Abstract
Glutathione-S-transferase(s) (E.C.2.5.1.18, GSTs) have been investigated in parasitic protozoans with respect to their biochemistry and they have been identified as potential vaccine candidates in protozoan parasites and as a target in the synthesis of new antiparasitic agents. In a search towards the identification of novel biochemical targets for antimalarial drug design, the area of Plasmodium glutathione metabolism provides a number of promising chemotherapeutic targets. GST activity was determined in various subcellular fractions of malarial parasites Plasmodium yoelii and was found to be localized mainly in the cytosolic fraction (specific activity, c. 0.058 ± 0.016 μmol/min/mg protein). Hemin, a known inhibitor of mammalian GST(s), maximally inhibited this enzyme from P. yoelii to nearly 86%. In a search towards synthetic modulators of malarial GST(s), 575 compounds belonging to various chemical classes were screened for their effect on crude GST from P. yoelii and 92 compounds belonging to various chemical classes were studied on recombinant GST from P. falciparum. Among all the compounds screened, 83 compounds inhibited/stimulated the enzyme from P. yoelii/P. falciparum to the extent of 40% or more.
Introduction
Malaria is one of the most important widespread diseases in the world, with about 300–500 million clinical cases and 1.7–2.5 million deaths every year. Plasmodium falciparum is the causative agent of the most severe form of malaria and is a disease prevalent in underdeveloped countries. Therefore inexpensive and safe drugs, vaccines and insecticides are needed to combat this disease. The rapidly developing resistance to existing drugs used for prophylaxis and treatment makes the identification of novel drug targets necessary [Citation1]. Important biochemical pathways in the Plasmodium life cycle (particularly blood stage) are the main targets of the present day antimalarials that can be exploited further as promising targets for antimalarial drug development. One such promising target is the area of glutathione metabolism in Plasmodium parasites.
Glutathione (GSH), a tripeptide consisting of glutamic acid, cysteine and glycine is known to protect malarial parasites from oxidative stress and attack by reactive oxygen species (ROS) as well as electrophilic toxic compounds. Its role in modulation of drug resistance has also been currently established [Citation2]. Glutathione is synthesized both in eukaryotes and prokaryotes by sequential action of γ-glutamyl cysteine synthetase (γ -GCase) and GSH synthetase [Citation3,Citation4]. Pharmacological depletion of GSH in the cells is known to be useful in the development of anticancer drugs and the increased vulnerability of the infected cells due to internally generated oxidants is useful in the development of antiparasitic agents Citation5-7. Many inhibitors of enzymes involved in GSH metabolism are known to be associated with antiparasitic activities and are potential chemotherapeutic targets. One such vulnerable target is the enzyme glutathione-S-transferase.
The Glutathione-S-transferase(s); GSTs [EC 2.5.1.18] represent a large family of enzymes found in organisms ranging from prokaryotes to mammals. They detoxify both endogenous and exogenous xenobiotic compounds via the nucleophilic addition of GSH to a large variety of electrophilic substrates. Besides catalyzing conjugation reactions, GST(s) possess selenium-independent glutathione peroxidase (GPx) activity towards organic hydroperoxides. This activity is protective because it prevents organic hydroperoxides of phospholipids, fatty acids and DNA becoming engaged in free radical propagation reactions ultimately leading to the destruction of macromolecules during oxidative stress [Citation8]. GST(s) are involved in the sequestering and transport of exogenous hydrophobic compounds such as pesticides, herbicides and antibiotics; furthermore, they bind a large variety of endogenous compounds such as steroids, bilirubin, bile acids and ferriprotoporphyrin IX with varying affinities. This ‘ligandin’ activity may sequester toxic substances, effectively decreasing their concentration in the cell. The possible dual function of the GST(s), i.e. catalytic and storage/transport is interesting, since the non-substrate ligands can also act as inhibitors of enzymatic activity Citation9-14. The roles of the GST(s) in the regulation of stress response, detoxification of lipid peroxidation products, the sequestration of potentially toxic compounds and furthermore in drug resistance [Citation15] are especially important considerations within a parasitic context.
GST activity has been detected in rodent (P. berghei, P. yoelii), simian (P. knowlesi) and human (P. falciparum) malarial parasites. In chloroquine-resistant parasites GST activity significantly increases compared to sensitive strains [Citation16]. Furthermore, the increase in enzyme activity is directly related to drug pressure of resistant P. berghei [Citation17].GST activity in P. knowlesi cell extracts has been shown to be inhibited by micromolar concentrations of chloroquine and it has been suggested that this inhibition might potentiate the accumulation of chloroquine metabolites in the parasite, thus enhancing its antiparasitic effects [Citation17]. The antimalarial drug chloroquine is, however, also considered as an inducer of oxidant damage to the parasitized red cell due to its role in preventing heme polymerization. Therefore, the reduction in hemozoin content is a feature of chloroquine-resistant parasites. As an alternative to polymerization, heme degradation based on the interaction with GSH has been suggested to represent an important intraparasitic principle for detoxifying heme [Citation18,Citation19].This hypothesis is supported by the fact that chloroquine-resistant parasites exhibit increased glutathione concentrations Citation16-19. Interestingly, GST activity in parasite extracts from P. knowlesi has been shown to be inhibited by micromolar concentrations of hemin [Citation17] and an inverse relationship between GST activity and hemozoin content has been reported for chloroquine-resistant and chloroquine-sensitive P. berghei Citation17-19.
