Abstract
Recently we found that 1-methyldodecanoylindole-2-carboxylic acid (1) and 1-[2-(4-carboxyphenoxy)ethyl]-3-dodecanoylindole-2-carboxylic acid (4) were inhibitors of the cytosolic phospholipase A2α (cPLA2α)-mediated arachidonic acid release in calcium ionophore A23187-stimulated human platelets with IC50-values of 4.8 μM (1) and 0.86 μM (4). We have now replaced the 3-acyl residue of these compounds by alkylated sulfinyl-, sulfony-, sulfinamoyl-, sulfamoyl-, carbonylamino-, or carbonylaminomethyl-substituents. Structure–activity relationship studies revealed that the pronounced cellular activity of 4 strongly depends on the presence of the 3-acyl moiety. Surprisingly, when testing 4 and its derivatives in an assay with the isolated cPLA2, none of these compounds showed an inhibitory potency at 10 μM indicating that they do not inhibit cPLA2α in the cells by a direct interaction with the active site of the enzyme.
Introduction
Phospholipase A2 (PLA2) are a class of esterases that catalyze the hydrolysis of membrane phospholipids at the sn-2 position to release fatty acids and lysophospholipids. When the fatty acid is arachidonic acid, further action by cyclooxygenase and lipoxygenase enzymes results in the production of eicosanoids, including prostaglandins and leukotrienes. In addition, lysophospholipid metabolism can lead to the formation of platelet-activating factor (PAF). Prostaglandins, leukotrienes, lysophospholipids and the PAF are potent mediators of inflammation Citation1-3. Thus, inhibition of PLA2 is considered as an interesting target for the design of new anti-inflammatory drugs Citation4-8. The special attraction of this approach is based on the evidence, that, unlike cyclooxygenase inhibitors, inhibitors of PLA2 do not only reduce the formation of prostaglandins, but also suppress the generation of leukotrienes, lysophospholipids and PAF. Therefore, it can be expected that inhibitors of PLA2 will possess improved therapeutic activities in comparison to the cyclooxygenase inhibitors therapeutically applied today, such as aspirin, indomethacin or celecoxib.
One problem associated with the in vitro search for PLA2 inhibitors is the selection of the appropriate enzyme, since many different PLA2s are present in the mammalian organism [Citation9]. They can be divided in PLA2s utilizing a catalytic histidine and in PLA2s having a serine in the active site. The small molecular weight (approx. 14 kDa) secretory PLA2s (sPLA2) are members of the first group. The second group consists of the cytosolic PLA2s (cPLA2), the calcium-independent PLA2s (iPLA2) and the lipoprotein-associated PLA2s, which have higher molecular weights than the sPLA2s. From all these PLA2s the α-subtype of cPLA2 (cPLA2α) seems to play the central role in the arachidonic acid cascade and during the inflammatory response as supported by experiments with cells overexpressing cPLA2α [Citation10] and with cPLA2α knockout animals Citation11-13.
Recently, we have found that several 3-acylindole-2-carboxylic acid derivatives are inhibitors of the cPLA2α-mediated arachidonic acid release induced by calcium ionophore A23187 in bovine platelets [Citation14]. One of these compounds was the indole derivative 1, which showed an IC50 of 8 μM in this in vitro assay ().
Figure 1 Structures of indole-2-carboxylic acid inhibitors of cPLA2α-mediated arachidonic acid release in intact platelets.
In the course of structure-activity relationship investigations an increase of inhibitory potency could be achieved by replacement of the N-methyl group of 1 by a longer residue with a terminal carboxylic acid moiety. For compounds 2 and 3 IC50-values of 1.6 μM were evaluated. The replacement of the carboxylic acid side chain of 2 or 3 in position 1 of the indole by a 2-(4-carboxyphenoxy)ethyl residue led to a further increase of activity [Citation14]. With an IC50 of 0.50 μM compound 4 was about threefold more active than 2 and 3 in the assay using bovine cells. Applying human platelets a similar discrepancy in activity was assessed [Citation15]. The IC50-value of 4 was 0.86 μM in this assay. Introduction of a fluoro atom in the meta-position of the benzoic acid moiety again resulted in an increase of activity. With an IC50 of 0.44 μM compound 5 was about twice as potent as 4 [Citation16].
In this paper we report the effects of structural variations of the keto group of compounds 1, 4 and 5 on the inhibition of the cPLA2α-mediated arachidonic acid release in intact human platelets as well as the inhibition of the isolated cPLA2α enzyme.
Material and methods
Chemical synthesis
General
Column chromatography was performed with silica gel 60 (70–230 mesh) (Merck, Darmstadt, Germany). Melting points were determined on a Büchi B-540 apparatus and are uncorrected. 1H-NMR spectra were recorded on a Varian Mercury Plus 400 spectrometer (400 MHz) with tetramethylsilane (TMS) as internal standard. Mass spectra were obtained on Finnigan GCQ and LCQ equipment applying electron beam ionization (EI) and electrospray ionisation (ESI), respectively. The purity of the target compounds was determined using two diverse HPLC systems with UV detection at 254 nm. The first one applied a reversed phase C18 column (Nucleosil 100 RP18, 7 μm, 4.6 mm (I.D.) × 300 mm, Macherey & Nagel, Düren, Germany) eluting the compounds isocratically with CH3CN/H2O/H3PO4 (70:30:0.1) at a flow rate of 1 mL/min. In the second system separation was performed under normal phase conditions using a cyano phase (LiChrospher 100 CN, 5 μm, 3.0 mm (I.D.) × 250 mm, Merck, Darmstadt, Germany) with an isohexane/THF gradient (16–50% THF) containing 0.1% trifluoroacetic acid at a flow rate of 0.5 mL/min. With the exception of 13 all target compounds showed purities greater than 98.5%. The purity evaluated for 13 was 92% (reversed phase system) and 96% (normal phase system), respectively.
