Abstract
The antagonistic activities of derivatives of spiroethyl phenyl(substituted)piperazine at the 5-HT1A and adrenergic α1d receptors is quantitatively analyzed employing physicochemical and structural parameters. The derived correlation equation revealed that a substituent, other than 2-CH3 in the phenyl ring, having higher molar refraction, MR, and a substituent producing higher positive field effect at the 3-position are beneficial in increasing the binding affinity at the 5-HT1A receptor. In addition, a less hydrophobic substituent at the 4-position is also helpful in augmenting the binding affinity. The 5-R substituents which have higher MR values, however, elicit a detrimental effect. Two disubstituted compounds which are not present in the original data-set and have higher theoretical binding affinities are designed from the correlation equation. These compounds consisting of 2-OCH(CH3)2, 3-Cl and 2-C3H7, 3-Cl in the phenyl ring, have theoretical pKi values 10.57 and 10.12 respectively. For the adrenergic α1d receptor, a less bulky group at the 3-position with 5-Cl (or simply a 3-Cl) is advantageous in increasing the binding affinity. Likewise, a substituent exhibiting a less negative resonance effect at the 4-position and the substituent with low polarizability and showing more a negative resonance effect at the 5-position are suitable for enhancement of the binding affinity. The analysis provides the grounds for rationalizing substituent selection in designing better potency antagonists in the series.
Introduction
Three subtypes of the human α1 adrenergic receptor (AR) have been identified at the molecular level and are cloned as α1a, α1b, and α1d which correlate with the pharmacologically defined receptors α1A, α1B, and α1D [Citation1,Citation2]. The α1A subtype is mainly present in rat submaxillary gland, human liver and different tissues namely, prostatic vas deferens, rabbit prostate and prostatic urethra Citation3-6. The α1B adrenoceptor is concerned with rat liver and spleen, on the other hand, the contractility of rat aorta is mediated through the α1D subtype. However, the α1d mRNA is shown to be the dominant α1 subtype present in the human bladder detrusor [Citation7]. In addition, it was reported Citation8-10 that the α1D adrenergic receptor is involved in detrusor instability, secondary to bladder outlet obstruction and mediates constriction of rat skeletal muscles arterioles and protein synthesis by arterial smooth muscles. Therefore, the selective α1D antagonists could be very useful in the treatment of urinary incontinence, vasoconstriction and atherosclerosis without affecting normal blood pressure. In this respect, among others, BMY 7378 first described in 1983 [Citation11] was later shown to be a selective α1d antagonist [Citation12]. However, the selectivity profile of this compound is limited by high affinity for the 5-HT1A serotonergic receptor.
More recently, a synthetic study was performed [Citation13] with a view to identifing highly selective antagonists at the adrenergic receptor. These compounds were tested in radioreceptor binding assays and the most significant compounds were further investigated for functional activity at the three α1 adrenoceptor subtypes and 5-HT1A serotonergic receptor. The initial structure-activity relationship (SAR) study on these compounds was, however, directed only to alteration of the substituents at different positions of the structure but no rationale has been provided to reduce the trial-and-error factors. Hence, a quantitative SAR (QSAR) on these analogues was conducted since QSAR not only provides the rationale for drug design but also illuminates their possible mechanism of action at the molecular level.