The identification of a gene encoding a protein with a sequence identity of upto 37% with known GST(s) on chromosome 14 of malarial parasite P. falciparum has paved the way for the characterization of a potential drug target for the effective chemotherapy of malaria [Citation20,Citation21]. Plasmodium species require an efficient antioxidant defence mechanism, since they have to survive in a pro-oxidant environment, the red blood cell. Even though they appear to enhance oxidative stress within the infected red blood cell, Plasmodium species are also known to be sensitive to oxidant killing [Citation22].
Therefore antioxidant enzymes have been potential targets for the development of antimalarial drugs. GST(s) play a central role in detoxification systems since the resulting S-conjugated products are often less toxic and more easily excreted than the original compounds. In parasites, this detoxification function may be particularly utilized, in an adapted manner, to aid in surviving host-induced stress. Natural substrates of parasitic GST(s) include the cytotoxic products of lipid peroxidation. Damage due to these products, in the absence of GST, would leave the parasites more vulnerable. GST(s) are therefore attractive targets for chemo- and immunoprophylaxis. The present study is an attempt towards the search for effective synthetic modulators of malarial GST(s) and lists the effect of 83 synthetic compounds on this enzyme from P. yoelii and 41 synthetic compounds on recombinant GST from P. falciparum.
Materials and methods
Chemistry
Synthesis of diglycosylated diaminoalkanes; (DGDAs) (1) ()
Figure 1 Core structure of DGDAs.
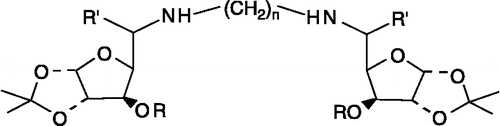
DGDAs were synthesized starting from sugar-derived aldehydes using reductive amination technique already reported by us [Citation23].
Synthesis of glycosyl amino acids; GAAs (2–3) and glycosyl amino ester; GAE (4)
These were prepared following earlier methods Citation24-26.
Synthesis of glycosyl hydroxamates (5–12)
This class of compounds was prepared from respective glycosyl amino esters through glycosyl amino acids [Citation24] by reaction of hydroxylamine hydrochloride in presence of dicyclohexyl carbodiimide (DCC) and triethyl amine in anhydrous acetonitrile following the procedure reported earlier [Citation26].
Solid phase synthesis of compound 15L: 3-[{N-(3-pyridyl acetyl)-isoleucinamid-1-yl}]-3-[(1R,2R,3S,4R)-3-O-benzyl-1,2-O-isopropylidene-1,4-pentafuranos-4-yl]-propanamide
This compound was synthesized as a library on Sieber amide resin as per an earlier protocol [27]. Sieber amide resin was placed in a solid phase reaction vessel and treated with appropriate amount of dimethyl formamide (DMF) at least twice for 5 min at room temperature under N2 agitation. The resin was further swelled in dichloromethane (DCM), twice for 5 min at room temperature under N2 agitation.
F-moc-3-amino-[(1R, 2R, 3S, 4R)-3-O-benzyl-1,2-O-isopropylidene-1,4-tetrahydrofuranos-4-yl)]-propionic acid was then added to the reaction vessel followed by addition of diisopropyl carbodiimide (DIC), dimethyl amino pyridine (DMAP) and hydroxy benzotriazole (HOBt) in DMF and the reaction mixture was agitated with a slow stream of N2 gas for 12 h at room temperature. The solvents and excess of reagents were removed by suction and the process of loading of the above compound was repeated in order to gain complete loading. The latter was washed with DMF, CH3OH and DCM and dried in vacuo to give amide resin. This resin was treated with 20% piperidine: DMF twice for 5 min and 25 min respectively. After the completion of the reaction, the resin was washed with DMF, CH3OH and DCM and dried. To this 3-amino- [(1R, 2R, 3S, 4R)-3-O-benzyl-1,2-O-isopropylidene-1,4-tetrahydrofuranos-4-yl)]-propagandize resin, dry DMF and F-moc isoleucine amino acid were added followed by addition of HOBt, diisopropyl ethylamine (DIPEA), N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl) uronium tetrafluoroborate (TBTU) and DIC in DMF with N2 agitation for 4 h at room temperature. The solvents and excess of reagents were removed by suction and the resin-loaded compound was washed with DMF, CH3OH and DCM. This resin-bound compound was further treated with 20% piperidine: DMF twice for 5 min and 25 min respectively for the deprotection of resin-bound compound.
To the resin-bound glycosyl carboxamine in dry DMF, 3-pyridyl acetic acid was added followed by addition of HOBt, DIPEA, TBTU and DIC in DMF with N2 agitation for 4 h at room temperature. After this, the solvents and excess reagents were purged and the resin-bound compound was washed with DMF, CH3OH and DCM. The solid phase reaction vessel was cooled up to 15°C by passing liquid N2 through the block, cleavage vials containing 2% trifluoroacetic acid (TFA) were placed below the reaction vessel and the compound was obtained in DCM. DCM was evaporated under N2 and the compound was lyophilized by dissolving in t-butanol/water (4:1) and freeze drying to give the above compound. The compound was characterized using FABMS, HPLC and 1H NMR spectroscopy. The glycosyl carboxamide was obtained in 80% purity, and formation of product was evidence by its FABMS 569 (M+H]+and it was identical to the product obtained earlier [Citation27].
