted to 11 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (98/2) as eluent to yield 11 as white crystals (100 mg); Yield 17%; TLC Rf (CHCl3:CH3OH = 95:5) 0.45;17%; IR (KBr), cm− 1: 3383 (NH), 3082, 2929, 1715 (C = O), 1685 (C = O), 1593, 1558, 1491, 1466, 1377, 1261, 1210, 1153, 1109, 1089, 1034, 936; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.64 (br s, 1H, NH), 777* (s, 1H, H-6), 7.25-7.07 (m, 5H, Ph), 6.94* (s, 1H, H-1’), 6.82 (br s, 1H, NHCOCF3), 6.65 (br s, 1H, CONH-linker), 6.27* (d, 1H, H-3’, 5.8), 5.85* (d, 1H, H-2’, 5.8), 5.27 (m, 1H, NH-Ala), 4.96 (m, 1H, H-4’), 4.41-4.22 (m, 2H, H-5’), 3.98 (m, 1H, CH-Ala), 3.61* (s, 3H, OCH3-Ala), 3.24-3.06 (m, 6H, 5-CH2 and CH2NHCOCF3 and CONHCH2), 1.50-1.17 (m, 23H, CH2:linker and CH3-Ala); 13C-NMR (CDCl3), δ, ppm: 174.4* (CO-Ala), 170.1 (5-CH2CONH), 164.3* (C-4), 157.3 (q, COCF, 37.0), 150.7* (C-2), 150.6* (Ph-ipso), 139.0* (C-6), 133.6* (C-2’), 129.6 (Ph-meta), 127.09 (C-3’), 125.1* (Ph-para), 120.6* (Ph-ortho), 115.0 (q, COCF3, 286.3), 109.3* (C-5), 90.0* (C-1’), 85.5* (C-4), 66.8* (C-5’), 52.3 (OCH3-Ala), 50.6* (CH-Ala), 40.0* (5-CH2), 34.9-34.6-29.4-29.4-29.3-29.2-29.1-28.9-26.9-26.7 (CH2:linker), 20.6* (CH3-Ala); 31P-NMR (CDCl3), δ, ppm: 4.09, 3.57.
5-(Methoxycarbonylmethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine 5′-(phenyl methoxyalaninyl phosphate) (12)
Compound 2 (400 mg, 1.42 mmol) was converted to 12 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (97/3) as eluent to yield 12 as white crystals (200 mg); Yield 27%; TLC Rf (CHCl3:CH3OH = 95.5) 0.68; IR (KBr), cm− 1: 3242 (NH), 3072, 2955, 1742 (C = O), 1686 (C = O), 1591, 1565, 1491, 1466, 1377, 1345, 1251, 1211, 1154, 1109, 1088, 1035, 1005, 934; 1H-NMR (CDCl3), δ, ppm, J, Hz: 8.91* (s. 1H, NH), 7.57* (s, 1H, H-6), 7.33*-7.20* (m, 5H, Ph), 7.10 (s, 1H, H-1’), 6.34* (dt, 1H, H-3’, 5.9 and 1.5). 5.96 (dt, 1H, H-2’, 5.9), 5.06* (m, 1H, H-4’), 4.38* (m, 2H, H-5’), 4.02-3.86 (m, 2H, CH-Ala and NH-Ala), 3.69 (s, 3H, OCH3-Ala), 3.65 (s, 3H, OCH3), 3.41-3.23 (m, 2H, 5-CH2), 1.34 (d, 3H, CH3-Ala, 6.8); 13C-NMR (CDCl3), δ, ppm: 173.9* (CO-Ala), 171.4 (COOCH3), 162.8 (C-4), 150.5 (C-2), 150.2 (Ph-ipso), 138.7* (C-6), 133.4 (C-2’), 129.8* (Ph-meta), 127.4* (C-3’), 125.3* (Ph-para), 120.2* (Ph-ortho), 108.6* (C-5), 90.1* (C-1’), 84.9* (C-4’), 67.1* (C-5’), 52.6 (OCH3-Ala), 52.2* (OCH3), 50.3* (CH-Ala), 31.5* (5-CH2), 21.0* (CH3-Ala), 31P-NMR (CDCl3), δ, ppm: 4.63, 3.92.
5-(2-Hydroxyethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (13)
Lithium borohydride (169 mg, 7.7 mmol, 10 equiv.) was added to a solution of the protected nucleoside 1 (250 mg, 0.77 mmol) in anhydrous THF (10 mL) under an argon atmosphere. The reaction mixture was heated under reflux for 4 h and cooled to room temperature. Methanol was then added dropwise while the temperature was maintained at 18°C. The salts were removed by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica-gel column chromatography using dichloromethane:methanol (90/10) as eluent to yield 13 as a while solid (150 mg); Yield 76%; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.32; IR (KBr), cm− 1: 3420 (OH), 1698 (C = O), 1684 (C = O), 1470, 1260, 1105, 1075, 1039, 801; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.30 (br s, 1H, NH lactam), 7.53 (s, 1H, H-6), 6.81 (s, 1H, H-1’), 6.40 (d, 1H, H-3’, 5.9), 5.89 (d, 1H, H-2’, 5.9), 4.98 (t, 1H, OH-5’, 5.0), 4.75 (m, 1H, H-4’), 4.55 (t, 1H, 5-CH2CH2OH, 5.2), 3.57 (m, 2H, H-5’), 3.38 (m, 2H, 5-CH2CH2OH), 2.28 (t, 2H, 5-CH2, 5.5); 13C-NMR (d6-DMSO), δ, ppm: 163.6 (C-4), 150.7 (C-2), 137.7 (C-6), 135.0 (C-2’), 125.8 (C-3’), 110.4 (C-5), 88.9 (C-1’), 87.3 (C-4’), 64.9 (C-5’), 59.4 (5-CH2CH2OH), 30.0 (5-CH2).
5′-O-Acetyl-5-(methyoxycarbonylmethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine (14) was prepared by the method of Delbederi et al [Citation6]
5-{2-[6-(Trifluoroacetylamino)hexylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine (15). Compound 14 (300 mg, 0.93 mmol) was converted to 15 by the procedure described for 3. The residue was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 15 as white crystals (258 mg); Yield 60%; m.p. 132°C; TLC Rf (CHCl3:CH3OH = 90:10) 0.41; IR (KBr), cm− 1: 3514 (OH), 3310 (NH), 3186 (NH), 3111, 2935, 2853, 1720 (C = O), 1704 (C = O), 1662 (C = O), 1631 (C = O), 1553, 1472, 1437, 1374, 1084; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.36 (s, 1H, NH), 9.40 (br s, 1H, NHCOCF3), 7.77 (t, 1H, CONH-linker, 5.5), 7.60 (s, 1H, H-6), 6.82 (m, 1H, H-1’), 6.40 (dd, 1H, H-3’, 6.0, 1.5), 5.88 (dd, 1H, H-2’, 6.0, 1.5), 4.97 (t, 1H, OH, 5.5), 4.75 (m, 1H, H-4’), 3.55 (t, 2H, H-5’, 4.5), 3.41 (s, 2H, 5-CH2), 3.14 (m, 2H, CH2NHCOCF3), 2.95 (m, 2H, CONHCH2), 1.44 (t, 2H, CH2CH2NHCOCF3, 6.5), 1.33 (t, 2H, CONHCH2CH2, 6.5), 1.21 (m, 4H, CH2:linker); 13C-NMR (d6-DMSO), δ, ppm: 168.9 (CO), 163.3 (C-4), 156.1 (q, COCF3, 36.0), 150.8 (C-2), 138.6 (C-6), 135.1 (C-2’), 125.7 (C-3’), 116.0 (q, COCF3, 278.1), 108.6 (C-3), 89.2 (C-1’), 87.4 (C-4’), 62.6 (C-5’), 39.1-38.6-33.1-29.0-28.2-25.9-25.9 (5-CH2 and CH2-linker).