Ethyl 3-dodecylsulfinyl-1-methylindole-2-carboxylate (8)
A mixture of ethyl 1-methylindole-2-carboxylate (1.4 g, 6.9 mmol) and thionyl chloride (5 mL) was stirred at room temperature under nitrogen for 5 min. The reaction mixture was allowed to stand for 1 h. The formed precipitate was filtered off, washed several times with dry ether and dried to give the unstable compound 7 (1.7 g, 86%) [Citation25]. A solution of 7 (1.2 g, 4.2 mmol) in dry THF was treated with a 1 M solution of dodecylmagnesium bromide in diethyl ether (1.5 mL) at − 70°C under nitrogen. The mixture was stirred at − 70°C for 45 min and then allowed to warm to room temperature. After addition of water and dilute HCl, it was extracted with diethyl ether. The organic layer was washed with sodium carbonate solution (10%), dried (Na2SO4) and concentrated. The residue was purified by chromatography on silica gel eluting with hexane/ethyl acetate (4:1) to afford a waxy solid 8 (0.22 g, 13%). 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7 Hz, 3H), 1.24 (m, 18H), 1.44 (t, J = 7 Hz, 3H), 1.8 (quint., J = 7 Hz, 2H), 3.07–3.29 (m, 2H), 4.06 (s, 3H), 4.37–4.50 (m, 2H), 7.24 (t, J = 8 Hz, 1H), 7.42–7.44 (m, 2H), 8.60 (d, J = 8 Hz, 1H).
3-Dodecylsulfinyl-1-methylindole-2-carboxylic acid (9)
A mixture of 8 (83 mg, 0.20 mmol), EtOH (12 mL) and 10% aqueous KOH (4 mL) was heated under reflux for 10 min, cooled, acidified with dilute HCl and extracted with diethyl ether. The organic phase was dried (Na2SO4) and the solvent evaporated. The residue was dissolved in a small amount of CH2Cl2. After addition of petroleum ether 9 (50 mg, 61%) precipitated; mp 167–169°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.83 (t, J = 6 Hz, 3H), 1.19–1.70 (m, 20H), 3.07–3.38 (m, 2H), 4.01 (s, 3H), 7.11 (t, J = 8 Hz, 1H), 7.40 (t, J = 8 Hz, 1H), 7.64 (d, J = 8 Hz, 1H), 8.33 (d, J = 8 Hz, 1H). MS (ESI): m/z 390 (M − H)+.
Ethyl 3-dodecylsulfonyl-1-methylindole-2-carboxylate (10)
A solution of KMnO4 (20 mg, 0.17 mmol) in water (1 mL) was treated with a solution of 8 (107 mg, 0.26 mmol) in acetone (3 mL). The mixture was stirred for 1 h at room temperature and then treated with KMnO4 (120 mg). The resulting mixture was stirred for 2 h and then filtered over a cotton pad. The pad was washed with acetone and the combined filtrates were concentrated. The residue was extracted with diethyl ether. The organic layer was dried (Na2SO4) and evaporated. The residue was purified by silica gel chromatography (hexane/ethyl acetate 4:1) to give 10 as an oil (70 mg, 66%). 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7 Hz, 3H), 1.22–1.30 (m, 18H), 1.47 (t, J = 7 Hz, 3H), 1.83 (m, 2H), 3.38 (t, J = 8 Hz, 2H), 3.93 (s, 3H), 4.50 (q, J = 7 Hz, 2H), 7.30–8.44 (m, 3H), 8.20 (d, J = 8 Hz, 1H). MS (EI): m/z 435 (M)+.
3-Dodecylsulfonyl-1-methylindole-2-carboxylic acid (11)
A mixture of 10 (66 mg, 0.15 mmol), EtOH (6 mL) and 10% aqueous KOH (2 mL) was heated under reflux for 10 min, cooled and acidified with dilute HCl. The resulting precipitate was filtered, washed with water and dried to yield 11 (40 mg, 66%); mp 88–90°C. 1H-NMR (CDCl3): δ (ppm) = 0.80 (t, J = 7 Hz, 3H), 1.28–1.30 (m, 18H), 1.70 (quint, J = 8 Hz, 2H), 3.28 (t, J = 8 Hz, 2H), 4.06 (s, 3H), 7.18–7.43 (m, 3H), 8.13 (d, J = 8 Hz, 1H). MS (ESI): m/z 406 (M − H)+.