Solid phase synthesis of compound 16L: 3-[(N-(Nicotinyl)-leucinamid-1-yl)]-3-[(1R,2R,3S,4R)-3-O-benzyl-1,2-O-isopropylidene-1,4-pentafuranos-4-yl]-propanamide
This compound was obtained by the procedure described above by replacing F-moc isoleucine amino acid with Fmoc leucine and 3-pyridyl acetic acid with nicotinic acid. FABMS: 555[M+H]+
Solid phase synthesis of compound 17L (glycosyl peptide): 3-[N-{(Phenylacetyl)-valinamid-1-yl}]-3-[(1R,2R,3S,4R)-3-O-benzyl-1,2-O-isopropylidene-1,4-pentafuranos-4-yl]-propanamide
This compound was also obtained as described above by replacing F-moc isoleucine amino acid with F-moc valine and 3-pyridyl acetic acid with phenyl acetic acid. FABMS: 554 [M+H]+
Solid phase synthesis of compound 18L: 3-[N-{(Pyridylacetyl)-nor-valinamid-1-yl}]-3-[(1R,2R,3S,4R)-3-O-benzyl-1,2-O-isopropylidene-1,4-pentafuranos-4-yl]-propanamide
This compound was also obtained by the procedure described for compound 15L by using F-moc norvaline and nicotinic acid in place of F-moc isoleucine and 3-pyridyl acetic acid respectively. FABMS: 569[M+H]+
Synthesis of N-protected glycopeptide (19)
The glycosyl amino ester [Citation24] was reacted with N-F-moc proline using standard amino acid coupling reagents (DCC, HOBt, DMAP in anhydrous DCM) to get N-protected glycosylpeptides. The Fmoc group was deprotected via 30% piperidine in acetonitrile to afford N-protected glycopeptide (19). The method has been patented [Citation28a,b].
Synthesis of glycosyl thiourea (20) and glycosyl ureas (21–24)
Sugar-derived N-substituted and N-unsubstituted amino esters were reacted with substituted phenyl isocyanate/isothiocyanate in anhydrous DCM to give N-glycosyl N-substituted urea/thiourea respectively and the method has been patented both on solid phase as well as by conventional method of synthesis and the chemistry of nucleosides has been published by us Citation28aCitationbCitation30-32
Solid Phase synthesis of glycosyl ureas (25L–31L) ()
Figure 2 Core structure of glycoconjugates (3–31L).
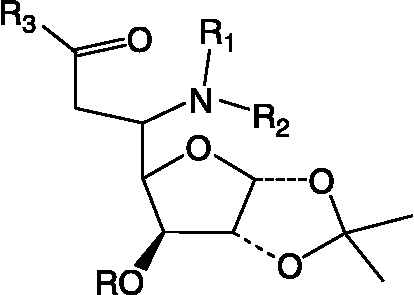
These compounds were synthesized as a Library on Sieber amide resin in an Advanced Chemtech Robotic synthesizer in vacuo by the method reported earlier by us [Citation33,Citation34].
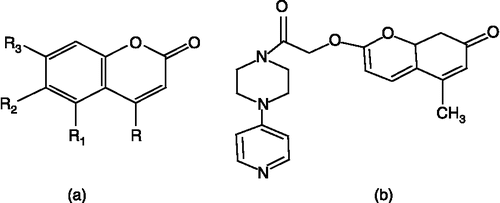
Synthesis of coumarin derivatives (33) ()
The Compound 33 was synthesized as reported earlier [Citation35,Citation36].
Figure 3 Core structure of sugar (32).
Figure 4 (a) Core structure of coumarins. (b) Structure of compound 33 (coumarin).
Synthesis of glycosylated hydantoin (34) ()
Figure 5 Structure of glycosyl hydantoin (34); R = CH2Ph, R1 = Leucine, R2 = 3-Acetyl Ph.
The synthesis of this compound has been reported earlier [Citation34].
Synthesis of nucleosides (35–42) by conventional method and C-nucleosides (43L–57L) on solid phase ()
Figure 6 Core structure of nucleosides.
A combinatorial library of C-Nucleosides was synthesized as reported in our previous paper [Citation37].
Synthesis of substituted isoxazole derivatives (58–79) ()
Figure 7 Core Structure of isoxazole derivatives.
This series of compounds was synthesized by the methods previously reported [Citation38,Citation39,Citation40a].
Synthesis of 2-hydroxymethyl-acrylic acid methyl ester (80)
This compound was synthesized by the method previously reported [Citation40a].
Synthesis of substituted 2-(hydroxymethyl)-acrylonitrile derivatives (81–82)
These compounds were synthesized by the method previously reported [Citation40b]. Analogous compounds synthesized have been reported to possess antimalarial activity [Citation40c].
Synthesis of 2-Methyl-5-phenyl-pyrrolidine-2, 3, 4-tricarboxylic acid-3, 4-dimethyl ester (83)
This compound was synthesized by the method previously reported [Citation41].
Biological
Materials
Reduced glutathione (GSH), trisodium citrate, saponin, hemin and bovine serum albumin (BSA) were purchased from Sigma Chemical Co., USA. All reagents used in PCR, cloning and expression were purchased from Sigma Chemical Co., USA. 1-chloro-2,4-dinitrobenzene (CDNB) was purchased from Spectrochem Pvt. Ltd. Mumbai, India. Folin & Ciocalteu's phenol reagent was purchased from Sisco Research Laboratories, Mumbai, India. Giemsa stain was from Qualigens India Ltd. and phosphate buffered saline (PBS) was from Himedia laboratories Pvt. Ltd. Mumbai, India and fibrous cellulose powder (CF11) was from Whatman International Ltd. (England). All other chemicals used were of analytical grade.