5-{2-[12-(Trifluoroacetylamino)dodecylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine (16)
Compound 14 (300 mg, 0.93 mmol) was converted to 16 by the procedure described for 3. The residue was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 16 as white crystals (325 mg); Yield 64%; m.p. 132°C; TLC Rf (CHCl3:CH3OH = 90:10) 0.60; IR (KBr), cm− 1: 3514 (OH), 3310 (NH), 3186 (NH), 3111, 2925, 2853, 1720 (C = O), 1704 (C = O), 1662 (C = O), 1631 (C = O), 1553, 1472, 1408, 1374, 1180, 1084; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.34 (s, 1H, NH), 9.40 (s, 1H, NHCOCF3), 7.84 (t, 1H, CONH-linker, 5.4), 7.60 (s, 1H, H-6), 6.82 (m, 1H, H-1’), 6.40 (d, 1H, H-3’, 6.0), 5.90 (d, 1H, H-2’, 6.0), 4.95 (t, 1H, OH, 5.4), 4.75 (m, 1H, H-4’), 3.56 (s, 2H, H-5’), 3.34 (s, 2H, 5-CH2), 3.16 (m, 2H, CH2NHCOCF3), 2.97 (m, 2H, CONHCH2), 1.43 (t, 2H, CH2CH2NHCOCF3, 6.2), 1.33 (t, 2H, CONHCH2CH2, 6.2), 1.22 (m, 16H, CH2:linker); 13C-NMR (d6-DMSO), δ, ppm: 168.9 (CO), 163.2 (C-4), 156.1 (q, COCF3, 35.3), 150.7 (C-2), 138.4 (C-6), 135.0 (C-2’), 125.7 (C-3’), 116.0 (q, COCF3, 284.6), 108.6 (C-5), 89.3 (C-1’), 87.4 (C-4’), 62.7 (C-5’), 39.0* (5-CH2), 29.0*-28.8*-28.1-26.2* (CH2-linker).
5-{2-[6-(Trifluoroacetylamino)hexylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine 5′-(phenyl methoxyalaninyl phosphate) (17)
Compound 15 (250 mg, 0.54 mmol) was converted to 17 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (97/3) as eluent to yield 17 as white crystals (60 mg); Yield 16%; TLC Rf (CHCl3:CH3OH = 95:5) 0.54; 1H-NMR (CDCl3), δ, ppm, J, Hz: 7.91* (s, 1H, H-6), 7.26-7.07 (m, 6H, Ph and NHCOCF3), 7.01 (s, 1H, H-1’), 6.92 (br s, 1H, CONH-linker), 6.29 (m, 1H, H-3’), 6.14 (m, 1H, H-2’), 5.82 (m, 1H, NH-Ala), 4.95 (br s, 1H, H-4’), 4.41-3.89 (m, 3H, CH-Ala and H-5’), 3.62 (s, 3H, OCH3-Ala), 3.28-2.88 (m, 6H, 5-CH2 and CH2NHCOCF3 and CONHCH2), 1.43-1.12 (m, 11H, CH2:linker and CH3-Ala); 13C-NMR (CDCl3), δ, ppm: 175.2* (CO-Ala), 171.0 (5-CH2CONH), 164.8* (C-4), 157.3 (q, COCF3, 39.5), 150.7* (C-2), 150.6* (Ph-ipso), 140.1* (C-6), 134.2* (C-2’), 129.7* (Ph-meta), 126.3 (C-3’), 125.0 (Ph-para), 120.9* (Ph-ortho), 116.0 (q, COCF3, 286.3), 109.3* (C-5), 89.8* (C-1’), 85.3 (C-4’), 66.3* (C-5’), 52.2* (OCH3-Ala), 50.3* (CH-Ala), 39.7* (5-CH2), 39.3*-34.8*-28.9*-26.6*-26.1*-25.7* (CH2:linker), 20.4* (CH3-Ala); 31P-NMR (CDCl3), δ, ppm: 6.70, 5.51.
5-{2-[12-(Trifluoroacetylamino)dodecylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine 5′-(phenyl methoxyalaninyl phosphate) (18)
Compound 16 (280 mg, 0.51 mmol) was converted to 18 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (97/3) as eluent to yield 18 as white crystals (50 mg); Yield 12%; TLC Rf (CHCl3:CH3OH) 0.76; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.27 (br s, 1H, NH), 7.76 (s, 1H, H-6), 7.26-7.07 (m, 6H, Ph and NHCOCF3), 6.93 (s, 1H, H-1’), 6.65 (m, 1H, CONH-linker), 6.15 (d, 1H, H-3’, 5.8), 5.80 (d, 1H, H-2’, 5.8), 5.40 (t, 1H, NH-Ala, 10.8), 4.96 (s, 1H, H-4’), 4.29 (s, 2H, H-5’), 3.92 (m, 1H, CH-Ala), 3.58 (s, 3H, OCH3-Ala), 3.29-3.05 (m, 6H, 5-CH2 and CH2NHCOCF3 and CONHCH2), 1.49-1.12 (m, 23H, CH2:linker and CH3-Ala); 13C-NMR (CDCl3), δ, ppm: 174.1* (CO-Ala), 170.1 (5-CH2CONH), 164.1 (C-4), 157.8 (q, COCF3, 38.5), 150.7 (C-2), 150.6* (Ph-ipso), 139.2 (C-6), 133.2 (C-2’), 129.6 (Ph-meta), 127.0 (C-3’), 125.0 (Ph-para), 120.3* (Ph-ortho), 116.0 (q, COCF3, 286.3), 109.2 (C-5), 89.9 (C-1’), 85.2* (C-4’), 66.3* (C-5’), 52.2 (OCH3-Ala), 50.3 (CH-Ala), 40.0* (5-CH2), 34.2*-29.3*-29.1*-28.9-26.7* (CH2:linker), 20.5* (CH3-Ala); 31P-NMR (CDCl3), δ, ppm: 4.09, 3.57.
5-(Methoxycarbonylmethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine (19)
The protected nucleoside 14 (400 mg, 1.23 mmol) was treated with a solution of sodium methoxide (66 mg, 1 equiv.) in anhydrous methanol (10 mL) at room temperature for 45 min. The reaction mixture was carefully neutralized by addition a solution of 1M HCl and concentrated to dryness in vacuo. The crude product was purified by silica-gel column chromatography eluted with methanol in dichloromethane (from 0–10%) to yield 19 (60 mg) as a white solid; Yield 17%; m.p. 150°C; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.43; IR (KBr), cm-1: 3550 (OH), 2961, 1730 (C = O), 1706 (C = O), 1681 (C = O), 1260, 1086, 1037; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.46 (s, 1H, NH), 7.73 (s, 1H, H-6), 6.82 (s, 1H, H-1’), 6.41 (d, 1H, H-3’, 6.1), 5.92 (d, 1H, H-2’, 6.1), 4.99 (m, 1H, H-4’), 4.76 (s, 2H, H-5’), 3.57 (m, 3H, OCH3), 3.21 (s, 2H, 5-CH2); 13C-NMR (d6-DMSO), δ, ppm: 167.8 (C(O)OCH3), 160.0 (C-4), 147.6 (C-2), 135.8 (C-6), 132.1 (C-2’), 122.7 (C-3’), 104.5 (C-5), 86.1 (C-1’), 84.4 (C-4’), 59.4 (C-5’), 48.6 (OCH3), 28.6 (5-CH2).
5-(Methoxycarbonylmethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine 5′-(phenyl methoxyalaninyl phosphate) (20)
Compound 19 (200 mg, 0.71 mmol) was converted to 20 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (97/3) as eluent to yield 20 as white crystals (50 mg); Yield 14%; m.p. 92-94°C; TLC Rf (CHCl3:CH3OH = 95:5) 0.45; IR (KBr), cm− 1: 3245 (NH), 3070, 2960, 1741 (C = O), 1685 (C = O), 1591, 1565, 1490, 1466, 1376, 1344, 1261, 1210, 1155, 1089, 1035, 1005, 934; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.59* (s, 1H, NH), 7.54* (s, 1H, H-6), 7.26*-7.16* (m, 5H, Ph), 6.95 (s, 1H, H-1’), 6.25* (m, 1H, H-3’), 5.85 (m, 1H, H-2’), 4.94* (s, 1H, H-4’), 4.23* (m, 2H, H-5’), 4.18-3.83 (m, 2H, CH-Ala and NH-Ala), 3.61 (s, 3H, OCH3-Ala), 3.65 (s, 3H, OCH3), 3.41-3.23 (2H, 5-CH2), 1.34 (d, 3H, CH3-Ala, 6.9); 13C-NMR (CDCl3), δ, ppm: 174.3* (CO-Ala), 171.6* (CO ester), 163.0* (C-4), 150.7* (C-2), 150.3 (Ph-ipso), 138.7* (C-6), 133.3* (C-2’), 129.6* (Ph-meta), 127.1 (C-3’), 125.1* (Ph-para), 120.2* (Ph-ortho), 108.5* (C-5), 89.9*(C-1’), 84.8* (C-4’), 66.8* (C-5’), 52.4* (OCH3-Ala), 52.0* (OCH3), 50.0* (CH-Ala), 31.5* (5-CH2), 20.8* (CH3-Ala)); 31P-NMR (CDCl3), δ, ppm: 4.70, 4.53.