Ethyl 3-dodecylsulfinamoyl-1-methylindole-2-carboxylate (12)
A solution of dodecylamine (1.70 g, 6.0 mmol) in dry diethyl ether (220 mL) was treated batchwise over 10 min at 0°C with 7 (2.20 g, 12 mmol). The mixture was stirred at 0°C for 30 min and then at room temperature for 15 min. The solvent was removed, water was added and the precipitate formed was filtered off. The residue was recrystallized from petroleum ether to yield 12 (2.0 g, 78%); mp 78–80°C. 1H-NMR (CDCl3): δ (ppm) = 0.87 (t, J = 6 Hz, 3H), 1.22 (m, 18H), 1.40 (t, J = 7 Hz, 3H), 1.55 (m, 2H), 3.04 (dt, J = 19 Hz and 6 Hz, 1H), 3.24 (dt, J = 19 Hz and 7 Hz, 1H), 4.00 (s, 3H), 4.35 (t, J = 6 Hz, 1H), 4.40–4.50 (m, 2H), 7.20–7.28 (m, 1H), 7.30–7.42 (m, 2H), 8.52 (d, J = 8 Hz, 1H).
3-Dodecylsulfinamoyl-1-methylindole-2-carboxylic acid (13)
A mixture of 12 (0.25 g, 0.58 mmol), EtOH (30 mL) and 10% aqueous KOH (10 mL) was heated under reflux for 10 min, cooled in an ice bath and acidified with dilute HCl. The resulting precipitate was filtered, washed with cold water and dried to yield 13 (0.21 g, 91%); mp 77–79°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.80 (t, J = 6 Hz, 3H), 1.24 (m, 20H), 3.26 (q, J = 6 Hz, 2H), 3.76 (s, 3H), 7.19 (t, J = 8 Hz, 1H), 7.33 (t, J = 8 Hz, 1H), 7.56 (d, J = 8 Hz, 1H), 8.08 (d, J = 8 Hz, 1H), 8.80 (t, 1H). MS (ESI): m/z 405 (M-H)+.
Ethyl 3-dodecylsulfamoyl-1-methylindole-2-carboxylate (14)
A solution of 12 (0.50 g, 1.2 mmol) in acetone (12 mL) was treated with a solution of KMnO4 (0.12 g, 0.76 mmol) in water (3 mL). The mixture was stirred for 1.5 h at room temperature and then treated with KMnO4 (240 mg). The resulting mixture was stirred for 1 h and then filtered over a cotton pad. The pad was washed with acetone and the combined filtrates were concentrated. The residue was extracted with diethyl ether. The organic layer was dried (Na2SO4) and evaporated. The residue was purified by silica gel chromatography (hexane/ethyl acetate 9:1 and 4:1) to give 14 as a waxy solid (0.24 g, 46%). 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 6 Hz, 3H), 1.23 (m, 20H), 1.45 (t, J = 7 Hz, 3H), 2.97 (q, J = 7 Hz, 2H), 3.98 (s, 3H), 4.53 (q, J = 7 Hz, 2H), 5.34 (t, J = 7 Hz, 1H), 7.28–7.44 (m, 3H), 8.28 (d, J = 8 Hz, 1H). MS (EI): m/z 450 (M+).
3-Dodecylsulfamoyl-1-methylindole-2-carboxylic acid (15)
A mixture of 14 (0.11 g, 0.25 mmol), EtOH (15 mL) and 10% aqueous KOH (5 mL) was heated under reflux for 10 min, cooled in an ice bath and acidified with dilute HCl. The resulting precipitate was filtered, washed with a cold mixture of ethanol and water (2:1) and dried to yield 15 (70 mg, 68%); mp 154–156°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.84 (t, J = 7 Hz, 3H), 1.09–1.33 (m, 20H), 2.70 (t, J = 7 Hz, 2H), 3.40 (s, 1H), 3.86 (s, 3H), 7.19 (t, J = 8 Hz, 1H), 7.29 (t, J = 8 Hz, 1H), 7.55 (d, J = 8 Hz, 1H), 7.94 (d, J = 8 Hz, 1H). MS (ESI): m/z 421 (M − H)+.
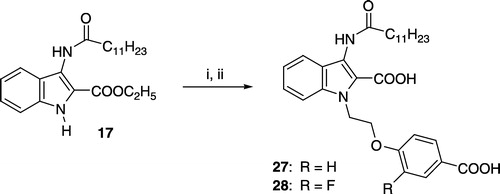
Ethyl 3-dodecanoylaminoindole-2-carboxylate (17)
A solution of ethyl 3-aminoindole-2-carboxylate (16) in dry CH2Cl2 (20 mL) was treated with a solution of dodecanoyl chloride (0.48 g, 2.2 mmol) in dry CH2Cl2 (5 mL). After addition of triethylamine (1 mL) and pyridine (5 drops) the mixture was stirred at room temperature for 1 h. Then CHCl3 was added. The organic solution was washed subsequently with dilute HCl and dilute NaOH, dried (Na2SO4) and evaporated. The residue was recrystallized from MeOH to yield 17 (0.54 g, 70%); mp 140–141°C. 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7 Hz, 3H), 1.20–1.39 (m, 16H), 1.44 (t, J = 7 Hz, 3H), 1.80 (quint, 2H, J = 7 Hz), 2.50 (t, J = 7 Hz, 2H), 4.43 (q, J = 7 Hz, 2H), 7.11–7.15 (m, 1H), 7.29–7.34 (m, 2H), 8.23 (d, J = 8 Hz, 1H), 8.39 (broad, 1H), 9.00 (broad, 1H).