Table I. Structural details of N1, Nn- bis-Xylofuranosylated diaminoalkane (1).
Table II. Structures of glycosyl amino acid (2), glycosyl hydroxamate (12), glycoconjugates (13, 14), glycosyl peptides (15L–18L), glycosyl thiourea (20), glycosyl urea (31L).
Table III. Structual details of glycosyl amino acid (3) (), glycosyl amino ester (4) (), glycosyl hydroxamic acids (5–11) (), N-protected glycopeptide (19) (), glycosyl ureas (21–30L) () and sugar (32) synthesized ().
Table IV. Structural detail of C-Nucleosides (35–57L) (see ) synthesized by a conventional method & on solid phase.
Table V. Structures of isoxazole derivatives (58–79).
Table VI. Structures of 2-hydroxymethylacrylic acid methyl ester (80), substituted hydroxymethyl acrylonitrile derivatives (81 and 82) and pyrrolidine derivative (83).
P. yoelii nigeriensis infection in Swiss albino mice
In vivo maintenance of rodent strain of malarial parasites P. yoelii was carried out using Swiss albino mice (Mus musculus, out-bred strain). The laboratory isolates of P. yoelii were routinely maintained by serial blood passages in mice. Blood from infected mice was collected through cardiac puncture. The course of parasitemia was monitored daily by microscopic examination of Giemsa-stained thin blood smears.
Isolation of malarial parasites
Blood of heavily P. yoelii infected albino mice was collected in sterile citrate buffer. White cells, platelets and plasma were removed by the passage of infected blood through a CF-11 column [Citation42]. The malarial parasites were isolated from the erythrocytes by lysis of infected erythrocytes with 0.15% saponin and the freed parasites were collected as a pellet after centrifugation at 10,000 × g for 10 min [Citation43]. The parasites were washed several times with cold PBS (pH 7.4) and stored at − 80˚C until further use.
Preparation of GST from malarial parasites P. yoelii nigeriensis
For preparation of malarial GST, mixed stages of malarial parasites P. yoelii isolated from the blood of P. yoelii infected Swiss albino mice in the previous step were thoroughly washed with PBS and then homogenized with a Potter Elvehjem homogenizer in a minimum volume of PBS. The homogenate was centrifuged at 1000 × g for 15 min, 10 000 × g for 30 min and subsequently at 100 000 × g for 60 min to obtain mitochondrial, post mitochondrial, cytosolic and microsomal fractions respectively. All the fractions obtained were analyzed for the activity of GST.
GST activity determination
GST activity in dialyzed cytosolic fractions was determined spectrophotometrically at 340 nm with the standard substrate CDNB and co-substrate GSH [Citation44]. All reactions were corrected for non-enzymatic conjugation, with reaction mixtures without enzyme serving as controls.
Under standard assay conditions, the reaction mixture contained 100 mM phosphate, pH 6.5, 1.0 mM CDNB in 20 μL ethanol, 1.0 mM GSH and enzyme protein unless stated otherwise. A unit of enzyme activity was expressed as the amount that catalyzes the formation of 1 μmol S-2, 4-dinitrophenyl-GSH adduct per minute, using a molar extinction coefficient of 9.6 mM− 1 cm− 1 for CDNB. Protein was estimated by the method of Lowry et al. [Citation45] using BSA as standard.
Expression and purification of recombinant P. falciparum GST (rPf-GST1)
For further validation of parasitic GST(s) as potential drug targets, the recombinant protein from P. falciparum was cloned, expressed and purified according to the method reported earlier [Citation21]. This work was done in collaboration with the Department of Biochemical Parasitology, Bernhard Nocht Institute for Tropical Medicine (BNI), Hamburg, Germany.
Enzyme inhibition studies
Effect of synthetic compounds on GST activity of P. yoelii and recombinant GST from P. falciparum was studied by adding them directly to the assay systems at 100 μM concentration 10 min prior to addition of the substrate. Reaction in all cases was initiated by the addition of 1.0 mM CDNB and the absorbance at 340 nm was monitored for 5 min at 30 s intervals. The percentage inhibition/stimulation of the enzyme activity by compounds was calculated by comparing with controls. For finding the IC50 values, the dose-dependent effect of synthetic compounds on recombinant GST from P. falciparum was studied by incubating the enzyme with varying concentrations (50–200 μM) of the synthetic compound for 10 min at room temperature.
In order to study the dose-dependent effect and Ki of the standard mammalian GST(s) inhibitor hemin on GST activity from cytosolic fraction of P. yoelii and on recombinant GST from P. falciparum, the enzyme was incubated with varying concentrations of inhibitor for 10 min at room temperature in the presence of 100 mM potassium phosphate buffer and 1.0 mM GSH. For determining the type of inhibition, the enzyme was incubated with varying concentrations of inhibitor at different substrate (GSH) concentrations. The test compounds as well as the standard inhibitor hemin were dissolved in 100% DMSO but the final concentration in the maintenance medium/assay system for the enzyme activity determination was always kept below 0.1%.