5-(2-Hydroxyethyl)-2′,3′-didehydro-2′,3′-dideoxy-β-L-uridine (21)
Compound 14 (250 mg, 0.77 mmol) was converted to 21 by the procedure described for 13. The residue was purified by silica-gel chromatography using dichloromethane:methanol (90/10) as eluent to yield 21 as a white solid (120 mg); Yield 61%; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.32; IR (KBr), cm− 1: 3405 (OH), 2957, 2928, 2874, 1700 (C = O), 1679 (C = O), 1470, 1401, 1260, 1104, 1087, 1074, 1043, 850, 802, 580; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.30 (br s, 1H, NH), 7.56 (s, 1H, H-6), 6.81 (m, 1H, H-1’), 6.38 (dd, 1H, H-3’, 5.9, 1.6), 5.88 (dd, 1H, H-2’, 5.9, 1.6), 4.98 (br s, 1H, 5’-OH), 4.75 (m, 1H, H-4’), 4.53 (br s, 1H, 5-CH2CH2OH), 3.56 (m, 2H, H-5’), 3.38 (t, 2H, 5-CH2CH2OH, 6.7), 2.30 (t, 2H, 5-CH2, 6.4); 13C-NMR (d6-DMSO), δ, ppm: 163.7 (C-4), 150.8 (C-2), 137.7 (C-6), 135.1 (C-2’), 125.8 (C-3’), 110.4 (C-5), 89.0 (C-1’), 87.4 (C-4’), 62.4 (C-5’), 59.4 (5-CH2CH2OH), 30.1 (5-CH2).
2′,3′,5′-Tri-O-acetyl-β-D-uridine (22)
Acetic anhydride (12.82 mL, 3.33 equiv.) was added slowly to a solution of uridine (10 g, 41 mmol) in anhydrous pyridine (100 mL) at room temperature under an argon atmosphere. The reaction mixture was heated at 60°C for 23 h and concentrated to dryness in vacuo. The residue was co-evaporated several times with toluene, dissolved in chloroform (200 mL), washed with water (200 mL), dried (MgSO4) and evaporated to dryness in vacuo to afford 22 as white crystals (14.52 g); Yield 96%; m.p. 127°C; TLC Rf (CH2Cl2:CH3OH = 85:15) 0.75; IR (KBr), cm− 1: 1723 (C = O), 1693 (C = O), 1469, 1441, 1422, 1383, 1251, 1105, 862; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.47 (s, 1H, NH), 7.70 (d, 1H, H-6, 8.0), 5.87 (d, 1H, H-1’, 5.1), 5.70 (d, 1H, H-5, 8.0), 5.43 (t, 1H, H-2’, 5.7), 5.31 (t, 1H, H-3’, 5.7), 4.25 (m, 3H, H-4’ and H-5’), 2.06 (s, 3H, CH3), 2.04 (s, 3H, CH3), 2.03 (s, 3H, CH3).
5-Iodo-2′,3′,5′-tri-O-acetyl-β-D-uridine (23)
A solution of compound 22 (10 g, 27 mmol) and iodine chloride (6.67 g, 3 equiv.) in anhydrous dichloromethane (250 mL) was heated at reflux for 7 h, allowed to cool to room temperature and the stirring continued for 12 h. The stirred mixture was treated with 5% NaHSO3/H2O necessary to reduce the excess of ICl. The organic phase was separated and washed with H2O (2 × 100 mL), dried (MgSO4) and evaporated to dryness in vacuo to yield the title compound 23 as white crystals (13 g); Yield 97%; m.p. 168°C; TLC Rf (CH2Cl2:AcOEt = 20:80) 0.73; IR (KBr), cm− 1: 1751 (C = O), 1691 (C = O), 1614, 1548, 1442, 1376, 1232, 1095, 1047; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.84 (s, 1H, NH), 8.17 (s, 1H, H-6), 5.85 (d, 1H, H-1’, 4.8), 5.44 (t, 1H, H-2’, 5.5), 5.32 (t, 1H, H-3’, 5.5), 4.28 (m, 3H, H-4’ and H-5’), 2.06 (s, 3H, CH3), 2.05 (s, 3H, CH3), 2.04 (s, 3H, CH3); 13C-NMR (d6-DMSO), δ, ppm: 169.9 (C = O), 162.0 (C-4), 150.0 (C-2), 145.4 (C-6), 106.0 (C-5), 88.0 (C-1’), 79.0 (C-4’), 72.0 (C-2’), 69.4 (C-3’), 62.9 (C-5’), 20.6 (CH3), 20.2 (CH3).
5-[3-N-(Trifluoroacetyl)amino-prop-1-ynyl]-2′,3′,5′-tri-O-acetyl-β-D-uridine (24)
To a deoxygenated solution of compound 23 (3.5 g, 7.06 mmol) in anhydrous DMF (40 mL) was added successively the 3-N-(trifluoroacetyl)amino-prop-1-yne (2.1 g, 2 equiv.), tetrakis(triphenylphosphine)palladium(0) (814 mg, 0.1 equiv.), copper(I)iodide (268 mg, 0.2 equiv.) and anhydrous deoxygenated N,N-diisopropylethylamine (2.45 mL, 2 equiv.). The resulting suspension was stirred at room temperature for 24 h under an argon atmosphere [monitored by TLC CH2Cl2:AcOEt (20/80)] until no starting material remained, mixed with diethyl ether (20 mL), filtered and thoroughly evaporated to dryness in vacuo. The residue was purified by silica-gel chromatography using dichloromethane:ethyl acetate (20/80) as eluent to yield 24 as a yellow crystals (3 g); Yield 82%; m.p. 100°C; TLC Rf (CH2Cl2:AcOEt = 20:80) 0.66: IR (KBr), cm-1: 3300 (NH), 1750 (C = O), 1723 (C = O), 1459, 1375, 1227, 1180, 1159, 1096, 1048; 1H-NMR (CDCl3), δ, ppm, J, Hz: 10.01 (br s, 1H, NH), 8.02 (br s, 1H, NH), 7.83 (s, 1H, H-6), 6.00 (d, 1H, H-1’, 4.4), 5.44 (t, 1H, H-2’, 5.2), 5.33 (t, 1H, H-3’, 5.2), 4.38 (m, 3H, H-4’ and H-5’), 4.33 (d, 2H, CH2COCF3, 5.9), 2.18 (s, 3H, CH3), 2.11 (s, 6H, 2 CH3); 13C-NMR (CDCl3), δ, ppm: 170.6 (CH3CO), 170.0 (CH3CO), 169.9 (CH3CO), 162.1 (C-4), 157.8 (q, COCF3, 36.6), 149.3 (C-2), 143.3 (C-6), 116.2 (q, COCF3, 287.4), 99.8 (C-5), 88.3 (C-1’), 88.2 (C ≡ CCH2), 80.1 (C4’), 74.9 (C ≡ CCH2), 73.4 (C-2’), 69.8 (C-3’), 62.8 (C-5’), 30.4 (CH2COCF3), 21.1 (CH3), 20.8 (CH3), 20.7 (CH3); MS m/z 520 (MH+).