Ethyl 3-dodecanoylamino-1-methylindole-2-carboxylate (18)
A mixture of 17 (0.23 g, 0.60 mmol), methyl p-toluenesulfonate (0.12 g, 0.66 mmol), tetrabutylammonium bromide (52 mg, 0.16 mmol), powdered NaOH (64 mg, 1.6 mmol), dry diethyl ether (20 mL) and dry CH2Cl2 (20mL) was stirred at room temperature for 4 h. After addition of diethyl ether, the organic phase was washed with brine, dried (Na2SO4) and evaporated. The residue was chromatographed on silica gel eluting with petroleum ether/ethyl acetate (4:1) and recrystallized from MeOH to yield 18 (0.16 g, 68%); mp 124–126°C. 1H-NMR (CDCl3): δ (ppm) = 0.87 (t, J = 7 Hz, 3H), 1.22–1.38 (m, 16H), 1.45 (t, J = 7 Hz, 3H), 1.79 (quint, J = 7 Hz, 2H), 2.47 (t, J = 7 Hz, 2H), 3.97 (s, 3H), 4.45 (q, J = 7 Hz, 2H), 7.11 (t, J = 8 Hz, 1H), 7.26–7.39 (m, 2H), 8.07 (d, J = 8 Hz, 1H), 9.05 (broad, 1H).
3-Dodecanoylamino-1-methylindole-2-carboxylic acid (19)
A mixture of 18 (65 mg, 0.16 mmol), EtOH (15 mL) and 10% aqueous KOH (5 mL) was heated under reflux for 1 h, cooled, acidified with dilute HCl and extracted with diethyl ether. The organic phase was dried (Na2SO4) and the solvent evaporated. The residue was dissolved in a small amount of diethyl ether. After addition of petroleum ether 19 precipitated (40 mg, 67%); mp 164–166°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.85 (t, J = 6 Hz, 3H), 1.25–1.37 (m, 18H), 3.43 (t, J = 7 Hz, 2H), 3.95 (s, 3H), 7.05 (t, J = 7 Hz, 1H), 7.31 (t, J = 7 Hz, 1H), 7.51–7.57 (m, 2H), 9.51 (broad, 1H). MS (ESI): m/z 371 (M-H)+.
3-(Dodecanoylaminomethyl)-1-methylindole-2-carboxylic acid (20)
Compound 20 was synthesized in the same way as the corresponding octadecanoyl derivative, whose synthesis has already been described [Citation27]. Mp 156–157°C. 1H-NMR (CDCl3): δ (ppm) = 0.87 (t, J = 7 Hz, 3H), 1.09–1.41 (m, 16H), 1.56 (m, 3H), 2.17 (t, J = 8 Hz, 2H), 3.99 (s, 3H), 4.72 (d, J = 6 Hz, 2H), 6.57 (broad, 1H), 7.17–7.21 (m, 1H), 7.36–7.39 (m, 2H), 7.68 (d, J = 8 Hz, 1H).
Ethyl 3-dodecylsulfanylindole-2-carboxylate (22)
A solution of ethyl 3-thiocyanatoindole-2-carboxylate [Citation26] (21) (1.6 g, 6.4 mmol) and dodecan-1-ol (1.2 g, 6.4 mmol) in dry THF (30 mL) was treated at − 20°C dropwise during 20 min with a solution of triphenylphosphine (1.4 g, 7.0 mmol) in dry THF (30 mL). The mixture was stirred for a further 20 min at the same temperature. After addition of ice and water the mixture was extracted with diethyl ether. The organic layer was dried (Na2SO4) and evaporated. The residue was purified by silica gel chromatography (hexane/ethyl acetate 9:1) to give 22 (1.5 g, 60%) as solid; mp 52–54°C. 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7 Hz, 3H), 1.21–1.42 (m, 20H), 1.46 (t, J = 7 Hz, 3H), 2.90 (t, J = 7 Hz, 2H), 4.47 (q, J = 7 Hz, 2H), 7.21 (t, J = 8 Hz, 1H), 7.35 (t, J = 8 Hz, 1H), 7.39 (d, J = 8 Hz, 1H), 7.88 (d, J = 8 Hz, 1H), 9.05 (broad, 1H). MS (EI): m/z 390 (M+).
Ethyl 3-dodecylsulfinyl-1-[2-(4-ethoxycarbonylphenoxy)ethyl]indole-2-carboxylate (23)
Sodium hydride (21 mg, 0.53 mmol, 60% dispersion in oil) was suspended in dry DMF (15 mL) and treated under nitrogen with a solution of 22 (200 mg, 0.51 mmol) in dry DMF (10 mL). The mixture was stirred for 1 h at room temperature. Then a solution of ethyl 4-(2-bromoethoxy)benzoate (0.14 g, 0.51 mmol) in dry DMF (5 mL) was added and the mixture was stirred at room temperature for a further 6 days. The mixture was poured into cold water and extracted with diethyl ether. The organic phase was dried (Na2SO4) and evaporated. The residue was dissolved in acetic acid (5 mL) and treated with Pb(IV)acetate (0.18 g, 0.41 mmol). The mixture was stirred for 24 h at room temperature. The solvent was removed and the residue was purified by silica gel chromatography (hexane/ethyl acetate 4:1) to yield 23 as an oil (0.21 g, 70%). 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7 Hz, 3H), 1.23–1.28 (m, 18H), 1.36 (t, J = 7 Hz, 3H), 1.45 (t, J = 7 Hz, 3H), 1.72–1.85 (m, 2H), 3.01–3.35 (m, 2H), 4.30–4.46 (m, 6H), 4.82–5.25 (m, 2H), 6.77 (d, J = 9 Hz, 2H), 7.23 (t, J = 8 Hz, 1H), 7.45 (t, J = 8 Hz, 1H), 7.59 (d, J = 8 Hz, 1H), 7.92 (d, J = 9 Hz, 2H), 8.53 (d, J = 8 Hz, 1H). MS (ESI): m/z 598 (M+H)+.