Assessment of antimalarial activity of test compounds
In vitro antimalarial efficacy of the respective enzyme modulators was determined against P. falciparum in culture at 100 nM concentration.
Results
GST(s) of P. yoelii/P. falciparum
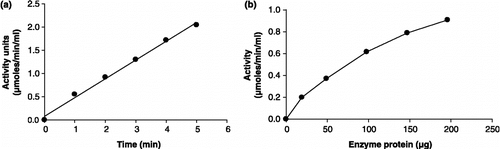
GST activity in various subcellular fractions of P. yoelii homogenate is depicted in . GST activity in malarial parasites was found to be mainly associated with the cytosolic fraction. Cytosolic P. yoelii GST reaction was found to be linear with respect to time (30 s–5 min) and the amount of enzyme protein (4.6–30.4 μg) (,b). The recombinant enzyme was purified using affinity chromatography followed by gel filtration. ,b show that the reaction of recombinant GST from P. falciparum was fairly linear with respect to time (30 s–5 min) as well as the amount of enzyme protein (19.6–98 μg).
Table VII. Subcellular distribution of GST activity in P. yoelii.
Figure 8 Linearity of P. yoelii cytosolic GST assay with respect to (a) time and (b) enzyme protein.
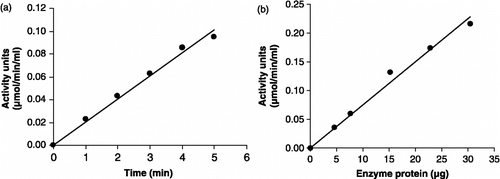
Figure 9 Linearity of P. falciparum recombinant GST assay with respect to (a) time and (b) enzyme protein.
Inhibition studies
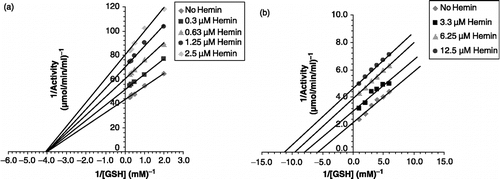
As reported earlier, hemin inhibited P. yoelii GST activity in a concentration-dependent manner showing maximum inhibition of c. 86% at 5.5 μM concentration with an apparent Ki c. 4.0 μM ( and ). The IC50 value was 4.0 μM. Hemin exhibited non-competitive inhibition kinetics with respect to the substrate GSH ().
Figure 10 Inhibition profile of GST from (a) P. yoelii and (b) P. falciparum by hemin.
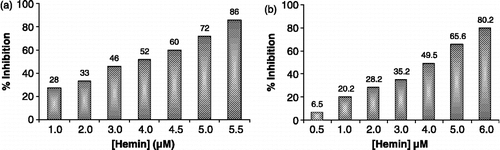
Figure 11 Type of inhibition of hemin on the activity of GST from (a) P. yoelii and (b) P. falciparum with respect to GSH.
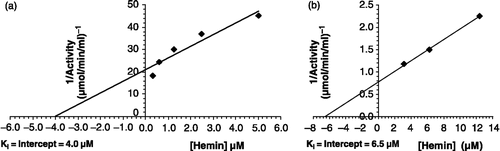
Hemin maximally inhibited Pf-GST to c. 80.2% at 6.0 μM when studied in the concentration range 0.5–6.0 μM (). Interestingly, for hemin an uncompetitive type of inhibition was determined () with a Ki value of 6.5 μM (). The IC50 value was about 4.0 μM ().
Figure 12 Ki of GST from (a) P. yoelii and (b) P. falciparum with respect to hemin.
Modulatory effect of synthetic compounds on native GST from P. yoelii and recombinant GST from P. falciparum
shows the inhibition potential of the synthetic compounds against native GST from P. yoelii and recombinant GST from P. falciparum. A total number of sixteen compounds inhibited native GST from P. yoelii to the extent of 40–100% of which one glycosyl peptide (compound 17L); one glycosyl urea (compound 31L); three isoxazole derivatives (compounds 58, 59 and 61), one 2-hydroxymethylacrylic acid methyl ester (compound 80) and one substituted hydroxymethyl acrylonitrile derivative (compound 81) showed 50% or more inhibition of intact GST. Seven compounds which inhibited GST from P. yoelii to the extent of 40–100% also showed antimalarial efficacy to the extent of 25.8–100% when tested in vitro, of which two glycoconjugates (compounds 13 and 14), one 2-hydroxymethylacrylic acid methyl ester (compound 80), two substituted hydroxymethyl acrylonitrile derivatives (compounds 81 and 82) and one pyrrolidine derivative (compound 83) showed antimalarial efficacy of 50% or more.
Table VIII. Modulatory effect of synthetic compounds on GST from Plasmodium yoelii and Plasmodium falciparum.