5-[3-N-(Trifluoroacetyl)aminopropyl]-2′,3′,5′-tri-O-acetyl-β-D-uridine (25)
A solution of compound 24 (3 g, 5.78 mmol) in anhydrous methanol (100 mL) was hydrogenated in the presence of catalyst (3 g of wet 10% palladium on carbon). The mixture was stirred at room temperature for approximately 6 h. The catalyst was removed by filtration, and the filtrate concentrated to give compound 25 as white crystals (2.86 g); Yield 95%; m.p. 74°C; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.73; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.42 (br s, 1H, NH), 7.49 (m, 1H, NH), 7.33 (s, 1H, H-6), 6.03 (d, 1H, H-1’, 5.4), 5.33-5.39 (m, 2H, H-2’ and H-3’), 4.43-4.47 (m, 2H, H-4’ and H-5’a), 4.33-4.36 (m, 1H, H-5’b), 3.30-3.46 (m, 2H, CH2COCF3), 2.40 (t, 2H, 5-CH2, 6.6), 2.14 (s, 6H, 2 CH3), 2.10 (s, 3H, CH3), 1.81-1.87 (m, 2H, CH2COCF3); 13C-NMR (CDCl3), δ, ppm: 170.7 (CH3CO), 170.2 (CH3CO), 170.0 (CH3CO), 164.1 (C-4), 157.8 (q, COCF3, 36.6), 150.4 (C-2), 137.2 (C-6), 116.2 (q, COCF3, 287.4), 114.3 (C-5), 87.9 (C-1’), 80.3 (C-4’), 72.9 (C-2’), 70.5 (C-3’), 63.5 (C-5’), 38.5 (CH2COCF3), 28.2 (5-CH2), 24.0 (CH2CH2COCF3), 21.1 (CH3), 20.8 (CH3), 20.7 (CH3).
5-[3-N-(Trifluoroacetyl)aminopropyl]-β-D-uridine (26)
To a solution of the protected nucleoside 25 (3 g, 5.74 mmol) in methanol (100 mL) was added sodium cyanide (140 mg, 0.5 equiv.). The reaction mixture was stirred at room temperature [monitored by TLC dichloromethane:methanol (90/10)] until no starting material remained and filtered on silica-gel. The filtrate was concentrated to dryness in vacuo to afford the free nucleoside 26 as white crystals (1.96 g); Yield 86%; m.p. 176°C; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.36; IR (KBr), cm− 1: 3412 (OH), 1693 (C = O), 1476, 1436, 1207, 1140; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.30 (br s, 1H, NH), 9.43 (m, 1H, NH), 8.30 (s, 1H, H-6), 5.76 (d, 1H, H-1’, 5.1), 5.37 (br s, 1H, OH-5’), 5.17 (br s, 2H, OH-2’ and OH-3’), 4.02 (t, 1H, H-2’, 4.9), 3.96 (t, 1H, H-3’, 4.9), 3.82 (d, 1H, H-4’, 2.8), 3.63 (d, 1H, H-5’a, 11.9), 3.54 (d, 1H, H-5’b, 117), 3.14 (m, 2H, CH2COCF3), 2.20 (m, 2H, 5-CH2), 1.66 (m, 2H, CH2CH2COCF3); 13C-NMR (d6-DMSO), δ, ppm: 163.5 (C-4), 156.2 (q, COCF3, 35.5), 150.7 (C-2), 136.7 (C-6), 116.0 (q, COCF3, 288.2), 112.6 (C-5), 87.7 (C-1’), 84.7 (C-4’), 73.4 (C-2’), 69.8 (C-3’), 60.8 (C-5’), 38.7 (CH2COCF3), 27.2 (5-CH2), 23.8 (CH2CH2COCF3).
5-[3-(N-(Trifluoroacetyl)aminopropyl)]-3′,5′-di-O-acetyl-2′-bromo-2′-deoxy-β-D-uridine (27)
Acetyl bromide (3.68 g, 50.4 mmol, 10 equiv.) was added dropwise to a boiling suspension of compound 26 (2 g, 5.04 mmol) in anhydrous acetonitrile (50 mL) under an argon atmosphere. The reaction mixture was stirred at 80°C for 15 min and then allowed to cool to room temperature. After evaporation to dryness under reduced pressure, the residue was dissolved in dichloromethane (200 mL) and the solution was washed with water (2 × 200 mL), saturated aqueous NaHCO3 solution (2 × 200 mL) and water (2 × 200 mL). The organic layer was separated, dried over MgSO4 and evaporated in vacuo to yield an oil which was crystallized from dry diethyl ether as a beige crystalline solid (1.54 g); Yield 56%; m.p. decomposed at 80°C TLC Rf (CH2Cl2:CH3OH = 95:5) 0.46; IR (KBr), cm− 1: 3365, 1750 (C = O), 1716 (C = O), 1686 (C = O), 1559, 1462, 1381, 1223, 1181, 1157, 1092, 1076, 1042; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.01 (br s, 1H, NH), 7.35 (s, 1H, H-6), 7.32 (br s, 1H, NHC(O)CF3), 6.22 (d, 1H, H-1’, 5.4), 5.18 (t, 1H, H-3’, 6.1), 4.62 (t, 1H, H-2’, 6.1), 4.49-4.33 (m, 3H, H-4’ and H-5’), 3.34-3.28 (m, 2H, CH2COCF3), 2.33 (t, 2H, 5-CH2, 6.83), 2.19 (s, 3H, CH3), 1.87-1.80 (m, 2H, CH2CH2COCF3); 13C-NMR (CDCl3), δ, ppm: 170.4 (COCH3), 169.7 (COCH3), 163.5 (C-4), 156.4 (q, COCF3, 37.0), 149.9 (C-2), 136.5 (C-6), 115.6 (q, COCF3, 286.1), 114.0 (C-5), 90.7 (C-1’), 80.4 (C-4’), 71.1 (C-3’), 62.8 (C-5’), 47.9 (C-2’), 38.2 (CH2COCF3), 27.9 (5-CH2), 23.8 (CH2CH2COCF3), 20.8 (CH3), 20.6 (CH3).
5-[3-(N-(Trifluoroacetyl)aminopropyl)]-5′-O-acetyl-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (28)
A solution of compound 27 (1 g, 1.84 mmol) in anhydrous ethanol (100 mL) under an argon atmosphere was mixed with freshly activated zinc dust (360 mg, 3 equiv.) and acetic acid (1 mL). The heterogeneous reaction mixture was stirred at room temperature for 1 h [the reaction was monitored by TLC in dichloromethane:methanol (95/5) until no starting material remained], filtered on Celite and the filtrate evaporated to dryness in vacuo. The residual oil was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 28 as a white solid (390 mg); Yield 52%; TLC Rf (CH2Cl3:CH3OH = 95:5) 0.34, Rf (AcOEt) 0.50; IR (KBr), cm− 1: 3346 (NH), 1717 (C = O), 1685 (C = O), 1559, 1465, 1384, 1258, 1228, 1181, 1156, 1108, 1086, 1038; 1H-NMR (CDCl3), δ, ppm, J, Hz: 9.90 (br s, 1H, NH), 7.76 (br s, 1H, NHCOCF3), 7.25 (s, 1H, H-6), 6.92 (t, 1H, H-1’, 1.70), 6.22 (d, 1H, H-3’, 5.97), 5.88 (d, 1H, H-2’, 5.97), 4.99 (br s, 1H, H-4’), 4.37 (dd, 1H, H-5’a, 12.4, 5.3), 4.12 (dd, 1H, H-5’b, 12.4, 2.9), 3.27 (m, 2H, CH2COCF3), 2.32 (t, 2H, 5-CH2, 6.83), 2.02 (s, 3H, CH3), 1.75 (m, 2H, CH2CH2COCF3); 13C-NMR (CDCl3), δ, ppm: 170.6 (COCH3), 164.2 (C-4), 157.4 (q, COCF3, 37.0), 150.5 (C-2), 137.0 (C-6), 133.1 (C-2’), 127.3 (C-3’), 115.9 (q, COCF3, 286.1), 113.6 (C-5), 90.3 (C-1’), 84.5 (C-4’), 64.8 (C-5’), 38.3 (CH2COCF3), 28.3 (5-CH2), 23.8 (CH2CH2COCF3), 20.8 (CH3).