Ethyl 3-dodecylsulfinyl-1-[2-(4-ethoxycarbonyl-2-fluorophenoxy)ethyl]indole-2-carboxylate (24)
For the preparation of 24 the synthesis sequence described for the preparation of 23 was applied starting with 22 and ethyl 4-(2-bromoethoxy)-3-fluorobenzoate except that the reaction time of the oxidation with Pb(IV)acetate was 48 h. The product 24 was afforded as an oil (0.23 g, 73%). 1H-NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7 Hz, 3H), 1.23–1.27 (m, 18H), 1.36 (t, J = 7 Hz, 3H), 1.45 (t, J = 7 Hz, 3H), 1.83–1.86 (m, 2H), 3.06–3.29 (m, 2H), 4.30–4.49 (m, 6H), 4.87–5.20 (m, 2H), 6.84 (t, J = 8 Hz, 1H), 7.25 (t, J = 8 Hz, 1H), 7.45 (t, J = 8 Hz, 1H), 7.61 (d, J = 8 Hz, 1H), 7.65–7.74 (m, 2H), 8.54 (d, J = 8 Hz, 1H).
1-[2-(4-Carboxyphenoxy)ethyl]-3-dodecylsulfinylindole-2-carboxylic acid (25)
A mixture of 23 (80 mg, 0.14 mmol), EtOH (30 mL) and 10% aqueous KOH (10 mL) was heated under reflux for 15 min, cooled, acidified with dilute HCl and extracted with diethyl ether. The organic phase was dried (Na2SO4) and the solvent evaporated. The residue was dissolved in a small amount of ethyl acetate. After addition of petroleum ether 25 (70 mg, 96%) precipitated; mp 185–187°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.83 (t, J = 7 Hz, 3H), 1.11–1.70 (m, 20H), 3.01–3.08 (m, 2H), 4.20–4.40 (m, 2H), 4.70–5.15 (m, 2H), 6.81 (d, J = 9 Hz, 2H), 7.16 (t, J = 8 Hz, 1H), 7.39 (t, J = 8 Hz, 1H), 7.55–7.78 (m, 3H), 8.32 (d, J = 8 Hz, 1H). MS (ESI): m/z 540 (M-H)+.
1-[2-(4-Carboxy-2-fluorophenoxy)ethyl]-3-dodecylsulfinylindole-2-carboxylic acid (26)
Compound 24 (100 mg, 0.17 mmol) was saponified in a similar manner as described for the synthesis of 25. The product 26 (50 mg, 53%) was precipitated from CH2Cl2 by addition of petroleum ether; mp 185–186°C. 1H-NMR (CDCl3): δ (ppm) = 0.87 (t, J = 7 Hz, 3H), 1.22–1.77 (m, 20H), 3.06–3.38 (m, 2H), 4.45–4.66 (m, 2H), 5.08–5.10 (m, 2H), 6.94 (t, J = 8 Hz, 1H), 7.30 (t, J = 8 Hz, 1H), 7.53 (t, J = 8 Hz, 1H), 7.64–7.82 (m, 4H).
1-[2-(4-Carboxyphenoxy)ethyl]-3-dodecanoylaminoindole-2-carboxylic acid (27)
A mixture of 17 (116 mg, 0.30 mmol), potassium tert-butylate (37 mg, 0.33 mmol) and dry DMSO (2 mL) was heated under nitrogen at 110°C for 5 min. After addition of ethyl 4-(2-bromoethoxy)benzoate (80 mg, 0.33 mmol), the mixture was heated for an additional 5 min at the same temperature. The mixture was cooled, diluted with brine and extracted with diethyl ether/CH2Cl2 (3:1). The organic phase was dried and the solvent evaporated. The residue was chromatographed on silica gel (petroleum ether/ethyl acetate 9:1 to 4:1) and the ester intermediate obtained was saponified using a similar method as described for the synthesis of 19. The product was dissolved in a small amount of THF. After addition of petroleum ether 27 (40 mg, 26%) precipitated; mp 203–205°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.83 (t, J = 7 Hz, 3H), 1.23–1.30 (m, 16H), 1.60 (quint. J = 7 Hz, 2H), 2.33 (t, J = 7 Hz, 2H), 4.30 (t, J = 5 Hz, 2H), 4.91 (t, J = 5 Hz, 2H), 6.88 (d, J = 9 Hz, 2H), 7.05 (t, J = 8 Hz, 1H), 7.30 (t, J = 8 Hz, 1H), 7.51 (d, J = 8 Hz, 1H), 7.61 (d, J = 8 Hz, 1H), 7.80 (d, J = 9 Hz, 2H), 9.58 (broad, 1H). MS (ESI): m/z 521 (M-H)+.