Fifty nine compounds stimulated native GST from P. yoelii to the extent of +13.8 to +216.4% of which one DGDA (compound 1); one GAA (compound 3); one GAE (compound 4); two glycosylated hydroxamic acids (compounds 5 and 10); one N-protected glycopeptide (compound 19); one glycosyl thiourea (compound 20); five glycosyl ureas (compounds 21, 25L–27L and 29L); one glycosylated hydantoin (compound 34); six nucleosides (compounds 35–40); two S-containing nucleosides (compounds 41 and 42); six C-nucleosides (compounds 49L, 50L, 52L–54L and 57L); one isoxazole derivative (compound 60) and seventeen substituted isoxazole derivatives (compounds 63–79) showed 50% or more stimulation on native GST from P. yoelii. Twenty five compounds which stimulated GST from P. yoelii to the extent of 50.3–216.4% also showed antimalarial efficacy to the extent of 16.8–70.0% when tested in vitro, of which one glycoconjugate (compound 5); one N-protected glycopeptide (compound 19); two glycosyl ureas (compounds 26L and 29L); one glycosylated hydrantoin (compound 34); three nucleosides (compounds 35, 36 and 40); two S-containing nucleosides (compounds 41 and 42); six C-nucleosides (compounds 49L, 50L, 52L–54L and 57L) and one substituted isoxazole derivative (compound 71) showed antimalarial efficacy of 50% or more.
On the other hand, fifteen compounds inhibited recombinant GST from P. falciparum to the extent of 27.1–100% of which one GAA (compound 2); two glycoconjugates (compounds 13 and 14); four glycosyl peptides (compounds 15L–18L); one glycosyl urea (compound 31L), two C-nucleosides (compounds 43L and 45L) and one isoxazole derivative (compound 61) showed 50% or more inhibition on recombinant GST from P. falciparum. Two glycoconjugates (compounds 13 and 14); one N-protected glycopeptide (compound 19) and one isoxazole derivative (compound 59) which inhibited GST from P. falciparum to the extent of 22.8–90.7%, also showed antimalarial efficacy to the extent of 25.8–100% when tested in vitro.
Sixteen compounds stimulated recombinant GST from P. falciparum to the extent of +16.9 to +119.3% of which one glycosylated amino ester (compound 4), six glycosylated hydroxamic acids (compounds 6–11), three C-nucleosides (compounds 47L, 48L and 51L) and three substituted isoxazole derivatives (compounds 64–66) showed 50% or more stimulation on recombinant GST from P. falciparum (). One C- nucleoside (compound 53L) and one glycosyl amino ester (compound 4) which stimulated GST from P. falciparum to the extent of 16.9 and 84.0%, respectively, also showed antimalarial efficacy of 54.4% and 42.8%, respectively, when tested in vitro.
Noteworthy in this collection are twenty two compounds; eleven of which showed simultaneous inhibition of P. yoelii and P. falciparum GST and the remaining eleven compounds showed simultaneous stimulation of P. yoelii and P. falciparum GST. These compounds are highlighted in . summarizes the IC50 values for the synthetic compounds which showed significant inhibition of GST from P. yoelii/P. falciparum.
Table IX. IC50 values of standard GST inhibitor hemin and synthetic compounds with respect to recombinant GST from P. falciparum.
Discussion
Malaria has plagued humans throughout recorded history and results in the death of over 2 million people per year. The protozoan parasite Plasmodium falciparum causes the most severe form of malaria in humans. Chemotherapy has become one of the major control strategies for this parasite; however, the development of drug resistance to virtually all of the currently available drugs is causing a crisis in the use and deployment of these compounds for prophylaxis and treatment of this disease. The genome sequence of P. falciparum is providing the informational base for the use of whole-genome strategies such as bioinformatics, micro arrays and genetic mapping. Various strategies have been employed to identify novel genes involved in the molecular mechanisms used by the parasite to circumvent the lethal effect of current chemotherapeutic agents [Citation46].
Advances in a malarial vaccine and drug development have been hindered in part by the complex multistage life cycle of the parasite, much of which is inaccessible to study, and by a large genome encoding over 5000 genes. Two human models of immunity to malaria, however, suggest that the development of an effective vaccine is within reach. A strategy has been outlined to identify the expression of hundreds to thousands of potential vaccine targets employing recently developed technologies for gene and protein expression. Combined with the exciting developments of malaria DNA vaccine technologies, these approaches form the basis for malaria subunit vaccines that may mimic the protective efficacy of human model systems and provide the foundation for novel approaches to vaccine development for a range of pathogens [Citation47].
Ongoing projects have characterized the glutathione metabolism and polyamine synthesis of Plasmodium falciparum. These studies demonstrate that the plasmodicidal effect of an inhibitor of glutathione synthesis does not depend on its specificity towards its target enzyme in the parasite, but on the changed physiological needs for the metabolite glutathione in the P. falciparum-infected red blood cells. Therefore, the depletion of glutathione has been proposed as a chemotherapeutic strategy for malaria, and γ-glutamylcysteine synthetase (GCS) and glutathione-S-transferase (GST) have been proposed as potential drug targets.
GST(s) are primarily involved in the detoxification of cytotoxic products of xenobiotic metabolism, the protection of tissues against oxidative damage and as ligandins, in the intracellular transport of hydrophobic compounds. In a parasitic context it is especially important to consider their function in the regulation of the oxidative stress response, in drug resistance and possibly in the modulation of host immune defence mechanisms.
In ongoing research programmes towards the development of biologically active compounds from sugars, glycosylated amino acid derivatives have been shown to possess various biological activities including parasitic DNA topoisomerase-II, as well as glutathione-S-transferase inhibitory, antitubercular and immunomodulatory activities [Citation25,Citation27,Citation48,Citation49]. Recognition of certain glycosylated amino acid derivatives by merozoites of the malarial parasite [Citation50], which divide in the red blood cells of the host, has also been reported. The role of sugars in drug targeting and their good pharmacokinetic properties [Citation51] as well as the chelating ability of hydroxamates [Citation52] has prompted the synthesis of certain glycosylated hydroxamic acids and their evaluation for antimalarial activities.
