Abstract
The ability of phosphoramidates Me2NP(O)(Cl)(p-NHC6H4NO2) 1, Me2NP(O)(p-NHC6H4NO2)2 2, (CH3C6H4O-p)P(O)(p-NHC6H4NO2)2 3 and (CH3C6H4O-p)2P(O)(p-NHC6H4NO2) 4 to inhibit human acetylcholinesterase (hAChE) has been evaluated by a modified Ellman's method and spectrophotometric measurements. Results showed that compounds 1 and 2 do not have any inhibitory potency, whereas compounds 3 and 4 were reversible mixed inhibitors. The IC50 values for inhibitors 3 and 4 were 0.143 and 0.581 mM, respectively. The previously unknown compounds 3 and 4 were synthesized and characterized by 1H, 13C, 31P NMR and IR spectroscopy and elemental analysis.
Introduction
Many phosphoramidate compounds inhibit human acetylcholinesterase (hAChE, EC 3.1.1.7) by phosphorylation of a serine hydroxyl group in the active site of this enzyme leading to its inactivation. This inhibition increases acetylcholine's levels in cholinergic synapses of both peripheral and central nervous systems Citation1-8. During the inhibition, phosphorylated enzyme undergoes a postinhibitory process and its spontaneous reactivation is very slow [Citation5,Citation8]. The mechanism of inhibition by these compounds has been widely studied. Many papers have reported the irreversible inhibition of human acetylcholinesterase by phosphoramidates Citation2-4Citation7Citation9–19 and some authors have reported reversible inhibitors Citation1Citation5Citation6Citation8Citation20-26. Drug design is a matter of great interest based on reactivation of hAChE with site-directed nucleophiles such as 2-pralidoxime (2-PAM) and its analogs. The nucleophile binds to the active site and reacts with the phosphorylated hydroxyl group to release free and active enzyme which in turn becomes phosphorylated [Citation1,Citation5,Citation8,Citation21,Citation22].
Bollinger et al. studied the inhibitory effect of (Me2N)2P(O)(p-NHC6H4NO2), 5, and Me2P(O) (p-NHC6H4NO2), 6, on acetylcholinesterase [Citation27]. They found that only molecule 5 with a (O)PN3 moiety has inhibitory potency and acts as reversible inhibitor. To extend investigation in this area, we designed and synthesized compounds 1–4, which have a XY(O)P(p-NHC6H4NO2) skeleton and examined their inhibitory potency and inhibition mechanism with hAChE using the spectrophotometric method based on Ellman's procedure [Citation28].
Materials and methods
All reactions for synthesis of the phosphoramidates were carried out under an argon atmosphere. Melting points were determined on a Gallenkamp apparatus. 1H, 13C and 31P NMR spectra were recorded on a Bruker (Avance DRS) 500 spectrometer and chemical shifts were determined relative to TMS and 85% H3PO4, respectively, as external standards. IR spectra (KBr pellets) were obtained with a Shimadzu, IR-60 model spectrometer. Elemental analysis was performed using a Heraeus CHN-O-RAPID instrument. UV measurements were performed on a Shimadzu UV-2100 spectrophotometer. All chemicals and solvents for syntheses were from Merck. Phosphoramidodichloridicacid-4-methylphenylester was prepared by the literature method [Citation29].
Synthesis
N,N-(dimethyl)-N′-(4-nitrophenyl)phosphoramidochloride, 1 and N,N-(dimethyl)-N′,N″-bis(4-nitrophenyl)phosphoramide, 2
were synthesized and characterized by 1H, 13C, 31P NMR and IR spectroscopy and elemental analysis [Citation30].
