Abstract
This article presents in brief the development of farnesyltransferase inhibitors (FTIs) and their preclinical and clinical status. In this review the mechanism of action of FTIs is discussed and their selectivity issue towards tumor cells is also addressed. The significant efficacy of FTIs as single or combined agents in preclinical studies stands in contrast with only moderate effects in Clinical Phase II–III studies. This suggests that there is a need to further explore and understand the complex mechanism of action of FTIs and their interaction with cytotoxic agents.
Introduction
Though there have been major breakthroughs in many areas of medicine over the past decades, the successful treatment of cancer still remains a significant challenge at the start of the twenty-first century. The need for new chemotherapeutics in cancer is evident as there are very few drugs able to cure or significantly prolong the survival of patients with disseminated tumors. The explosion of the biological complexities of cancer and the molecular genetic defects underlying tumorogenesis has offered new opportunities for drug discovery and development for the treatment of cancer [Citation1,Citation2]. The goal is to produce novel agents that can selectively kill tumor cells or inhibit their proliferation without the general toxicity that limits traditional cancer therapy.
The most frequently occurring mutations in cancer are alterations of the Ras genes [Harvey (Ha), Kirtsen (Ki) and N-Ras] [Citation3,Citation4]. The Ras genes encode 21-kDa proteins, called p21 or Ras, which are localized to the inner face of the plasma membrane. Ras binds GTP and GDP and serves as a molecular switch, interfacing between receptors and intracellular effector proteins. When Ras is stimulated by receptor activation to bind GTP, it promotes cell proliferation. The GTPase activity of Ras then turns off the biological event. The mutations in Ras genes most often found in cancer inhibit Ras GTPase activity, so Ras remains bound to GTP and constitutively stimulates cell growth. Alteration in the function of Ras gene products, particularly Ki4B-Ras and N-Ras, are found in many different tumors, including carcinomas of the colon, pancreas and lung, neurofibrosarcomas, and various leukemias, including both adult and juvenile chronic myelogenous leukemia [Citation3,Citation5,Citation6].
Many efforts have been made to inhibit the function of oncogenic Ras and subsequent cellular transformation induced by mutant Ras [Citation7,Citation8]. Emphasis has been towards developing novel chemotherapeutics against Ras-induced cell transformation and has centered on inhibiting the enzyme farnesyl transferase (FTase) [Citation9]. The discovery that Ras required prenylation for activity led to the targeting of the enzyme FTase [Citation10,Citation11]. In this regard, posttranslational modifications have attracted much attention because they are required for appropriate sub-cellular localization of Ras proteins in the plasma membrane [Citation10]. Thus FTIs were conceived as a rational way to treat cancer by inhibiting the function of the oncogene Ras.
Structure and function of farnesyltransferase
Protein FTase is a metalloenzyme that catalyzes the reaction between FPP and the cysteine residue of a polypeptide's C-terminal CaaX motif where C is Cys; a is usually an aliphatic amino acid; X is the C-terminal residue, (typically Met) to give a farnesyl thioether () [Citation12]. The enzyme is a heterodimer consisting of 48 kDa α- and 46 kDa β subunits. The former subunit is also a component of the closely related enzyme geranylgeranyltransferase type I (GGTase-I), which utilizes geranylgeranyl diphosphate (GGPP) as a prenyl donor and has different CaaX specificity (X is typically Leu) [Citation12]. Farnesylation is usually followed by additional modification of the prenylated protein, including proteolytic removal of the last three residues of the ‘CaaX box’ and carboxymethylation of the new C-terminal farnesylated cysteine [Citation13]. The initial prenylation step is crucial and required for the correct membrane localization and function of Ras.
Figure 1 Post translational modification of Ras.