It has been shown that certain sugar derivatives possess very good activity against filarial glutathione metabolizing enzymes in vitro [Citation48,Citation53,Citation54]. These compounds possess immunomodulatory activity [Citation50] and in vitro antifilarial activity too. As sugars are known to offer better pharmacokinetics, better transport and above all less toxicity to drugs Citation55-57, it is presumed that amino acid analogs if grafted on a sugar backbone may serve as substrate for GSH biosynthesis and may hamper its de novo synthesis. C-nucleosides having a C–C linkage instead of a C–N linkage between the aglycone and the sugar moiety are known mainly for their anticancer [Citation57], antiviral [Citation58] and antileukemic [Citation59] activities. A C–C bond between the sugar and aglycone makes the nucleosides more stable towards enzymatic degradation, and therefore their half-lives in the body are far greater than the corresponding N-nucleosides. In addition, sugar derivatives are known to offer better stability, better pharmacokinetic parameters, facilitate the transport of drugs and at the same time are less toxic. Keeping in view the above facts, the above mentioned compounds were synthesized and evaluated for their effect on GSTs from Plasmodium yoelii/Plasmodium falciparum.
Conclusion
In conclusion, the present study lists the effect of 83 synthetic compounds on crude GST from Plasmodium yoelii and 41 compounds on recombinant GST from Plasmodium falciparum. The present study will determine the potential for selective inhibition/stimulation of malarial GST, prove whether or not modulation of GST activity in malarial parasites has any drastic consequences on their survival and also determine whether or not the effective modulators of malarial GST enhance the antimalarial activity of the presently available antimalarial drugs when used in combination. If the present strategy i.e. modulation of GST activity in malarial parasites, affects the survival of the parasites or helps in the enhancement of antimalarial activity of presently available antimalarial drugs, it will help in the design of new target-based chemotherapeutic agents which will benefit millions of people suffering from this widespread disease.
Acknowledgements
This investigation received financial support from Volkswagen Stiftung, Germany. We are also grateful to Dr S.K. Puri, Head, Parasitology Division, CDRI, Lucknow for providing the P. yoelii nigeriensis infected albino mice for the maintenance of infection in the present work.
Related Research Data
References
- Olliaro P. Pharmacol Ther 2001; 89: 207–219
- Charvet ED, Delarue S, Biot C, Schwobel B, Boehme CC, Musiigbrodt A, Maes L, Sergheraert C, Grellier P, Schirmer RH, Becker K. J Med Chem 2001; 44: 4268–4276
- Martin RJ, Robertson AP, Bjorn H. Parasitol 1997; 114: 111–124
- Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. Science 1985; 227: 1485–1487
- Arrick BA, Griffith OW, Cerami A. J Exp Med 1981; 153: 720–725
- Varki A. Glycobiology 1993; 3: 97–130
- Rademacher TW, Parekh RB, Dwek RA. Ann Rev Biochem 1988; 57: 785–838
- Hayes JD, Strange RC. Free Rad Res 1995; 22: 193–207
- Whalen R, Boyer TD. Semin Liver Dis 1998; 18: 345–358
- Eaton DL, Banunler TK. Toxicol Sci 1999; 49: 156–164
- Salinas AE, Wong MG. Curr Med Chem 1999; 6: 279–309
- Strange RC, Jones PW, Fryer AA. Toxicol Lett 2000; 112–113: 357–363
- van Bladeren PJ. Chem Biol Interact 2000; 129: 61–76
- van Bladeren PJ, van Ommen B. Pharmacol Ther 2000; 51: 35–46
- Hemingway J, Hawkes N, Prapanthadara L, Jayawardenal KG, Ranson H. Phil Trans R Soc Lond B Biol Sci 1998; 353: 1695–1699
- Dubois VL, Platel DE, Pauly G, Tribouley-Duret J. Exp Parasitol 1995; 81: 117–124
- Srivastava P, Puri SK, Kamboj KK, Pandey VC. Trop Med Int Health 1999; 4: 251–254
- Famin O, Krugliak M, Ginsburg H. Biochem Pharmacol 1999; 58: 59–68
- Platel DF, Mangou E, Tribouley-Duret J. Mol Biochem Parasitol 1999; 98: 215–223
- Harwaldt P, Rahlfs S, Becker K. Biol Chem 2002; 383: 821–830
- Liebau E, Bergmann B, Campbell AM, Teesdale-Spittle P, Brophy P, Lüersen K, Walter RD. Mol Biochem Parasitol 2002; 124: 85–90
- Flohe L, Hecht HJ, Steinert P. Free Radic Biol Med 1999; 27: 966–984
- Tiwari VK, Tewari N, Katiyar D, Tripathi RP, Arora K, Gupta S, Ahmad R, Srivastava AK, Khan MA, Murthy PK, Walter RD. Bioorg Med Chem 2003; 11: 1789–1800
- Tripathi RP, Tripathi R, Tiwari VK, Bala L, Sinha S, Srivastava A, Srivastava R, Srivastava BS. Eur J Med Chem 2002; 37: 773–781
- Khan AR, Tripathi RP, Tiwari VK, Mishra RC, Reddy VJM, Saxena JK. J Carbohyd Chem 2002; 21: 587–600
- Mishra RC, Tripathi R, Katiyar D, Tewari N, Singh N, Tripathi RP. Bioorg Med Chem 2003; 11: 5363–5734
- Tripathi RP, Rastogi SK, Kundu B, Saxena JK, Reddy VJM, Srivastava S, Chandra S, Bhaduri AP. Comb Chem High Throughput Screen 2001; 4: 237–244
- (a) Tripathi RP, Tiwari VK, Mishra RC, Srivastava R, Srivastava S, Chaturvedi V, Srivastava KK, Srivastava BS (b) Tripathi RP, Tiwari VK, Mishra RC,Srivastava R, Srivastava S, Chaturvedi V, Srivastava KK, Srivastava BS (NF/474/01), NF/476/01).