N,N′-bis(4-nitrophenyl)phosphoramidicacid-4-methylphenylester, 3
To a solution of phosphoramidodichloridicacid-4-methylphenylester (2.25 g, 10 mmol) in dry benzene (20 ml), the sodium salt of 4-nitroaniline (2.76 g, 20 mmol) was added under an argon atmosphere. After 4 h stirring, the precipitate was removed and washed with dry chloroform (20 ml). The purity of the product was up to 98%. (yield 65%), m. p. = 174–176°C. Anal. Calc. for C19H17N4O6P: C, 53.27; H, 3.97; N, 13.08. Found: C, 53.20; H, 3.93; N, 12.99%. 1H NMR (DMSO-d6), δ (ppm): 2.24 (s, 3H, p-CH3), 6.63 (d, 3JH–H = 8.28 Hz, 4Hortho),7.0 (d, 3JH–H = 7.97 Hz, 2Hmeta), 7.1 (d, 3JH–H = 7.93 Hz, 2Hortho), 7.86 (s, 2 N–H), 7.92 (d, 3JH–H = 8.28 Hz, 4Hmeta); 13C NMR (DMSO-d6), δ (ppm): 20.28 (s, p-CH3), 113.02 (s), 119.94 (s), 126.32 (s), 129.86 (s), 133.57 (s), 136.07 (s), 148.76 (s), 155.03 (s); 31P NMR (DMSO-d6), δ (ppm): − 19.09 (s); IR (KBr), ν (cm− 1): 2855 (w, N-H), 1962 (w), 1589 (s), 1514 (vs, NO2), 1342 (s, NO2), 1309 (m), 1198 (m, P = O), 1121 (m), 990 (m, P-N), 919 (m), 855 (m), 824 (m), 735 (m), 672 (m), 499 (m), 493 (s).
N-(4-nitrophenyl)phosphoramidicacid-bis(4-methylphenyl)ester, 4
The sodium salt of 4-nitroaniline (1.6 g, 10 mmol) was added to a solution of phosphoramidodichloridicacid-4-methylphenylester (2.25 g, 10 mmol) in dry benzene (20 ml) under an argon atmosphere and the mixture was stirred at room temperature for 5 h. The yellow precipitate obtained was washed with dry chloroform (20 ml). The purity of the product was up to 98%. (yield 58%), m.p. = 165–168°C. Anal. Calc. for C20H19N2O5P: C, 60.3; H, 4.77; N, 7.03. Found: C, 60.1; H, 4.72; N, 6.988%. 1H NMR (DMSO-d6), δ (ppm): 2.24 (s, 6H, p-CH3), 6.61 (d, 3JH–H = 7.58 Hz, 2Hmeta), 6.75 (s, NH), 7.01 (d, 3JH–H = 7.3 Hz, 4Hmeta), 7.07 (d, 3JH–H = 7.36 Hz, 4Hortho), 7.91 (d, 3JH–H = 7.47 Hz, 2Hortho); 13C NMR (DMSO-d6), δ (ppm): 20.74 (s, p-CH3), 112.93 (s), 120.44 (s), 126.82 (s), 130.12 (s), 133.37 (s), 136.06 (s), 149.82 (s), 156.25 (s); 31P NMR (DMSO-d6), δ (ppm): − 17.64 (s); IR (KBr), ν (cm− 1): 3400 (w, N–H), 2920 (m), 1598 (w), 1499 (s, NO2), 1337 (m, NO2), 1255 (s), 1203 (vs, P = O), 1163 (m), 1080 (m), 1031 (s), 990 (m), 949 (s, P–N), 920 (s), 820 (m), 705 (w), 548 (w), 499 (m), 475 (m).
Kinetic experiments
All reagents for enzymatic experiments were from Fluka. Human acetylcholinesterase (hAChE) from SIGMA (50 units/785 μl) was diluted 25-fold in phosphate buffer (Na2HPO4/NaH2PO4, 70 mM, pH = 7.8).
The activity of the enzyme was measured at 25°C by a modified Ellman's method [Citation28]. The reaction mixture for determination of IC50 values consisted of: DTNB solution, 50 μl; Inhibitor, x μl (5 < x < 400); acetylthiocholine (ASCh) solution, 15 μl; phosphate buffer, (835-x) μl; AChE solution, 100 μl. The concentrations of substrate (s0), DTNB and inhibitors 1–4 were 1.35 × 10− 4, 10− 4 and 0.019, 0.014, 0.012 and 0.013 M, respectively, and the enzyme concentration under the assay conditions was 33.4 × 10− 9 M. Km and Vmax were obtained in the absence and presence of inhibitor from double-reciprocal Linweaver-Burk plots [Citation31]. A control solution containing all of above materials except inhibitor was used to determine the activity of the enzyme.
Results
Synthesis and spectral data
The interesting point in compounds 1 and 2 is their 3JPNCH = 0. Usually, in compounds with the general formula Me2NP(O)XY (X = Y = halide or amine or X = halide, Y = amine) a doublet with 3JPNCH of about 10–14 Hz is observed Citation27Citation32-34.
The 1H NMR spectra of compounds 3 and 4 showed that 7JP–H is zero. This phenomena was has also been observed in some phosphoramidates [Citation32,Citation33]. The reason for the vanishing 7JP–H coupling was described as the formation of partial multiple bonds between phosphorus and nitrogen in phosphoramidates [Citation33].