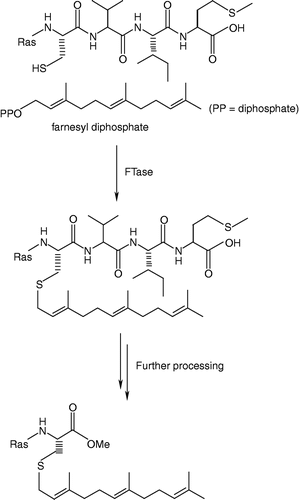
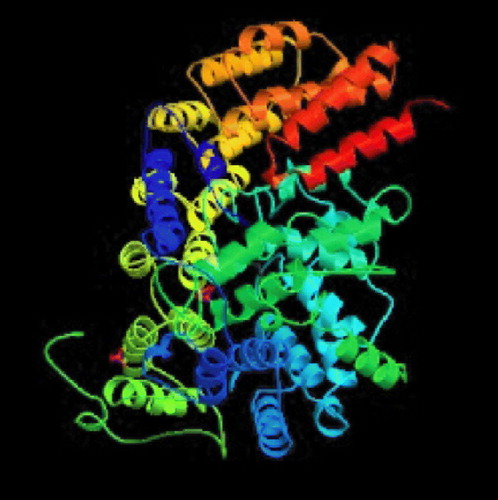
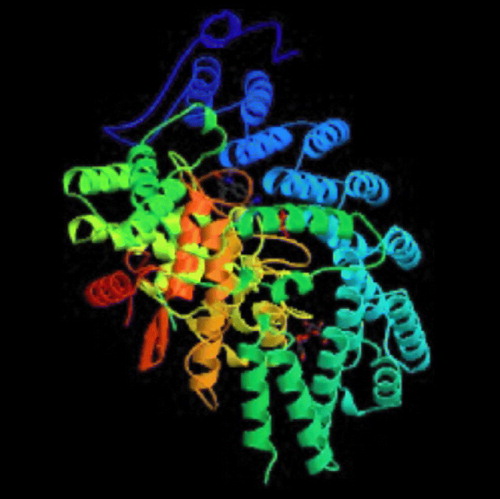
Crystallographic studies and enzymology has helped us to understand the mechanistic details of catalysis by FTase [Citation14]. Some of the enzymes deposited in the protein data bank with their PDB codes and active site residues are mentioned in . well displays the secondary structure of FTase bound to farnesyldiphosphate. Long and co-workers afforded an excellent overview of this enzyme-catalyzed process [Citation15]. The reaction proceeds via an ordered mechanism, with farnesyl diphosphate (FPP) binding first, followed by the CaaX substrate. The structures of ternary complexes using non-reactive FPP or peptide analogs provide an explanation for this ordering. The isoprenoid moiety forms a substantial part of the binding surface for the CaaX peptide. The peptide binds in an extended conformation with the cysteine sulfur coordinated to the active site zinc ion, which apparently lowers the pKa of the thiol, significantly increasing the local concentration of the reactive thiolate [Citation16,Citation17]. It is proposed that a rotation of the FPP backbone brings its C1 carbon into proximity with the thiolate, leading to a transition state that is consistent with previous mechanistic studies [Citation15]. Release of the farnesylated peptide product is slow and requires the addition of FPP [Citation18]. The FPP displaces the product isoprenoid group to a new binding site, the ‘exit groove’, with concomitant change in the CaaX backbone from an extended to a β-turn conformation. Product release thus regenerates the FTase/FPP complex. The published structures have revealed that FTIs may inhibit the enzyme by blocking the peptide substrate site or by occupying part of the peptide site and the exit groove in a manner similar to the farnesylated peptide product [Citation15].
Table I. Various FTase structures deposited in databank with their PDB codes, resolution and active site amino acid residues.
Figure 2 Farnesyltransferase enzyme complexed with farnesyldiphosphate (PDB code: 1FPP).
Farnesyltransferase inhibitors: Mechanism of action
Due to the functional role of Ras farnesylation, FTase inhibition was thought to be a strategy for interfering with Ras-dependent transformation Citation19-23. Farnesylation is catalyzed by FTase, which is responsible for catalyzing farnesylation of several cellular proteins characterized by a CaaX box, transferring a C-15 farnesyl moiety from FPP to Cysteine. The three main Ras proteins share high homology in the first 165 amino acids, but show difference in 25 amino acids of the carboxy-terminal region that constitutes the hypervariable region. The post-translational modifications, beginning from amino acid 186 (always cysteine), increase the hydrophobicity of the carboxy-terminal region of the protein. The post-translational modifications are the farnesylation of C-186 by the cleavage of the three downstream amino acids (aaX), followed by methylation of C-186 and finally a palmitoylation of cysteine residues in the region 165–186. Cysteine mutation in the CaaX box prevents farnesylation and Ras function. Mammalian N- and H-Ras proteins contain additional cysteine near their carboxy-termini (181 for N-Ras; 181 and 184 for H-Ras), and these serve as palmitoylation sites. The functional consequences of these palmitoylations are beginning to be understood. K-Ras4B lacks this upstream cysteines and is prenylated, proteolyzed and methylated but not palmitoylated. Instead, K-Ras4B has a cluster of 8 lysine residues near its carboxyl terminus that are thought to function by forming electrostatic contacts with negatively charged phospholipids Citation24-27.