Materials and methods
The QSAR analysis was made on a reported series of compounds [Citation13], the derivatives of spiroethyl phenyl (substituted) piperazine, having the general structure shown in . These compounds along with their activity values for 5-HT1A and α1d receptor subtype are compiled in . The most appropriate physicochemical, structural and indicator variables are also listed in this Table. Amongst them, the physicochemical parameters such as molar refraction, MR (scaled to 0.1), Field, F and resonance, R are taken from the literature [Citation14] whereas the structural parameter, the van der Waals volume for a given substituent was calculated according to the method discussed in one of our earlier publications [Citation15] and the usefulness of this structural parameter has already previously been established Citation16-21. Additionally, indicator variables are also employed to reflect upon some special structural features of a compound. The numerals, subscripted or within parentheses following these variables are indicative of the varying positions of title compounds. The affinities estimated were derived from displacement of [3H]prazosin for the α1 adrenoceptor and [3H]8-hydroxy-2-(di-n-propylamino)tetraline (3H-8-OH-DPAT) for the 5-HT1A receptor. For the present work, the reported affinity constants Ki for 5-HT1A and α1d receptors are expressed as pKi on a molar scale. The multiple regression analysis (MRA), employing the method of least squares, is used to derive significant correlation equations for further discussion. In addition, the final QSAR equations were also subjected to a validation test [Citation22] by the leave-one-out (LOO) method. This method generates a number of modified data sets by taking away one compound from the parent data set in such a way that each observation is taken away once and once only. Then one model is developed for each reduced data set and the response values of the deleted observations are predicted from the model. The squared differences between predicted and actual values are added to give the predictive residual sum of squares (PRESS). In this way, PRESS will contain one contribution from each observation. The cross-validated, q2 value may further be calculated as (SSY– PRESS)/SSY, where SSY denotes the variance of the observed activities of molecules around the mean value. For a reasonable QSAR model, q2 should be greater than 0.6, and a value of this statistical index greater than 0.9 indicates an excellent model.
Figure 1 Structure of spiroethyl phenyl(substituted)piperazine derivatives.
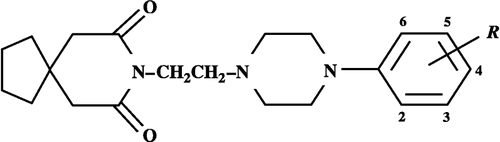
Table I. QSAR parameters and antagonistic activities of substituted phenylpiperazine derivatives at the 5-HT1A and α1d receptors (see for structure).
Results and discussion
lists the compounds where the alteration in substituents occurred at the phenyl ring linked to the piperazine ring. In order to account for effects produced by such substituents, a large number of descriptors related to hydrophobic, electronic and steric interactions were initially examined for the five varying positions of the phenyl ring in various possible permutations. The selected parameters for various substituents for each of these positions were hydrophobicity, π, hydrogen-bond donor, HD, electronic (meta and para), σ, field, F, resonance, R, dipole moment, μ, Taft's steric, ES, molar refraction, MR, molecular weight, MW and van der Waals volume, Vw. This resulted into a large number of QSAR equations, which were then subjected to different statistical tests. The correlation equations, which returned the highest correlation coefficient, r and F-statistic and lowest standard deviation, s were finally retained for further discussion. The highest significant correlation as shown by equation (1) is finally obtained. The steps of its development are described through the correlation i-iv in .
Table II. Stepwise development of equation (1) pKi = a0 + a1MR2 + a2 F3 + a3π4 + a4MR5 + a5I2 for n=34.