It has been demonstrated that the crystalline state of compound (MeO)2P(O)(p-NHC6H4NO2) exists as a network of linear hydrogen bonds [Citation35]. In compounds 3 and 4, we expected the formation of intermolecular hydrogen bonding. The existence of 4-nitroaniline as an electron withdrawing group in these molecules increased the acidity of the amine hydrogen. The highly acidity of these protons caused the exchange with the moisture of DMSO-d6 and appeared in the 1H NMR spectra as a singlet downfield peak. The 13C NMR spectra of molecules 3 and 4 did not show any coupling of carbons with phosphorus (nJP–C = 0). 31P NMR indicated that the phosphorus atom in compound 4 was more deshielded than in compound 3. This may be attributed to greater electronegativity of oxygen compared to nitrogen. The IR spectra showed a stronger νP = O and a weaker νPN in compound 4 than in compound 3.
Human acetylcholinesterase inhibition
Compounds 1 and 2: In experiments with these compounds, the activity of the enzyme showed negligible changes () and their inhibitory potency was negligible.
Figure 1 The plot of VI/V0 against log ([I] × 106) for inhibitors (Me2N)P(O)(Cl)(p-NHC6H4NO2), 1, and (Me2N)P(O)(p-NHC6H4NO2)2, 2. VI and V0 are the enzyme activity (OD min− 1) and [I] is the inhibitor concentration (μM).
![Figure 1 The plot of VI/V0 against log ([I] × 106) for inhibitors (Me2N)P(O)(Cl)(p-NHC6H4NO2), 1, and (Me2N)P(O)(p-NHC6H4NO2)2, 2. VI and V0 are the enzyme activity (OD min− 1) and [I] is the inhibitor concentration (μM).](/cms/asset/10f4246e-5e1d-45ae-a677-ae3a5920e43c/ienz_a_145652_f0001_b.gif)
Compounds 3 and 4: By plotting the VI/V0 (VI and V0 are the activity of the enzyme in the presence and absence of inhibitors, respectively) against log [I], where [I] is the inhibitor concentration, the IC50 values of compounds 3 and 4 were obtained as 0.143 and 0.581 mM, respectively (). Usually, a plot of remaining activity of the enzyme versus time for irreversible inhibiting phosphoramidates shows a linearly decrease from which the rate constant for inhibition of the enzyme may be obtained [Citation16,Citation36]. These plots for the inhibitors 3 and 4 indicated that the activity of the enzyme remains constant with time and demonstrating that the inhibition process is reversible.
Figure 2 The plot of VI/V0 against log ([I] × 106) for inhibitors: 1-(p-CH3C6H4O)P(O)(p-NHC6H4NO2)2, 3, 2-(p-CH3C6H4O)2P(O)(p-NHC6H4NO2), 4. VI and V0 are the enzyme activity (OD min− 1) and [I] is the inhibitor concentration (μM).
![Figure 2 The plot of VI/V0 against log ([I] × 106) for inhibitors: 1-(p-CH3C6H4O)P(O)(p-NHC6H4NO2)2, 3, 2-(p-CH3C6H4O)2P(O)(p-NHC6H4NO2), 4. VI and V0 are the enzyme activity (OD min− 1) and [I] is the inhibitor concentration (μM).](/cms/asset/83f3b210-1353-460b-bde0-2a03fc2d44a4/ienz_a_145652_f0002_b.gif)
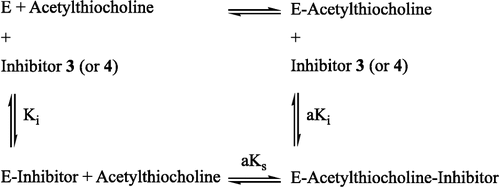
To further characterize the reversible process and to obtain the Km and Vmax values in the absence and in the presence of inhibitor, 1/V was plotted against 1/[S], where the V and [S] are the enzyme activity and substrate concentration, respectively. These Lineweaver-Burk plots [Citation31] indicated that compounds 3 and 4 were mixed inhibitors (Scheme ) and their values were 23.19 × 10− 5 and 10.79 × 10− 5 mol L− 1 and
values were 9.16 × 10− 6 and 9.97 × 10− 6 mol L− 1 min− 1, respectively. Also, the Km and Vmax values for enzyme were 108.20 × 10− 5 mol L− 1 and 10.03 × 10− 6 mol L− 1min− 1, respectively.
Scheme 1 The mechanism of human acetylcholinesterase inhibition by compounds 3 and 4.