Studies have shown that farnesylation of Ras is the obligatory, first step in a series of post-translational modifications that leads to membrane association that determines the switch from an inactive to an active Ras-GTP bound form. Upon receiving a signal output, Ras-GTP acts as a molecular switch that turns on downstream effectors Citation24-27.
Ras has guanine nucleotide-binding activity and GTPase activities. Thus, Ras has GDP-bound inactive and GTP-bound active forms, which are intercompatible by the GDP/GTP exchange and GTPase reactions. The GTPase reaction is regulated by GTPase-activating proteins (GAPs), such as p120, NF1, and GAP1. The GDP/GTP exchange is regulated by guanine exchange factors (GEFs). Ras is known to be the downstream molecule of receptor tyrosine kinases, such as EGF and PDGF receptors, and protein kinase C. The activation of these proteins results in the stimulation of GEFs and the conversion from the GDP-bound form to GTP-bound form of Ras. Other Ras effectors belonging to the GEFs family are RalGDS, RGL and Rlf/RGL2 that serve as activators of the Ral small monomeric GTPases [Citation27]. Ral-GTP activates the phospholipase-D (PLD) that, by hydrolyzing phosphatidylcholines, generates saturated and monounsaturated phosphatidates which are putative activator molecules of Rho. RAL also seems to interact with a Cdc42 and RAC GTpase activating proteins [Citation27]. Rho, RAC and Cdc42 constitute another family of monomeric G proteins that play an important role in cytoskeleton remodeling and activate the kinases regulating the activity of various transcriptional factors. It has been shown that Rho represses expression of a cyclins inhibitor p21WAFI [Citation27].
The third Ras effector is the phosphatidylinositol 3-kinase (PI3-K). Ras-GTP can bind and activate the catalytic subunit of this enzyme that generates PI Citation3-5 P3 by phosphorylation the PI [Citation4,Citation5] P2 in 3-position. The PI Citation3-5 P3 acts directly like a second messenger, binding several cytoskeleton kinase proteins and modulating the activity by conformational changes and/or their membrane translocation. PKB/AKT is an enzyme indirectly activated by PI3-K, which inactivates BAD, pro-apoptotic factor, by phosphorylation [Citation27].
These observations guide us in understanding the complex relationship between Ras activation, cellular proliferation and apoptosis. Thus, Ras can induce simultaneously anti/pro apoptotic pathways as a function of quality and intensity of activating signals, as a function of cell type and metabolic conditions, and by activation of other Ras-independent or dependent pathways. Further understanding of these processes would help us identify the factors directly responsible for cell cycle de-regulation in several tumors and would help in the design of specific strategies for the control on the proliferation of neoplastic cells.
Mutations in Ras genes have all been found in human tumors and the frequency of Ras mutations were found to be highest among genes in human cancers [Citation28]. Activating mutations of Ras are found in humans in nearly all pancreatic cancers, one half of colon and thyroid tumors and one-third of lung tumors [Citation28]. A divergent association of Ras mutations with distinct neoplasm is also seen when comparing humans and rodents. For example, Ras mutations are strongly associated with the carcinogen-induced mammary carcinomas in mice and rats [Citation29], while such mutations are rare in human breast cancers [Citation28]. Activating point mutations of Ras genes occur in approximately 30% of human cancers, with certain malignancies having a particular high incidence [Citation28]. The incidence of K-Ras mutations in pancreatic cancers approaches 90%, while mutations in K-Ras are detected in 50% colorectal carcinomas and 30% Non Small Cell Lung Cancers (NSCLC). Hematologic malignancies harbor N-Ras mutations [Citation28]. H-Ras transformed cells appeared to be more sensitive to FTIs than those transformed either by K-Ras or N-Ras Citation19-23Citation30Citation31.