As given above, n is the number of data points, F-statistic is the F-ratio between the variances of calculated and observed activities, and the ± data within the parentheses are the 90% confidence intervals. From above equation it appears that the substituents at the 4-positions are engaged in hydrophobic interaction while those at the 3-position are involved in electronic interaction. Furthermore, the MR variable, accounting for molecular bulk and/or polarizability, appears to be the important aspect for the substituents at the 2- and 5-positions. The arbitrarily chosen indicator variable, I2, in addition, accounts for a methyl substituent at the 2-position. Its value, either 1 or 0 in that order, indicates the presence or absence of a 2-CH3 in the phenyl ring. The statistical parameters, obtained for equation (1), do not indicate significant results as the r2 value accounts for 75% of the variance and q2 is nearer to a specified significant level. However, the F-value remained significant at 99% [F5,28(0.01) = 3.754]. These observations reflect upon the parametric requirements for the subtituents in a compound that leads to binding affinity at the 5-HT1A serotonergic receptor. In order to improve upon the significance levels of equation (1), all data points in , were further analyzed for their deviation from a regular trend. Compounds that show large difference between observed and calculated pKi values are underlined for this abnormality and are treated as ‘outliers’. Compounds 9 and 17 are such congeners. Compound 9 having a 2-OH substituent seems to involved in hydrogen bonding with some site on to the receptor and elicits a higher observed binding affinity compared to the calculated value ( = 7.68) using the QSAR equation (2). Similarly the 2-COOC2H5 substituent in compound 17 appears to undergo hydrolysis prior to reaching its receptor site and reveals lesser pKi value than the calculated one ( = 9.55) obtained from the model equation. Paying no attention to these two compounds, QSAR reveals correlation equation (2)
Now both the r- and F-values were increased to account for 82% (r2 = 0.821) of variance in the observed activities and 99% level of significance [F5,26(0.01) = 3.818]. Also, the s-value and 90% confidence intervals ( ± data within parentheses) associated with regression coefficients were significantly lowered. Additionally, the higher value obtained for q2 expressed a reasonable QSAR model. That the variables used in deriving equation (2) had no mutual correlation is shown in . The calculated activity values, using this equation and listed in , are in close agreement with the observed ones. The predicted activity values from the data set of equation (2) and various model equations, discussed earlier, were also listed in this table for comparison sake. From equation (2), it appeared that a substituent, other than a methyl, present at 2-position and having a higher value of MR is advantageous to improve the pKi value pertaining to the 5-HT1A receptor. Similarly a substituent producing a higher positive field effect at the 3-position and a less hydrophobic substituent at the 4-position are also favorable to increase the binding affinity. The substituents which are present at the 5-position and have higher MR values, on the other hand, cause a detrimental effect. These guidelines may be used to design congeners which are more active than the compounds reported in the original data-set. Two such disubstituted compounds that possess 2-OCH(CH3)2, 3-Cl and 2-C3H7, 3-Cl in the phenyl ring have theoretical pKi values of 10.57 and 10.12 respectively and may, therefore, be explored in the future.
As mentioned earlier, compounds which are selective antagonists at the α1d adrenergic receptor could be very useful in the treatment of diseases such as urinary incontinence, vasoconstriction and atherosclerosis, with no effects on blood pressure. It is, therefore, pertinent to establish the quantitative relationship between binding affinity for the α1d and the quantifying parameters. Employing data from in MRA has revealed, through successive steps (), correlation equation (3)
Table III. Stepwise development of equation (3) pKi = b0 + b1Vw(3) + b2R4 + b3MR5 + b4R5 + b5I3,5 for n=37.
This equation analyzes the importance of 3-, 4- and 5-R substituents while the substituents at the 2-position make no contribution to pKi(α1d). The indicator variable I3,5 is selected to highlight the presence of a Cl group at the meta-position (3 or 5) of the phenyl ring (). Thus, for the presence of a 3-Cl or a 5-Cl, the value considered for I3,5 is 1 and for its absence the value assigned to this variable is 0. Compound 12, having 2,4-(CH3)2 substituents in the phenyl ring, has unusual behavior and, at present, there is no appropriate explaination. This compound, in addition to previously considered ‘outlier’ compounds 9 and 17, is ignored to yield a more significant correlation equation (4)
Table IV. The intercorrelation matrixa amongst the independent variables of equation (2).
All the statistical parameters, including 90% confidence intervals, of this equation have significantly improved over that of equation (3). The r value now accounts for 82% of the variance and the s value is lowered. In addition, the F value remained significant at 99% level, and the q2 index, explaining a satisfactory statistical model, are both increased. The calculated pKi values, using equation (4), and predicted pKi values, using the LOO method, listed in , are in close agreement with the observed ones. The required orthogonality conditions amongst the independent variables of this equation are evident in . From equation (4), it appears that a less bulky group at the 3-position, while a Cl group at the 5-position (or simply a 3-Cl) are advantageous in increasing the activity of a compound. Similarly a substituent, exhibiting less polarizability and a more negative resonance effect at the 5-position, leads to higher antagonistic activity at α1d receptor. Also, a substituent present at the 4-position and showing a less negative resonance effect is beneficial.