Discussion
Usually, the inhibition of acetylcholinesterase by phosphoramidates has an irreversible mechanism Citation2-4Citation7Citation9–19 and only a few show a reversible inhibitory effect such as (Me2N)2P(O)(p-NHC6H4NO2), 5 [Citation27]. Compounds 1–4 were designed and synthesized to further investigate the influence of these phosphoramidates on human acetylcholinesterase.
summarizes the spectral and enzymatic data for compounds 1–6. Compound 1 with a chlorine atom as a suitable leaving group was expected to be an irreversible inhibitor, but to our surprise this compound gave no significant change in the activity of the enzyme hAChE. The 31P NMR spectra of this compound in D2O indicated two peak at − 10.59 and 0.29 ppm with relative ratio 1:1. The P–Cl bond in molecule 1 is labile and hydrolysis in phosphate buffer perhaps produces a P–OH bond. This may be the reason for the negligible inhibitory potency of molecule 1.
Table I. The spectral and enzymatic data for compounds 1–6.
The behavior of compound 2 with hAChE was similar to that of compound 1. Although the structure of compound 2 is similar to that of molecule 5 of Bollinger et al. [Citation27] and contains a (O)PN3 moeity, their inhibition powers are different against hAChE.
Debord et al. [Citation37] showed that the (O)PN3 moeity in aliphatic phosphoramides interacts with the hydrophilic zone of the catalytic site of butyrylcholinesterase. Jarv et al. indicated [Citation38] that in acetylcholinesterase three hydrophobic regions bind to hydrocarbon substituents and surround this zone. In phosphoramide 2, the existence of two nitroaniline groups decrease the lipophilicity of the aromatic ring. The hydrophobic constants obtained by Rekker et al. [Citation39] are the measure of lipophilicity of a molecular fragment which is 1.69 for C6H4 and − 0.059 for an aromatic nitro group. Therefore, it is likely that two nitroaniline groups will decrease the lipophilicity of the molecule to a great extent, and this probably leads to the negligible inhibitory effect of this compound.
Surprisingly, compounds 3 and 4 revealed different results. Although the only structural difference between molecules 2 and 3 is the replacement of a Me2N group by p-cresol, the interaction of molecule 3 with hAChE indicated that it was a reversible mixed inhibitor.
To further investigate the relationship between the inhibitory effects of this molecule with a p-cresol substituent, we used molecule 4 which has two p-cresol groups. The inhibitory potency of this molecule is drastically decreased in comparison to molecule 3. The IC50 values for compounds 3 and 4 are 0.143 and 0.581 mM, respectively. These values show that the inhibitory potency of compound 3 is greater than that of compound 4.
The noticeable point in molecules 3 and 4 with phenolic substituents is that the P–O–ph groups are stable against hydrolysis and show only a singlet peak in 31P NMR spectra in D2O.
Finally, it is concluded that changing the substituents X and Y in phosphoramidates (X)(Y)P(O) (p-NHC6H4NO2) leads to compounds with different inhibitory potency. The effect of 4-nitroaniline within this research area is interesting. Compounds 1 and 2 showed no inhibitory effect while compounds 3 and 4 possesing (O)PN2O and (O)PNO2 moieties, respectively, are reversible mixed inhibitors. Spectral data showed that in compounds with a (O)P(p-NHC6H4NO2) moiety all phosphorus-hydrogen and phosphorus-carbon couplings disappeared.
Acknowledgements
We would like to express our thanks to Dr. M. Naderimanesh in the Genetics department of Tarbiat Modares University and for his helpful discussions. Also, we thank Miss. Z. Zarandi and Mrs. Kh. Khorramishad for their laboratory assistance.