Effect of FTIs on cell cycle progression
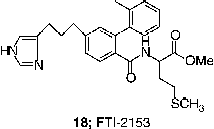
FTIs have been shown to interfere with cell cycle progression in human cancer cells [Citation32]. In most cells, FTIs induce accumulation of human cancer cells in the G2/M phase of the cell cycle, but in some cells FTIs could rather induce a Go/G1 block or have no effect on the cell cycle progression. Furthermore, it has been shown that the G2/M accumulation is due to the inhibition of the prophase/metaphase transition during mitosis Citation33-36. For example, after a 24-hour exposure to FTI-2153 (18), cancer cells attempted to cycle from prophase to metaphase in preparation for chromosomal alignment for mitosis, but were unable to form bipolar spindles and their chromosomes failed to align to form a metaphase plate [Citation33]. This inhibition of bipolar spindle formation correlated with an accumulation of cells in the prometaphase [Citation33,Citation34].
Further investigation into the mechanism of action, therefore involved the analysis of farnesylated proteins required for prophase/metaphase transition and bipolar spindle formation. Centromere associated proteins (CENP-E and CENP-F) were the first targets to be investigated, as they are critical to the processes of chromosome alignment and spindle formation Citation34-36. However, localization of farnesylated CENP-E and CENP-F to the kinetochore was unaffected by FTI-2153 (18) [Citation33] suggesting that these centromeric proteins may not be molecular targets for the inhibition of bipolar spindle formation by FTIs [Citation34]. Further investigations are still needed in order to elucidate the mechanism by which FTIs induce cell cycle arrest in human cancer cells.
Development of farnesyltransferase inhibitors
Various structurally diverse classes of FTIs have been reviewed Citation13Citation37-40. Here emphasis is given only to the efforts that lead to development of various clinical candidates.
The general approaches used for the development of FTIs are:
Design and synthesis of FDP analogs that compete with the substrate FDP for FTase.
Design and synthesis of peptide and non-peptide CaaX peptidomimetics that compete with the CaaX portion of Ras for FTase.
Design and synthesis of bisubstrate analogs that combine features of both FDP analogs and peptidomimetics.
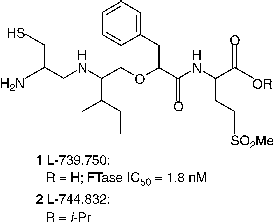
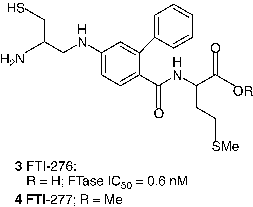
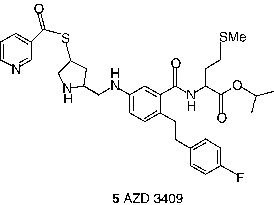
Even though potent FDP analogs have been discovered, much attention has been given to the analogs of the CaaX peptide [Citation13,Citation37]. Two of the most notable CaaX derived inhibitors are L-739,750 (1, Merck; FTase IC50 = 1.8 nM) [Citation41] and FTI-276 (3, Hamilton and Sebti group; FTase IC50 = 0.6 nM) [Citation42], and their ester prodrugs L-744,832 (2) and FTI-277 (4) respectively. L-744,832 was found to exhibit impressive efficacy in a transgenic mouse model of cancer [Citation41]. In this transgenic model 2 induced complete tumor regression in the absence of any obvious toxicity. In contrast, the cytotoxic agent doxorubicin failed to shrink tumors in this model at its maximum tolerated dose. Such observations suggested that FTIs might indeed fulfill the promise of effective cancer chemotherapy without toxicity. Despite encouraging in vivo data, skepticism prevailed about the clinical utility of 2 because of its potential for thiol-based toxicity. Nonetheless, the AstraZeneca compound AZD3409 (5), which has both the key thiol and carboxylate groups blocked in a double prodrug strategy advanced to clinical stage [Citation43].