1-[2-(4-Carboxy-2-fluorophenoxy)ethyl]-3-dodecanoylaminoindole-2-carboxylic acid (28)
Compound 17 (116 mg, 0.30 mmol) was reacted with ethyl 4-(2-bromoethoxy)-3-fluorobenzoate (96 mg, 0.33 mmol) applying the reaction sequence described for the synthesis of 27. The product 28 was recrystallized from ethanol/H2O to yield a white solid (30 mg, 19%); mp 197–199°C. 1H-NMR (DMSO-d6): δ (ppm) = 0.84 (t, J = 7 Hz, 3H), 1.20–1.38 (m, 16H), 1.61 (quint. J = 7 Hz, 2H), 2.35 (t, J = 7 Hz, 2H), 4.43 (t, J = 5 Hz, 2H), 4.93 (t, J = 5 Hz, 2H), 7.05 (t, J = 7 Hz, 1H), 7.21 (t, J = 9 Hz, 1H), 7.29 (t, J = 8 Hz, 1H), 7.49–7.66 (m, 4H), 9.51 (broad, 1H).
Biological assays
Cell assay
The ability of compounds to inhibit cPLA2α activity in intact cells was determined by measuring the calcium ionophore A23187-induced arachidonic acid release from human platelets with HPLC/UV-detection according to a procedure previously described [Citation15]. Deviating, the HPLC-separation of arachidonic acid was achieved on a RP18 multospher 100 column, 3 μm, 3.0 mm (I.D.) × 125 mm, with a RP18 multospher 100 guard column, 5 μm, 3.0 mm (I.D.) × 20 mm (CS-chromatographie service, Langerwehe, Germany). The mobile phase consisted of acetonitrile/10 mM (NH4)2HPO4 buffer adjusted to pH 7.4 with ortho-phosphoric acid (50:50, v/v). The flow rate was 0.33 mL/min and the injected sample volume was 300 μl. The detection wavelength was 200 nm applying a Waters 2487 UV-detector. After each run the column was washed with 0.6 mL methanol. 3-(4-Decyloxyphenyl)propanoic acid was applied as internal standard.
Assay with the isolated enzyme
The inhibition of cPLA2α isolated from human platelets was performed as previously described [Citation22]. Briefly, sonicated co-vesicles consisting of 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (0.2 mM) and 1,2-dioleoyl-sn-glycerol (0.1 mM) were used as enzyme substrate. The cPLA2α activity was determined by measuring the arachidonic acid released by the enzyme with reversed-phase HPLC and UV-detection at 200 nm after cleaning up the samples by solid phase extraction.
Results and discussion
Chemistry
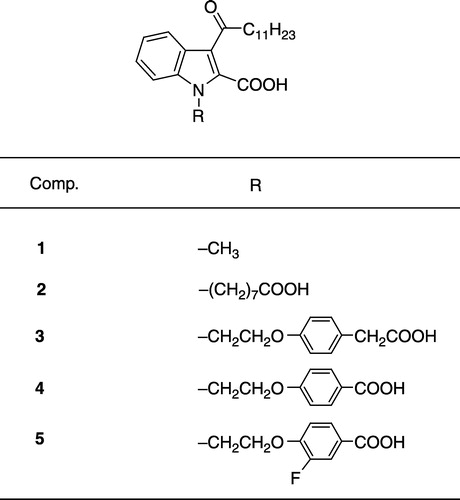
Scheme outlines the chemical approach used to synthesize derivatives of 1, which contain a sulfinyl- (9), sulfonyl- (11), sulfinamoyl- (13) and sulfamoyl-moiety (15), respectively, instead of the keto function. Thus, 1-methylindole-2-carboxylate was treated with SOCl2 to yield the chlorosulfinyl-derivative 7 [Citation25]. The chlorosulfinyl-moiety of 7 was then converted to an alkane-1-sulfinyl-residue by reaction with dodecylmagnesium bromide. Saponification of the obtained intermediate 8 with KOH yielded the target compound 9. The corresponding sulfonyl-derivative 11 was obtained by oxidation of the sulfinyl-group of 8 with KMnO4 in acetone/water followed by hydrolysis of the ester moiety. Treatment of 7 with dodecylamine afforded the 3-dodecylsulfinamoylindole-2-carboxylic ester 12. Ester hydrolysis led to the desired acid 13. Surprisingly, in solution 13 easily underwent decarboxylation at room temperature. The velocity of this reaction depended on the solvent. While in d6-DMSO about 50% of the decarboxylation product was formed after 7 days, in CDCl3 after 1 day the acid had already been transformed quantitatively to its decarboxylation product. In contrast, the sulfamoyl-derivative 15, which was obtained by KMnO4 oxidation of 12 followed by ester hydrolysis, did not show such a decarboxylation behaviour at room temperature.
Scheme 1 (i) SOCl2, room temp.; (ii) dodecylmagnesium bromide, diethyl ether, THF, − 70°C; (iii) EtOH, 10% aqueous KOH, reflux; (iv) aqueous KMnO4, acetone, room temp.; (v) dodecylamine, diethyl ether, 0°C/room temp.