Related Research Data
References
- Pang Y-P, Kollmeyer TM, Hong F, Lee J-C, Hammond PI, Haugabouk SP, Brimijoin S. Biochem Biol 2003; 10: 491–502
- Elhanany E, Ordentlich A, Dgany O, Kaplan D, Segall Y, Barak R, Velan B, Shafferman A. Chem Res Toxicol 2001; 14: 912–918
- Barak D, Ordentlich A, Kaplan D, Barak R, Mizrahi D, Kronman C, Segall Y, Velan B, Shafferman A. Biochemistry 2000; 39: 1156–1161
- Mallender WD, Szegletes T, Rosenberry TL. Biochemistry 2000; 39: 7753–7763
- Wong L, Radic Z, Bruggemann RJM, Hosea N, Berman HA, Taylor P. Biochemistry 2000; 39: 5750–5757
- Viragh C, Kovach IM, Pannell L. Biochemistry 1999; 38: 9557–9561
- Thompson CM, Suarez AI, Rodriguez OP. Chem Res Toxicol 1996; 9: 1325–1332
- Ordentlich A, Kronman C, Barak D, Stein D, Ariel N, Marcus D, Velan B, Shafferman A. FEBS Lett 1993; Vol. 334(2)215–220, 13242
- Lassiter TL, Marshall RS, Jackson LC, Hunter DL, Vu JT, Padilla S. Toxicology 2003; 186: 241–253
- Guliy OI, Ignatov OV, Makarov OE, Ignatov VV. Biosens Bioelectron 2003; 18: 1005–1013
- Kaur K, Adediran SA, Lan Martin JK, Pratt RF. Biochemistry 2003; 42: 1529–1536
- Haux JE, Lockridge O, Casida JE. Chem Res Toxicol 2002; 15: 1527–1533
- Massiah MA, Viragh C, Reddy PM, Kovach IM, Johnson J, Rosenberry TL, Mildvan AS. Biochemistry 2001; 40: 5682–5690
- Kaplan D, Ordentlich A, Barak D, Ariel N, Kronman C, Velan B, Shafferman A. Biochemistry 2001; 40: 7433–7445
- Viragh C, Harris TK, Reddy PM, Massiah MA, Mildvan AS, Kovach IM. Biochemistry 2000; 39: 16200–16205
- Haux JE, Quistad GB, Casida JE. Chem Res Toxicol 2000; 13: 646–651
- Chemnitius J-M, Sadowski R, Winkel H, Zech R. Chem-Biol Inter 1999; 119-120: 183–192
- Ordentlich A, Barak D, Kronman C, Benschop HP, De Jong Leo PA, Ariel N, Barak R, Segall Y, Velan B, Shafferman A. Biochemistry 1999; 38: 3055–3066
- Scanlan TS, Reid RC. Chem Biol 1995; 2(2)71–75
- Schulze H, Vorlova S, Villatte F, Bachmann TT, Schmid RD. Biosens Bioelectron 2003; 18: 201–209
- Ashani Y, Bhattacharjee AK, Leader H, Saxena A, Doctor BP. Biochem Pharmacol 2003; 66: 191–202
- Paci A, Martens T, Royer J. Bioorgan Medi Chem Lett 2001; 11: 1347–1349
- Shulman-Roskes EM, Noe DA, Gamcsik MP, Marlow AL, Hilton J, Hausheer FH, Colvin OM, Ludeman SM. J Med Chem 1998; 41: 515
- Lougestay-Madec R, Florent J-C, Monneret C, Nematti F, Poupan M-F. Anti-Cancer Drug Des 1998; 13: 995
- le Roux C, Madro AM, Madro TA. J Org Chem 1995; 60: 3832
- Chan KK, Hong PS, Tutsch K, Trump DL. Cancer Res 1994; 54: 6421
- Bollinger J-C, Levy-Serpier J, Debord J, Penicaut B. J Enz Inhib 1990; 3: 211–217
- Ellman GL, Courtney KD, Andres V, Featherstone RM. Biochem Pharmacol 1961; 7: 88–95
- Kronblum N, Lurie AP. J Am Chem Soc 1959; 81: 2710
- Gholivand K, Shariatinia Z, Tadjarodi A. Main Group Chem 2005; in press.
- Copeland RA. A practical introduction to structure, mechanism and data analysis. Wiley-VCH, New York 2000
- Gholivand K, Mahmoudkhani AH, Khosravi M. Phosphorus Sulfur Silicon Relat Elem 1995; 106: 173–177
- Gholivand K, Ghadimi S, Naderimanesh H, Forouzanfar A. Mag Reson Chem 2001; 39: 684–688
- Bollinger J-C, Yvernault G, Yvernault T. Spectrochim Acta 1985; 41A: 399–406
- Du Plessis MP, Modro TA, Nassimbeni LR. Acta Cryst 1982; B38: 1504–1507
- Quistad GB, Zhang N, Sparks SE, Casida JE. Chem Res Toxicol 2000; 13: 652–657
- Debord J, Labadie M, Bollinger JC, Yvernault T. Phosphorus. Sulfur Silicon Relat Elem 1985; 22: 121–130
- Jarv J, Aaviksaar A, Godovikov N, Lobanov D. Biochem J 1977; 167: 823–825
- Rekker RF, De Kort HM. Eur J Med Chem 1979; 14: 479–488