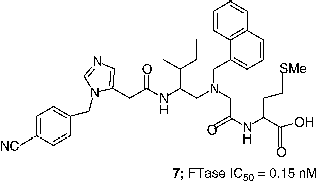
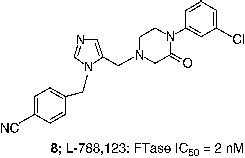
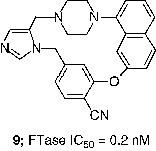
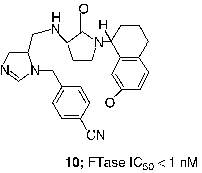
An extensive effort to address the concern about the peptidomimetic mercaptan and carboxylate groups was undertaken by scientists at Merck. Truncation of the CaaX tetrapeptide and subsequent re-engineering gave rise to the piperazine based non-peptide thiol 6 (FTase IC50 = 1 nM) [Citation44]. Another early success was the identification of a general thiol replacement in 4-cyanobenzyl imidazole. This was designed to simultaneously ligate the active site zinc ion and occupy a nearby hydrophobic binding site, which resulted in a potent peptidomimetic FTI like 7 (FTase IC50 = 0.15 nM) [Citation45]. Fusion of the non-peptide 8 with the thiol replacement in 7 and further optimization of the piperazine template led to piperazinone FTIs, which displayed significantly improved cell based activity compared with the earlier peptidomimetic FTIs [Citation46]. For example the Merck clinical candidate L-778,123 (8, FTase IC50 = 2 nM) inhibited the growth of H-Ras transformed cells with an IC50 value of 15 nM. Studies directed at optimization of such non-peptide cyanobenzylimidazoles utilized transferred Nuclear Overhauser Effect (NOE) data to design macrocyclic FTIs such as compound 9 (FTase IC50 = 0.2 nM) [Citation47]. Members of this class of FTIs exhibited the highest cell potency yet described for inhibitors of FTase. Compound 10, for example inhibited the processing of HDJ2 in PSN-1 cells with an EC50 value of 180 pM [Citation48].
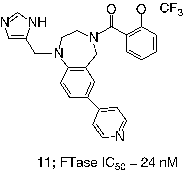
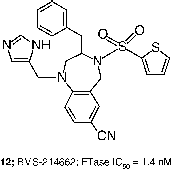
Researchers at Bristol-Myers Squibb sought analogues of thiol 6, in which the mercaptan was replaced by imidazole [Citation49] and evolved their initial micromolar leads into a tetrahydrobenzodiazepine-based series of compounds represented by 11 (FTase IC50 = 24 nM) [Citation50]. It was discovered that the potency of analogues such as 11 was enhanced by addition of a phenylmethyl moiety at the 3-position and replacement of the 4-position amide with a sulfonamide. Incorporation of a 7-cyano substituent simultaneously improved the potency and aqueous solubility and these modifications led to the clinical candidate BMS-214662 (12, FTase IC50 = 1.4 nM) [Citation21]. It seems likely that this moiety takes advantage of similar binding interactions to the cyanobenzyl in many Merck compounds.
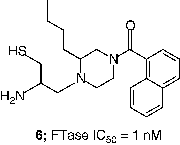
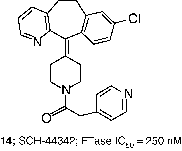
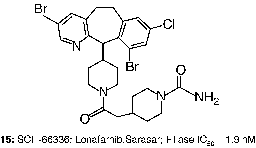
In contrast to the gradual evolution from CaaX peptides to small molecules such as 8 and 12, other researchers obtained attractive leads for the development of FTIs from screening of compound libraries. For example, screening at Schering-Plough identified micromolar FTIs such as SCH-37370 (13, FTase IC50 = 27000 nM) a close analog of loratidine [Citation40]. Exploration of structure-activity relationships led to SCH-44342 (14), which exhibited significantly enhanced potency (FTase IC50 = 250 nM). Alternatives to the pyridine side chain were explored to improve the pharmacokinetic profiles, leading to a clinical candidate SCH-66336 (15, Lonafarnib, Sarasar; FTase IC50 = 1.9 nM) [Citation51]. This tricyclic inhibitor was distinguished from those tested in humans by its lack of a ligand for the active site zinc ion in FTase.