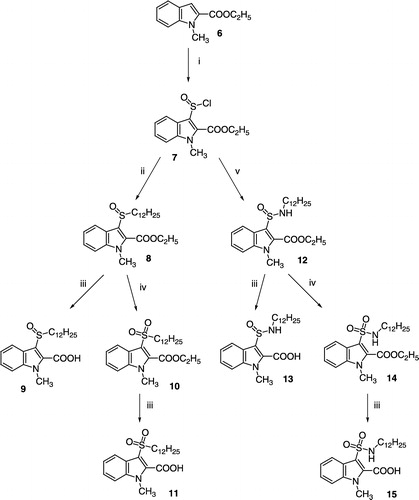
The 1-methylindole-2-carboxylic acid with a dodecanoylamino substituent in position 3 (19) was prepared from ethyl 3-aminoindole-2-carboxylate (16) as shown in Scheme . The 3-amino group of 16 was acylated with dodecanoyl chloride. Methylation of the indole nitrogen with methyl p-toluenesulfonate and ester hydrolysis with KOH gave the required compound 19. The 3-(dodecanoylaminomethyl)-1-methylindole-2-carboxylic acid (20) was prepared using the reaction sequence published for the synthesis of its octadecanoyl-derivative [Citation25].
Scheme 2 (i) Dodecanoyl chloride, CH2Cl2, triethylamine, pyridine, room temp.; (ii) methyl p-toluenesulfonate, (C4H9)4N+Br− , powdered NaOH, diethyl ether, CH2Cl2, room temp.; (iii) EtOH, 10% aqueous KOH, reflux.
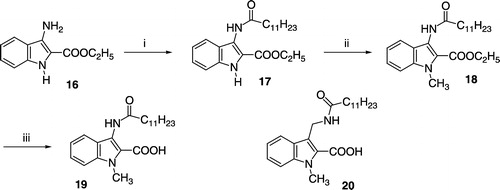
Compounds 25 and 26 were synthesized as outlined in Scheme . The 3-thiocyanate group of 21 was transformed to a dodecylsulfanyl residue by reaction with dodecan-1-ol and triphenylphosphine. Coupling of the obtained intermediate 22 with the appropriate 4-(2-bromoethoxy)benzoic acid ethylester and oxidation of the sulfanyl- to a sulfinyl-moiety with Pb(OAc)4 afforded the dicarboxylic acid esters 23 and 24, which were saponified to the target compounds 25 and 26. The 3-dodecanoylaminoindoles 27 and 28 were obtained from 17 as shown in Scheme .
Scheme 3 (i) Dodecan-1-ol, triphenylphosphine, THF, − 20°C; (ii) NaH, ethyl-4-(2-bromoethoxy)benzoate, DMF, room temp.; (iii) Pb(IV)acetate, acetic acid, room temp.; (iv) EtOH, 10% aqueous KOH, reflux.
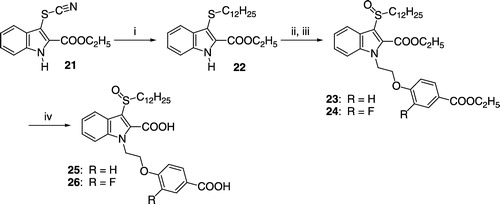
Scheme 4 (i) tert-BuOK, ethyl 4-(2-bromoethoxy)benzoate, DMSO, 110°C; (ii) EtOH, 10% aqueous KOH, reflux.
Biological evaluation
The assays for screening cPLA2α inhibitors in vitro can be divided in two categories, namely, into assays with the isolated enzyme, and into assays with intact cells.
Since for the development of the dual cyclooxygenase/5-lipoxygenase inhibitor licofelone Citation17-21 a cellular assay strategy had successfully been employed, we decided to screen cPLA2α inhibitors also in a cellular situation, initially. In our assay the cPLA2α-inhibitory potency of the test compounds was evaluated by measuring the calcium ionophore A23187-induced arachidonic acid release from human platelets with HPLC and UV-detection at 200 nm [Citation15]. To avoid metabolism of arachidonic acid via the cyclooxygenase-1 and the 12-lipoxygenase pathways, the dual cyclooxygenase/12-lipoxygenase inhibitor 5,8,11,14-eicosatetraynoic acid (ETYA) was added to the platelets in these experiments.
Although a cellular assay reflects the in vivo situation much better than a test system with the isolated enzyme, it also has some disadvantages. One weakness is the evidence that in the cells other isoforms of the enzyme may be present such as sPLA2s or iPLA2s. These could be involved in substrate cleavage. Furthermore, since cells are complex systems, a reduction of arachidonic acid release does not necessarily prove a direct interaction of a test compound with the enzyme. Therefore, in completion of our assay strategy we established a cPLA2α assay with the isolated cPLA2α [Citation22]. In this assay cPLA2α from human platelets was used. Like other lipases, cPLA2α has evolved to work optimally at a lipid-water interface. For this reason sonicated covesicles consisting of 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine and 1,2-dioleoyl-sn-glycerol were used as enzyme substrate. A possible problem with this screen is that a test compound could inhibit the enzyme not by binding to its active site but merely by altering the substrate assembly and hence causing the enzyme to desorb from the lipid-water-interface [Citation23,Citation24]. To exclude this way of action, the mole fraction of inhibitor in the interface was kept low (maximum concentration of the inhibitor 10 μM, concentration of the vesicle forming lipids 300 μM).
All newly synthesized indole-2-carboxylic acid derivatives were evaluated in the cellular assay. The most interesting of these compounds were also tested for inhibition of the isolated cPLA2α [Citation16]
Structure–activity relationships
The structure–activity relationships of the 3-acylindole-2-carboxylic acids discussed in preceding papers [Citation14,Citation16,Citation27,Citation28] were extended to the variation of the keto moiety of the 3-acyl residue. Replacing the 3-dodecanoyl substituent of 3-dodecanoyl-1-methylindole-2-carboxylic acid (1) by dodecylsulfinyl (9), dodecylsulfonyl (11), dodecylsulfinamoyl (13), dodecylsulfamoyl (15), dodecanoylamino (19), and dodecanoylaminomethyl (20), respectively, did not lead to a substantial change of inhibitory potency in the whole cell cPLA2α assay. The IC50-values of the compounds ranged from 5–10 μM ().