5-[3-(N-(Trifluoroacetyl)aminopropyl)]-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (29)
To a solution of the protected nucleoside 28 (1.25 g, 3.08 mmol) in methanol (10 mL) was added crystals of NaCN (75 mg, 0.5 equiv.). The reaction mixture was stirred at room temperature [monitored by TLC dichloromethane:methanol (95/5)] until no starting material remained. The solvent was evaporated under reduced pressure and the oily residue was purified by silica-gel column chromatography using dichloromethane:methanol (95/5) as eluent to yield 29 as white crystals (640 mg); Yield 57%; TLC Rf (AcOEt:CH3OH = 95:5) 0.52, Rf (AcOEt) 0.50; IR (KBr), cm− 1: 3428 (OH), 3104 (NH), 2961, 1722 (C = O), 1709 (C = O), 1691 (C = O), 1664, 1649, 1563, 1466, 1382, 1260, 1215, 1151, 1106, 1089, 1036, 976; 1H-NMR (CDCl3), δ, ppm, J, Hz: 8.99 (br s, 1H, NH), 7.61 (s, 1H, H-6), 7.35 (br s, 1H, NHCOCF3), 6.94 (s, 1H, H-1’), 6.29 (d, 1H, H-3’, 5.9), 5.81 (d, 1H, H-2’, 5.9), 4.87 (s, 1H, H-4’), 3.88 (d, 1H, H-5’a, 12.3, 2.1), 3.76 (d, 1H, H-5’b, 12.4, 2.2), 3.24 (m, 2H, CH2COCF3), 2.97 (br s, 1H, OH), 2.38-2.18 (m, 2H, 5-CH2), 1.73 (m, 2H, CH2CH2COCF3); 13C-NMR (CDCl3), δ, ppm: 164.2 (C-4), 157.7 (q, COCF3, 37.0), 150.6 (C-2), 138.4 (C-6), 134.7 (C-2’), 126.4 (C-3’), 115.9 (q, COCF3, 286.1), 112.7 (C-5), 90.1 (C-1’), 87.3 (C-4’), 63.1 (C-5’), 38.7 (CH2COCF3), 27.4 (5-CH2), 23.7 (CH2CH2COCF3).
5-[3-(N-(Trifluoroacetyl)aminopropyl)]-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine 5′-(phenyl methoxyalaninyl phosphate) (30)
Compound 29 (300 mg, 0.83 mmol) was converted to 30 by the procedure described for 8. The residue was purified by silica-gel chromatography using dichloromethane:methanol (98/2) as eluent to yield 30 as a colourless oil (230 mg); Yield 46%; TLC Rf (CHCl3:CH3OH = 95:5) 0.68; 1H-NMR (CDCl3), δ, ppm, J, Hz: 10.50 (br s, 1H, NH), 8.61 (br s, 1H, NHCOCF3), 7.37 (s, 1H, H-6), 7.05-7.28 (m, 5H, Ph), 6.80 (s, 1H, H-1’), 6.20 (dt, 1H, H-3’, 6.0, 1.6), 5.85 (m, 1H, H-2’), 4.96 (m, 1H, H-4’), 4.35-4.15 (m, 3H, H-5’ and NH-Ala), 4.26-4.18 (m, 1H, CH-Ala), 3.63* (s, 3H, OCH3-Ala), 3.27-3.17 (m, 2H, CH2COCF3), 2.31-2.17 (m, 2H, 5-CH2), 1.79-1.43 (m, 2H, CH2CH2COCF3), 1.28* (d, 3H, Me-Ala, 6.6); 13C-NMR (CDCl3), δ, ppm: 173.9* (CO-Ala), 164.2 (C-4), 157.5* (q, COCF3, 36.6), 151.0 (C-2), 150.2* (Ph-ipso), 136.7* (C-6), 133.0* (C-2’), 129.5* (Ph-meta), 127.5* (C-3’), 125.3* (Ph-para), 120.2* (Ph-ortho), 116.4* (q, COCF3, 286.1), 114.2* (C-5), 89.8* (C-1’), 84.7* (C-4’), 67.1* (C-5’), 52.5* (OCH3-Ala), 50.2* (CH-Ala), 39.0* (CH2COCF3), 27.3* (5-CH2), 23.7* (CH2CH2COCF3), 20.7* (CH3-Ala).
5-(3-Aminopropyl)-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (31)
To a solution of the protected nucleoside 28 (400 mg, 1.00 mmol) in methanol (50 mL) was added sodium hydroxide (60 mg, 1.5 equiv.). The reaction mixture was stirred at room temperature [monitored by TLC CH3OH:NH4OH (98/2)] until no starting material remained. The solvent was evaporated under reduced pressure and the oily residue was purified by silica gel column chromatography [CH3OH:NH4OH (98/2)] to give the free nucleoside 31 as a colourless oil (161 mg); Yield 61%; TLC Rf (CH3OH:NH4OH = 98:2) 0.35; 1H-NMR (CDCl3), δ, ppm, J, Hz: 11.54 (d, 1H, NH, 2.1), 7.67 (d, 1H, H-6, 8.2), 6.13 (d, 1H, H-1’, 7.3), 5.76 (dd, 1H, H-3’, 8.2, 2.1), -5.23 (dd, 1H, H-2’, 5.9, 3.6), 4.99 (dd, 1H, OH-5’, 7.3, 5.9), 4.22-4.33 (m, 3H, H-4’ and H-5’), 3.24 (m, 2H, CH2COCF3), 2.97 (br, s, 1H, OH), 2.38-2.18 (m, 2H, 5-CH2), 1.73 (m, 2H, CH2CH2COCF3).
5-{2-[8-(9-Acridinylcarbonylamino)octylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (32)
To a solution of the nucleoside 4 (250 mg, 0.51 mmol) in anhydrous methanol was added sodium hydroxide (31 mg, 1.5 equiv.) and the reaction mixture was stirred at room temperature for 1 h [monitored by TLC dichloromethane:methanol (90/10)] until no starting material remained. The solution was carefully neutralized by addition of Amberlite IRN-77 (H+) resin until moistened pH paper indicated pH ∼ 7. The mixture was filtered, and the resin was washed with methanol. The combined filtrate was evaporated to dryness in vacuo. Then the terminal amino group was linked to the acridinylcarbonyl moiety without further purification. To a suspension of 9-acridinecarboxylic acid hydrate (114 mg, 1 equiv.) in anhydrous DMF (5 mL) was added benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate (BOP, 225 mg, 1 equiv.), N,N-diisopropylethylamine (0.11 mL, 1.25 equiv.), and the free nucleoside, and the reaction mixture was stirred at room temperature for 8 h [monitored by TLC dichloromethane:methanol (90/10)] until no starting material remained. After removal of the solvent in vacuo, the residue was dissolved in AcOEt (300 mL), washed with saturated aqueous NaHCO3 solution (2 × 300 mL), dried over MgSO4, and concentrated in vacuo. The residue was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 32 as yellow crystals (70 mg); Yield 23%: TLC Rf (CH2Cl2:CH3OH = 90:10) 0.43; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.30 (br s, 1H, NH), 9.01 (br s, 1H, NHCO-acrid), 8.18 (d, 2H, H-4:acrid and H-5:acrid, 8.8), 7.96 (d, H-1:acrid and H-8:acrid. 8.5), 7.88 (t, 2H, H-2:acrid and H-7:acrid, 7.4), 7.78 (t, 1H, CONH-linker, 5.5), 7.67 (t, 2H, H-3:acrid and H-6:acrid, 7.4), 7.61 (s, 1H, H-6), 6.83 (s, 1H, H-1’), 6.40 (d, 1H, H-3’, 4.9), 5.90 (d, 1H, H-2’, 4.9), 4.95 (br s, 1H, OH), 4.76 (m, 1H, H-4’), 3.56 (m, 2H, H-5’), 3.48 (m, 2H, 5-CH2), 3.00 (m, 4H, CONHCH2-linker and linker-CH2NHCO-acrid), 1.64-1.38-1.15 (m, 12H, CH2:linker); 13C-NMR (d6-DMSO), δ, ppm: 169.3 (CONH-linker), 166.2 (CO-acrid), 163.7 (C-4), 151.1 (C-2), 148.6 (C-4a:acrid and C-10a:acrid), 142.9 (C-9:acrid), 139.0 (C-6), 135.5 (C-2’), 131.0 (C-2:acrid and C-7:acrid), 129.6 (C-4:acrid and C-5:acrid), 127.3 (C-3:acrid and C-6:acrid), 126.1 (C-3’), 126.0 (C-1:acrid and C-8:acrid), 122.2 (C-8a:acrid and C-9a:acrid), 109.0 (C-5), 89.5 (C-1’), 87.1 (C-4’), 63.0 (C-5’), 39.1-38.9-33.5-29.4-29.2-29.1-26.9-26.7 (5-CH2 and CH2:linker).