Table V. The intercorrelation matrixa amongst the independent variables of equation (4).
Equations (2) and (4) were further subjected to an external validation method (EVM). In this method, a few compounds were considered in the test set and were left out to derive a correlation equation on the remainder. The equation was then used to predict the activities of compounds in the test set. In this way, several equations were obtained and are listed in corresponding to the compounds left out of the test set. The predicted activities for these compounds are given in for comparison. These activities were found in close agreement with the observed ones. The conclusions deduced from equations (2) and (4) may be used as guidelines to obtain more potency antagonists in the further synthesis of similar compounds.
Table VI. QSAR model equations using external validation method.
Acknowledgements
We are grateful to the Department of Chemistry and the College Administration for providing the facilities to complete this work.
References
- Hancock AA. Drug Dev Res 1996; 39: 54–107
- Leonardi A, Testa R, Motta G, De Benedetti PG, Hieble JP, Giardinà D. Perspectives in receptor research. Elsevier Science B.V., Amsterdam 1996; 135–152
- Michel AD, Loury DN, Whiting RL. Br J Pharmacol 1989; 98: 883–889
- García-Sáinz JA, Romero-Avila Ma T, Tórres-Márquez Ma E. Eur J Pharmacol, Mol Pharmacol Sect 1995; 289: 81–86
- Eltze M, Boer R, Sanders KH, Kolassa N. Eur J Pharmacol 1991; 202: 33–44
- Testa R, Guarneri L, Ibba M, Strada G, Poggesi E, Taddei C, Simonazzi I, Leonardi A. Eur J Pharmacol 1993; 249: 307–315
- Malloy BJ, Price DT, Price RR, Bienstock AM, Dole MK, Funk BL, Donatucci CF, Schwinn DA. J Urol 1998; 160: 937–943
- Nagarathnam D, Wetzel JM, Miao SW, Marzabadi MR, Chiu G, Wong WC, Hong X, Fang J, Forray C, Branchek TA, Heydorn WE, Chang RSL, Broten T, Schorn TW, Gluchowski C. J Med Chem 1998; 41: 5320–5333
- Leech CJ, Faber JE. Am J Physiol 1996; 270: H710–H722
- Xin X, Yang N, Eckhart AD, Faber JE. Mol Pharmacol 1997; 51: 764–775
- Yevich JP, Temple DL, Jr, New JS, Taylor DP, Riblet LA. J Med Chem 1983; 26: 194–203
- Goetz AS, King HK, Ward SD, True TA, Rimele TJ, Saussy DL, Jr. Eur J Pharmacol 1995; 272: R5–R6
- Leonardi A, Barlocco D, Montesano F, Cignarella G, Motta G, Testa R, Poggesi E, Seeber M, De Benedetti PG, Fanelli F. J Med Chem 2004; 47: 1900–1918
- Hansch C, Leo A. Substituents constants for correlation analysis in chemistry and biology. John Wiley, New York 1979
- Gupta SP, Bhatnagar RP, Singh P. Res Commun Chem Pathol Pharmacol 1979; 25: 111–119
- Bondi A. J Phys Chem 1964; 68: 441–451
- Moriguchi I, Kanada Y, Komatsu K. Chem Pharm Bull 1976; 24: 1799–1806
- Kumar K, Bindal MC, Singh P, Gupta SP. Int J Quant Chem 1981; 20: 123–129
- Gupta SP, Prabhakar YS. J Sci Ind Res 1985; 44: 189–198
- Singh P. Arzneim-Forsch 1986; 36: 1437–1439
- Singh P, Sharma RC. Quant Struct-Act Relat 1990; 9: 29–32
- Wold S. Quant Struct-Act Relat 1991; 10: 191–193