Table I. Inhibition of the cPLA2α-mediated arachidonic acid release from human platelets stimulated with calcium ionophore A23187. 
Contrary, substitution of the keto moiety of 4 by sulfinyl and carbonylamino resulted in a significant decrease of activity. While the IC50 of 4 was 0.86 μM, 25 and 27 showed IC50-values of about 6 μM. A similar effect occurred when varying the fluoro-substituted derivative of 4 (5) in an analogous way (). The sulfinyl and carbonylamino derivatives of 5 (26 and 28) were about twenty times less active than 5. In conclusion, the pronounced activity of 4 and 5 in the cellular assay ( = inhibition of the arachidonic acid release at a submicromolar concentration) depends on the concurrent presence of the 3-acyl- and the 1-[2-(4-carboxyphenoxy)ethyl]-residue.
Table II. Inhibition of cPLA2α-activity. 
Surprisingly, in the assay with the isolated cPLA2α 4, 5 and their derivatives 25–28 did not cause inhibition of the enzyme at 10 μM (). These results show that the inhibition of the cellular cPLA2α activity by these compounds does not arise from a direct interaction with the active site of the enzyme. Therefore, the proposed model of the interaction of the indole-carboxylic acids with the active site of the enzyme [Citation14] cannot be sustained any longer. However, the marked inhibition of the cPLA2α-mediated arachidonic release in A23187-stimulated platelets by 4 and 5, respectively, and its susceptibility to small variations in the molecules indicates that at least these two substances do not only act in the cells in a non-specific way as described for several lipophilic acids [Citation23]. The exact mechanism of their inhibitory action against cellular cPLA2α-activity needs to be evaluated.
References
- Serhan CN, Haeggström JZ, Leslie CC. FASEB J 1996; 10: 1147–1158
- Huang YH, Schafer-Elinder L, Wu R, Claesson HE, Frostegard. J Clin Exp Immunol 1999; 116: 326–331
- Ryborg AK, Deleuran B, Sogaard HR, Kragballe K. Acta Derm Venereol 2000; 80: 242–246
- Connolly S, Robinson DH. Expert Opin Ther Pat 1993; 3: 1141–1155
- Connolly S, Robinson DH. Expert Opin Ther Pat 1995; 5: 673–683
- Tibes U, Friebe WG. Expert Opin Investig Drugs 1997; 6: 279–298
- Mayer RJ, Marshall LA. Emerging Drugs 1998; 3: 333–344
- Lehr M. Expert Opin Ther Pat 2001; 11: 1123–1136
- Six LA, Dennis EA. Biochim Biophys Acta 2000; 1488: 1–19
- Lin LL, Lin AY, Knopf JL. Proc Nat Acad Sci USA 1992; 89: 6147–6151
- Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Koma-gata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Nature 1997; 390: 618–622
- Bonventre JV, Huang Z, Taheri MR, O'Leary E, Li E, Moskowitz MA, Sapirstein A. Nature 1997; 390: 622–625
- Gijon MA, Spencer DM, Siddiqi AR, Bonventre JV, Leslie CC. J Biol Chem 2000; 275: 20146–20156
- Lehr M. J Med Chem 1997; 40: 2694–2705
- Lehr M, Schulze Elfringhoff A. Arch Pharm Pharm Med Chem 2000; 333: 312–314
- Lehr M, Klimt M, Schulze Elfringhoff A. Bioorg Med Chem Lett 2001; 11: 2569–2572
- Dannhardt G, Lehr M. J Pharm Pharmacol 1992; 44: 419
- Dannhardt G, Steindl L, Lehr M. Chem Abstr 1991; 114: 163999, EP 397175 1990
- Lehr M. PhD Thesis. University of Regensburg 1989
- Laufer SA, Augustin J, Dannhardt G, Kiefer W. J Med Chem 1994; 37: 1894–1897
- Laufer S. Inflammopharmacology 2001; 9: 101–112
- Schmitt M, Lehr M. J Pharm Biomed Anal 2004; 35: 135–142
- Connolly S, Bennion C, Botterell S, Croshaw PJ, Hallam C, Hardy K, Hartopp P, Jackson CG, King SJ, Lawrence L, Mete A, Murray D, Robinson DH, Smith GM, Stein L, Walters I, Wells E, Withnall WJ. J Med Chem 2002; 45: 1348–1362
- Ghomashchi F, Loo R, Balsinde J, Bartoli F, Apitz-Castro R, Clark JD, Dennis EA, Gelb MH. Biochim Biophys Acta 1999; 1420: 45–56
- Unangst PC, Connor DT, Stabler SR. J Heterocycl Chem 1987; 24: 817–820
- Tamura Y, Kwon S, Chun MW, Ikeda M. J Heterocycl Chem 1978; 15: 425–427
- Lehr M. Arch Pharm Pharm Med Chem 1996; 329: 386–392
- Griessbach K, Klimt M, Schulze Elfringhoff A, Lehr M. Arch Pharm Pharm Med Chem 2002; 335: 547–555