- Tewari N, Mishra RC, Tiwari VK, Tripathi RP. Synlett 2002; 11: 1779–1782
- Mishra RC, Katiyar D, Tewari N, Tripathi RP. Nucleosides Nucleotides Nucleic Acids 2004; 23: 531–544
- Tewari N, Tiwari VK, Mishra RC, Tripathi RP, Srivastava AK, Ahmad R, Srivastava R, Srivastava BS. Bioorgan Med Chem 2003; 11: 2911–2922
- Tripathi RP, Rastogi SK, Kundu B, Saxena JK, Reddy VJM, Srivastava S, Chandra S, Bhaduri AP. Comb Chem High Throughput Screen 2001; 4: 237–244
- Ahmad R, Mishra RC, Tewari N, Tripathi RP, Srivastava AK, Walter RD. Med Chem Res 2004; 13: 724–745
- Arora K, Mishra RC, Tripathi RP, Srivastava AK, Walter RD. Med Chem Res 2004; 13: 687–706
- Bhattacharya SM, Katiyar D, Bajpai P, Tripathi RP, Saxena JK. Parasitol Res 2004; 92: 177–182
- Katiyar D, Tiwari VK, Tripathi RP, Reddy VM, Bhattacharya SM, Saxena JK. Arzneimittelforschung/Drug Res 2003; 53: 857–863
- Mishra RC, Tewari N, Arora K, Ahmad R, Tripathi RP, Tiwari VK, Walter RD, Srivastava AK. Comb Chem High Throughput Screen 2003; 6: 37–50
- Patra A, Roy AK, Batra S, Bhaduri AP. Synlett 2002; 1819–1822
- Batra S, Srinivasan T, Rastogi SK, Kundu B, Patra A, Bhaduri AP, Dixit M. Bioorg Med Chem Lett 2002; 12: 1905–1908
- (a) Patra A, Batra S, Kundu B, Joshi BS, Roy R, Bhaduri AP. Synthesis 2001; 276–281. (b) Cai J, Zhou Z, Zhao G, Tang C. Org Lett 2002; 4: 4723–4725. (c) Kundu MK, Sundar N, Kumar SK, Bhat SV, Biswas S, Valecha N. Bioorg Med Chem Lett 1999; 9:731–736.
- Bajpai LK. PhD dissertation. RML Univ. Faizabad. 1997.
- Richards WH, Williams SG. Ann Trop Med Parasitol 1973; 67: 249–250
- Ferone R, Burchall JJ, Hitchings GH. Mol Pharmacol 1969; 5: 49–59
- Habig HW, Pabst MJ, Jakoby WB. J Biol Chem 1974; 249: 7130–7139
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem 1951; 193: 265–276
- Cowman AF. Int J Parasitol 2001; 31: 871–888
- Carucci DJ. Vaccine 2001; 19: 2315–2318
- Srivastava AK, Tripathi RP, Khan AR, Bhaduri AP. Helminthologia 1995; 32: 25
- Gupta S, Srivastava AK. Med Sci Monit 2006; 12: 1–9
- Perkins ME, Rocco LJJ. Immunol 1988; 141: 3190–3196
- Khan AR, Tripathi RP, Bhaduri AP, Sahai R, Puri A, Tripathi LM, Srivastava VML. Eur J Med Chem 2001; 36: 435–445
- Mishra RC, Tripathi R, Katiyar D, Tewari N, Singh D, Tripathi RP. Bioorg Med Chem 2003; 11: 5363–5374
- Tripathi RP, Singh V, Khan AR, Bhaduri AP, Bhatnagar S, Srivastava AK. Trends carbohydrate chem, PL Soni. Surya International, Dehradun, India 1995; 1
- Srivastava AK, Tripathi RP, Khan AR, Bhaduri AP, Singh SN, Chatterjee RK. Ind J Parasitol 1994; 18: 127–133
- Negre E, Chance ML, Hanboula SY, Monsigny M, Roche AC, Mayer RM, Homme lM. Antimicrob Agents Chemother 1992; 36: 2228–2232
- Fischer JF, Harrison AW, Bundy GL, Wilkinson KF, Rush BD, Ruwart MJ. J Med Chem 1991; 34: 3140–3143
- Burchenal JH, Ciovacco K, Kalhar K, Toole TO, Kiefner R, Dowing MD, Chu CK, Watanabe KA, Wempen I, Fox J. J Cancer Res 1976; 36: 1520–1523
- Schaeffer HJ, Beauchamp L, de Miranda P, Ellion GB, Bour DJ, Collins P. Nature 1978; 272: 583–585
- Robins RK, Srivastava PC, Narayanan VL, Plowman J, Paul KD. J Med Chem 1982; 25: 107–108