5-{2-[10-(9-Acridinylcarbonylamino)decylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (33)
Compound 5 (250 mg, 0.48 mmol) was converted to 33 by the procedure described for 32. The residue was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 33 as yellow crystals (100 mg); Yield 34%; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.45; IR (KBr), cm− 1: 3408 (OH), 3066 (NH), 2963, 2926, 2853, 1709 (C = O), 1689 (C = O), 1677 (C = O), 1657 (C = O), 1640 (C = O), 1547, 1516, 1462, 1440, 1382, 1261, 1090, 1038, 1022, 800, 758; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.33 (br s, 1H, NH), 9.00 (t, 1H, NHCO-acrid, 5.4), 8.18 (d, 2H, H-4:acrid and H-5:acrid, 8.7), 7.97 (d, 2H, H-1:acrid and H-8:acrid, 8.7). 7.87 (t, 2H, H-2:acrid and H-7:acrid, 7.7), 7.76 (t, 1H, CONH-linker, 5.7). 7.66 (t, 2H, H-3:acrid and H-6:acrid, 7.7), 7.61 (s, 1H, H-6), 6.83 (s. 1H, H-1’), 6.40 (d, 1H, H-3’, 6.0), 5.89 (d, 1H, H-2’, 6.0), 4.95 (t, 1H, OH, 5.4), 4.75 (m, 1H, H-4’), 3.56 (m, 2H, H-5’), 3.48 (m, 2H, 5-CH2), 3.00 (m, 4H, CONHCH2-linker and linker-CH2NHCO-acrid), 1.66-1.36-1.25 (m, 16H, CH2-linker); 13C-NMR (d6-DMSO), δ, ppm: 168.9 (CONH-linker), 165.9 (CO-acrid), 163.3 (C-4), 150.8 (C-2), 148.2 (C-4a:acrid and C-10a:acrid), 142.6 (C-9:acrid), 138.6 (C-6), 135.1 (C-2’), 130.6 (C-2:acrid and C-7:acrid), 129.3 (C-4:acrid and C-5:acrid), 126.7 (C-3:acrid and C-6:acrid), 125.7 (C-3’), 125.6 (C-1:acrid and C-8:acrid), 121.8 (C-8a:acrid and C-9a:acrid), 108.6 (C-5), 89.1 (C-1’), 87.4 (C-4’), 62.6 (C-5’). 40.1-38.7-33.1-29.3-29.1-29.1-29.0-28.8-28.7-26.4-26.4 (5-CH2 and CH2-linker).
5-{2-[12-(9-Acridinylcarbonylamino)dodecylamino]-2-oxoethyl}-2′,3′-didehydro-2′,3′-dideoxy-β-D-uridine (34)
Compound 6 (250 mg, 0.45 mmol) was converted to 34 by the procedure described for 32. The residue was purified by silica-gel chromatography using dichloromethane:methanol (95/5) as eluent to yield 34 as yellow crystals (70 mg); Yield 21%; TLC Rf (CH2Cl2:CH3OH = 90:10) 0.47; 1H-NMR (d6-DMSO), δ, ppm, J, Hz: 11.30 (br s, 1H, NH), 8.98 (br s, 1H, NHCO-acrid), 8.18 (d, 2H, H-4:acrid and H-5:acrid, 8.8), 7.97 (d, 2H. H-1:acrid and H-8:acrid, 8.8), 7.87 (t, 2H, H-2:acrid and H-7:acrid, 7.5), 7.73 (t, 1H, CONH-linker, 5.1), 7.65 (t, 2H, H-3:acrid and H-6:acrid, 7.5), 7.60 (s, 1H H-6), 6.82 (s, 1H, H-1’), 6.40 (d, 1H, H-3’, 5.8), 5.89 (d, 1H, H-2’, 5.8), 4.90 (br s, 1H, OH), 4.75 (br s, 1H, H-4’), 3.56 (m, 2H, H-5’), 3.48 (m, 2H, 5-CH2), 3.00 (m, 4H, CONHCH2-linker and CH2NHCO-acrid), 1.66-1.36-1.25 (m, 20H, CH2:linker); 13C-NMR (d6-DMSO), δ, ppm: 168.8 (CONH-linker), 165.8 (CO-acrid), 163.2 (C-4), 150.7 (C-2), 148.2 (C-4a:acrid and C-10a:acrid), 142.5 (C-9:acrid), 138.5 (C-6), 135.3 (C-2’), 130.5 (C-2:acrid and C-7:acrid), 129.2 (C-4:acrid and C-5:acrid), 126.6 (C-3:acrid and C-6:acrid), 125.6 (C-3’), 125.5 (C-1:acrid and C-8:acrid), 121.7 (C-8a:acrid and C-9a:acrid), 108.5 (C-5), 89.1 (C-1’), 87.4 (C-4’), 62.6 (C-5’), 41.7-39.1-38.7-33.1-29.0-29.0-28.9-28.9-28.7-28.6-26.5-26.3 (5-CH2 and CH2:linker).
Antiviral Test Procedures
The cultures of CEM-SS and MT4 cells were maintained at 37°C in a 5% CO2 atmosphere in RPMI-1640 medium supplemented with 10% complement-depleted foetal bovine serum (FBS). The antiviral HIV-1 activity of a given compound in CEM-SS cells was measured by quantification of the Reverse Transcriptase activity (RT) associated with particles released from HIV-1LAI-infected cells in the culture medium. CEM-SS cells were infected with 100 TCDI50 (the virus stock was titrated under the same experimental conditions); after 30 min. adsorption, free virus particles were washed out and cells were re-suspended in RPMI-1640 plus 10% calf foetal serum at a final concentration of 105 cells mL-1 in the presence of different concentrations of test compounds. After 5 days, virus production was measured by RT assay as previously described [Citation15]. The 50% inhibitory concentration (IC50) was derived from the computer generated median effect plot of the dose-effect data [Citation16]. The cytotoxicity of the drugs was evaluated in parallel by incubating uninfected cells in the presence of different concentrations of antiviral products. The cell viability was determined by a measure of mitochondrial dehydrogenase activity, enzymes reducing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) into formazan (whose quantity was measured by the absorbance at 540 nm) [Citation17]. The 50% cytotoxic concentration (CC50) is the concentration of drug which reduces cell viability by 50% and was calculated with the program used in the determination of the IC50. The assays using different cells and virus isolates were done according to previously published protocols [Citation15,Citation18]; virus production was quantified by the RT activity associated with virus particles released from the cells in the culture medium. Conditions in which the inhibitory properties were measured on HIV-1 Reverse Transcriptase in vitro have also been described [Citation15]. In vitro RT inhibition has also been described [Citation15]. The CEM-SS cells were obtained from P. Nara through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, Md., USA).
Results and discussion
Chemistry
The first part was focused on the synthesis of the arylphosphoramidates of the β-D- and β-L-d4T analogues bearing an ω-aminoalkylaminocarbonylmethyl tether at the C-5 position.
The synthetic method for the β-D-target arylphosphoramidate compounds 8-11 involved the formation of the key precursor (5’-O-acetyl-5-methoxycarbonylmethyl-2’,3’-didehydro-2’,3’-dideoxy-β-D-uridine) 1 as outlined in . This precursor, used as the starting material for the arylphosphoramidate derivatives reported herein, was prepared from D-arabinose via an arabinoamino-oxazoline in four steps according to our previously published method [Citation6]. This precursor 1 proved to be a key intermediate for introducing a linker arm at the C-5 position through an amide linkage [Citation19]. In the continuation of our study, we have found that the 5-methoxycarbonylmethyl-2’,3’-didehydro-2’,3’-dideoxy-β-D-uridine 2 can also be used as a suitable key component for the synthesis of modified nucleosides bearing a protected primary amine residue which derived from n-alkyldiamines (n = 6, 8, 10 and 12). Thus, deacetylation of 1 was performed using sodium cyanide in methanol at room temperature to yield the free nucleoside 2 after purification by silica-gel chromatography (82%) [Citation20]. The nucleoside 2 readily reacted with alkylenediamines to introduce an amine-linker at the C-5 position. After removal of the excess of diamine and solvent, the terminal amino group of the corresponding intermediates was protected by a trifluoroacetyl group without further purification by reaction with ethyl trifluoroacetate in methanol to afford the expected 5-[N-(n-trifluoroacetylaminoalkyl)aminocarbonylmethyl]-2’,3’-didehydro-2’,3’-dideoxy-β-D-uridine 3-6 [21]. These modified nucleosides were converted to novel phosphoramidates according to the method previously described by McGuigan [2–4]. In this way, the phenylmethoxyalaninyl phosphorochloridate 7 was prepared according to a previously reported procedure by the reaction of phenyl dichlorophosphate with β-alanine methyl ester hydrochloride in the presence of triethylamine [22]. This highly reactive phosphorochloridate intermediate 7 was allowed to react with modified nucleosides 3-6, in THF containing N-methylimidazole under standard conditions, to give diastereisomeric mixtures of the target blocked phosphoramidates 8-11 as shown by spectroscopic data, in moderate yields, after purification by chromatography (10–25%). As anticipated, these materials displayed two closely spaced signals by 31P-NMR corresponding to the diastereoisomers resulting from the mixed stereochemistry at the phosphate centre.
Figure 1 Chemical synthesis of novel β-D-arylphosphoramidate d4T analogues 8-12.
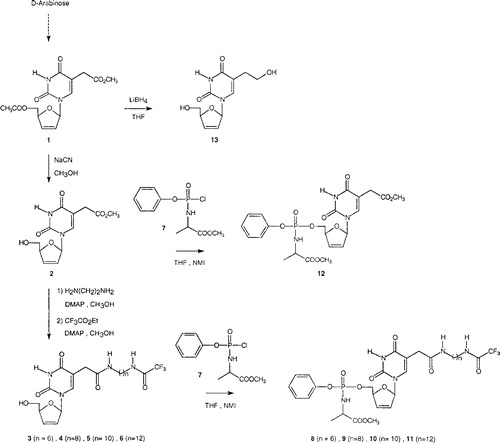
Similarly, we were also interested in preparing the phosphoramidate of 5-(methoxycarbonylmethyl)-β-D-d4T analogue 12. Thus, according to the synthetic method previously developed for phosphoramidate derivatives, reaction of the phosphorochloridate reagent 7 with the d4T analogue 2 in THF led to the production of the masked nucleoside phosphoramidate 12.
For purposes of comparison, we also prepared the 5-hydroxyethyl compound 13. Thus the parent nucleoside 1 was converted into the free corresponding 5-hydroxyethyl derivative 13 using lithium borohydride in anhydrous THF; 13 was isolated in 76% yield after purification by chromatography Citation23-24.
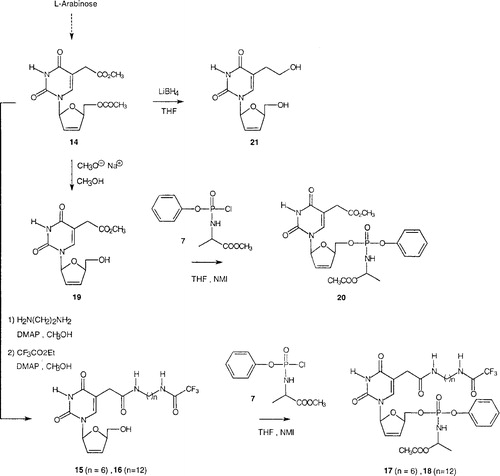
The β-L precursor 14 used as the starting material for the β-L-arylphosphoramidate derivatives was prepared from L-arabinose via an arabinoamino-oxazoline in four steps according to our previously published method [Citation6]. For comparative studies, the arylphosphoramidate ester approach has been successfully applied to the β-L-d4T analogues 15, 16 and 19 affording the corresponding 5-[N-(n-trifluoroacetylaminoalkyl)]carbamoylmethyl- and 5-(methoxycarbonylmethyl)-2’,3’-didehydro-2’,3’-dideoxy-β-L-uridine phosphoramidates 17-18 and 20, respectively, as summarized in . For purposes of comparison, we also prepared the 5-hydroxyethyl-β-L compound 21.
Figure 2 Chemical synthesis of novel β-L-arylphosphoramidate d4T analogues 17, 18 and 20.
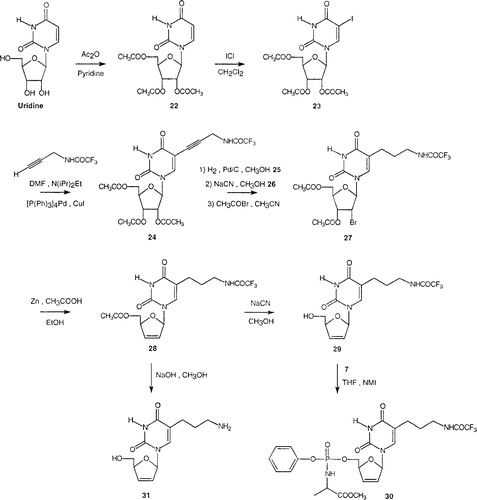
The second part was devoted to selective modification in the side chain at the C-5 position of the nucleobase. The proposed phosphoramidate 30 required a novel synthetic approach from commercially available uridine and we utilized the synthetic protocol outlined in , involving a carbon-carbon bond formation. Thus, uridine was converted into the corresponding tri-O-acetyl derivative 22 upon treatment with acetic anhydride in the presence of pyridine. Subsequently, the C-5 iodination of 22 to its 5-iodo counterpart 23 was further effected using iodine monochloride in dichloromethane [Citation25]. The 5-iodo-uridine nucleoside 23 underwent a smooth and efficient Pd-catalysed coupling reaction to introduce an alkynyl linker at the C-5 position via Sonogashira's procedure Citation26-31. Thus treatment of 23 with N-trifluoroacetylpropargylamine [Citation32] in anhydrous and deoxygenated DMF/N,N-diisopropylethylamine at 25°C, in the presence of tetrakis(triphenylphosphine)palladium(0) and copper(I)iodide, under an argon atmosphere afforded the desired 5-alkynyl nucleoside analogue 24, in 82% yield after purification by silica-gel chromatography. The protected 5-alkynyl nucleoside 24 was then hydrogenated in the presence of wet 10% palladium on carbon into the corresponding N-trifluoroacetylaminopropyl derivative 25, isolated in 95% yield. Deacetylation of 25, performed using sodium cyanide in methanol, afforded the free nucleoside 26 in 86% yield. To introduce the olefinic bond between the C-2’ and C-3’ positions of the uridine derivative, 26 was subjected to a bromoacetylation reaction, using acetyl bromide in anhydrous acetonitrile, to provide the bromoacetate 27 in 56% yield [Citation33]. The reductive β-elimination of the acetoxy-bromo nucleoside 27 proceeded by adding zinc dust in anhydrous ethanol in the presence of a catalytic amount of acetic acid to afford the olefinic nucleoside 28, in 52% yield after chromatography [Citation34]. The remaining 5’-O-acetyl group of 28 was conveniently eliminated by treatment of the protected nucleoside 28 with a methanolic solution of NaCN, to give the analogue 29. Finally, according to the synthetic method previously developed for phosphoramidate derivatives, reaction of the phosphorochloridate reagent 7 with the d4T analogue 29 in THF led to the production of the masked nucleoside phosphoramidate 30. For purposes of comparison, we also prepared the 5-(3-aminopropyl) d4T analogue 31 by treatment of the intermediate 28 with sodium hydroxide.
Figure 3 Chemical synthesis of the novel β-D-arylphosphoramidate d4T analogue 30.
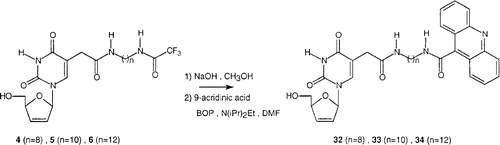
In a third part, a parallel study was carried out to conjugate the previous modified nucleosides 4–6 to a strongly fluorescent dye. In this way, we prepared the analogues lacking the arylphosphoramidate moiety and instead carrying an acridinyl dye, as outlined in . The first step was the deprotection of the protected nucleosides 4–6 by sodium hydroxide. Then 9-acridinecarboxylic acid hydrate, benzotriazol-1-yloxy(dimethylamino)-phosphonium hexafluorophosphate (BOP) and N,N-diisopropylethylamine (DIPEA) were added and the reaction was stirred for 1 hour. TLC revealed the presence of an intense fluorescent spot, corresponding to compounds 32–34, isolated in low yields (21 to 34%) [Citation35].
Figure 4 Chemical synthesis of fluorescence-tagged d4T analogues 32-34.
Biological
All of the synthesized phosphoramidates 8–12, 17–18, 20 and 30 were evaluated for their ability to inhibit replication of HIV and the results obtained using HIV-1-infected CEM-SS and MT-4 cells are displayed in . For the purpose of reference, data for parental nucleosides 2, 19 and 29 are also shown. The clinically used nucleoside analogue AZT was included as reference material. Surprisingly, all the phosphoramidates showed no notable elevation in antiviral potency against HIV-1 compared to the corresponding parental nucleosides. These results are particularly striking since we have previously noted that the β-D-d4T analogues and their β-L-enantiomers bearing an ω-aminoalkylaminocarbonylmethyl tether attached at the C-5 position containing twelve methylene units displayed a weak activity depending on the nature of the cells, at least in the CEM-SS cells (IC50 2.3 μM and 4.4 μM) [Citation6]. The simplest interpretation is that none of these novel prodrugs release the free monophosphate.
Table I. Antiviral and Cytotoxicity Evaluation of the β-D-4dT analogues (2, 13, 29 and 31), the β-D-arylphosphoramidate d4T analogues (8-12 and 30), the β-L-d4T analogues (19 and 21), the β-L-arylphosphoramidate d4T analogues (17, 18, 20) and the fluorescence-tagged d4T analogues (32-34)
In order to study the effect on activity of the nature of the spacer, we extended the tests to the enantiomers 13 and 21 bearing a side-chain with two methylenes including a terminal hydroxy group and the β-D-d4T analogue 31 bearing an aminopropyl linker. Unfortunately, these d4T analogues 13, 21 and 31 were devoid of anti-HIV activity in the CEM-SS and MT-4 cells.
Finally, the tests were also conducted for the nucleoside analogues 32–34 bearing a side-chain linked to a fluorescent moiety. Unfortunately, these chain-terminating fluorescence-tagged d4T analogues were also devoid of anti-HIV activity.
Acknowledgements
These investigations were supported by Grants from “Ensemble Contre le SIDA”.
References
- Meier C. Synlett 1998; 3: 233–242
- McGuigan C, Cahard D, Sheeka HM, De Clercq E, Balzarini J. J Med Chem 1996; 39: 1748–1753
- McGuigan C, Tsang HW, Cahard D, Turner K, Velàsquez S, Salgado A, Bidois L, Naesens L, De Clercq E, Balzarini J. Antiviral Res 1997; 35: 195–204
- Sidiqui AQ, McGuigan C, Ballatore C, Zuccotto F, Gilbert IH, De Clercq E, Balzarini J. J Med Chem 1999; 42: 4122–4128
- Siddiqui AQ, Ballatore C, McGuigan C, De Clercq E, Balzarini J. J Med Chem 1999; 42: 393–399
- Delbederi Z, Fossey C, Fontaine G, Benzaria S, Gavriliu D, Ciurea A, Lelong B, Laduree D, Aubertin A-M, Kirn A. Nucleosides, Nucleotides & Nucleic Acids 2000; 19: 1441–1461
- Spadari S, Maga G, Verri A, Focher F. Exp Opin Invest Drugs 7 1998; 1285
- Wang P, Hong JH, Cooperwood JS, Chu CK. Antiviral Res 1998; 40: 19–44
- Belleau B, Dixit D, Nguyen-Bu N, Kraus J-L. International Conference on AIDS. Montreal, Canada, 4-9 June, paper no T.C.O.1. 1989
- Schinazi RF, Chu CK, Peck A, McMillan A, Mathis R, Cannon D, Jeong LS, Beach JW, Choi WB, Yeola S. Antimicrob Agents Chemother 1992; 36: 672–676
- Mansuri MM, Farina V, Starrett JE, Jr., Benigni DA, Brankovan V, Martin JC. Bioorg Med Chem Lett 1991; 1: 65–68
- Gosselin C, Boudou V, Griffon J-F, Pavia G, Pierra C, Imbach J-L, Aubertin A-M, Schinazi RF, Faraj A, Sommadossi J-P. Nucleosides & Nucleotides 1997; 16: 1389–1398
- Rosenblum BB, Lee LG, Spurgeon SL, Khan SH, Menchen SM, Heiner CR, Chen SM. Nucleic Acids Research 1997; 25: 4500–4504
- Confalone PN. J Het Chem 1990; 27: 31–46
- Moog C, Wick A, Le Ber P, Kim A, Aubertin AM. Antiviral Res 1994; 24: 275–288
- Chou J, Chou TC. Computer Software for Apple II Series and IBM-PC and Instruction Manual. Elsevier-Biosoft, Cambridge 1985; 19–28
- Mosmann T. J Immunol Methods 65 1983; 55–63
- Lefebvre I, Perigaud C, Pompon A, Aubertin AM, Girardet JL, Kirn A, Gosselin G, Imbach JL. J Med Chem 1995; 38: 3941–3950
- Sawai H, Nakamura A, Sekiguchi S, Yumoto K, Endo M, Ozaki H. J Chem Soc 1994; 1997–1998
- Kozak J, Johnson C. Nucleosides & Nucleotides 1998; 17: 2221–2239
- Ozaki H, Nakamura A, Midori A, Endo M, Sawai H. Bull Chem Soc Jpn 1995; 68: 1981–1987
- McGuigan C, Pathirana RN, Mahmood N, Devine KG, Hay AJ. Antiviral Res 1992; 17: 311–321
- Bergstrom DE, Brattesani AJ, Ogawa MK, Schwaickert MJ. J Org Chem 1981; 46: 1423–1431
- Cuenoud B, Casset F, Hüsken D, Natt F, Wolf RM, Altmann K-H, Martin P, Moser HE. Angew Chem In Ed 1998; 37: 1288–1291
- Robins MJ, Barr PJ, Giziewicz J. Can J Chem 1982; 60: 554–557
- McGuigan C, Yarnold CJ, Jones G, Velázquez S, Barucki H, Brancale A, Andrei G, Snoeck R, De Clercq E, Balzarini J. J Med Chem 1999; 42: 4479–4484
- Hobbs FW. J Org Chem 1989; 54: 3420–3422
- Hashimoto H, Nelson MG, Switzer C. J Am Chem Soc 1993; 115: 7128–7134
- Osborne SE, Cain RJ, Glick GD. J Am. Chem Soc 1997; 119: 1171–1182
- Graham D, Parkinson JA, Brown T. J Chem Soc Perkin Trans I 1998; 1131–1138
- Nakatani K, Dohno C, Saito I. J Org Chem 1999; 64: 6901–6904
- Trybulski EJ, Zhang J, Kramss RH, Mangano RM. J Med Chem 1993; 36: 3533–3541
- Maramuto R, Honjo M. Chem Pharm Bull 1974; 22: 128–134
- Furniss BS, Hannaford AJ, Rogers V, Smith PWG, Tatchell AR. Vogel's Textbook of Practical Organic Chemistry. Longman, New York 1978; 315–318
- Kossanyi A, Mestre B, Perrée-Fauvet M. Synth Comm 1999; 29: 4341–4346