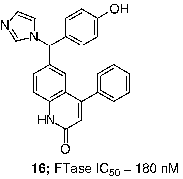
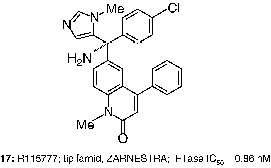
At the Janssen Research Foundation, screening for FTIs afforded lead quinolinone compounds such as 16 (FTase IC50 = 180 nM). It was found that attachment of the imidazole moiety via the 5-position, combined with N-methylation of the imidazole, increased potency against FTase and selectivity against cytochrome P450-dependent enzymes. Incorporation of a benzylic amino group was shown to improve cell potency, and additional optimization produced R115777 (17, tipifarnib, ZARNESTRA; FTase IC50 = 0.86 nM), the first FTI to advance to human clinical trials [Citation51]. Compound 17 inhibited the growth of H-Ras-transformed NIH 3T3 cells with an impressive IC50 value of 1.7 nM [Citation52]. Docked tipifarnib is well depicted in .
Figure 3 Farnesyltransferase enzyme complexed with tipifarnib (PDB code: 1SA4).
Monica et al. at Karolinska Institute, Stockholm in collaboration with Phillips University, Leibnz Institute and Ludwig-Maximilians University in Germany recently reported benzophenone-based FTase inhibitors as highly active against Trypanosoma Cruzi. These inhibitors can be helpful in the treatment of Chaga's disease [Citation53]. Researchers at the University of Washington also identified Tipifarnib as a potent inhibitor of Trypanosoma Cruzi with ED50 of 4 nM. They used homology models of T. Cruzi CYP51 (E.C 1.14.13.70) for the prediction of the binding modes of substrates, tipifarnib and lanosterol [Citation54]. Scientists at Princeton and Scheringh-Plough institute, New Jersey reported novel FTIs by screening ECLiPS library and also inferred that enhancement in potency can be afforded through interaction with the active site zinc [Citation55]. Qun Li and co-workers explored the tipifarnib scaffold and synthesized new FTIs by substituting the benzimidazolones and indoles for 2-quinoline of tipifarnib retaining considerable FTase inhibitory activity [Citation56]. Katja et al. were successful in synthesizing and screening FTase inhibitors for antimalarial activity, they also carried out docking studies in order to explore the binding interactions with the active site of Ftase [Citation57]. Guida and co-workers carried out flexible docking studies of various peptide, peptidomimetic and non-peptidomimetic inhibitors of the zinc metalloenzyme FTase and inferred from the study that various conformations for the methionine side chain can be accommodated by the enzyme suggesting that the methionine pocket can tolerate groups larger than methionine at the C-terminus of tetrapeptide [Citation58].
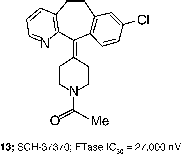
Efficacy of farnesyltransferase inhibitors
Inhibitors that are competitive with respect to CaaX protein substrate are more efficacious than compounds that are competitive with respect to FPP Citation59-63. In addition to inhibiting H-Ras protein, FTase inhibitors block farnesylation of a number of proteins including lamin A, lamin B, Pxf and perhaps as many as 15 as yet uncharacterized polypeptides [Citation64,Citation65]. Thus, it is incorrect to refer to FTase inhibitors as Ras inhibitors. However the biochemical specificity of FTase inhibitors is unquestioned, since these agents do not block geranylgeranylation of proteins [Citation61,Citation62,Citation66,Citation67].
Biological activity of FTase inhibitors against H-Ras transformed rodent fibroblast cells has been demonstrated in cell culture assays that monitor key phenotypes of cellular transformation: anchorage-independent growth [Citation44,Citation68], the rapidity of growth in monolayer, morphological transformation [Citation62,Citation69,Citation70], and alterations in the cytoskeleton [Citation70]. The concentrations of compounds necessary to illicit these effects are similar to those necessary to block intracellular farnesylation, suggesting that the observed efficacy is mechanism based.
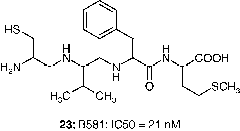
In addition to inhibiting the growth of transformed fibroblasts, FTase inhibitors also block the anchorage-independent growth of human tumor-derived cells Citation68Citation71-73. Human tumor cells with and without mutant ras alleles are sensitive to B581:IC50 (23) and L-744,832 (2), although the doses of these agents necessary to achieve this effect varied depending on the particular cell line tested [Citation68,Citation74]. Thus the action of FTase inhibitors may target a broader set of tumors than earlier anticipated.
Initially, the principle aim of these cell-based assays was not only to demonstrate efficacy against Ras-transfomed cells but also to distinguish the actions of FTase inhibitors from more traditional cytotoxic agents. To do this, activity of FTase inhibitors against Ras-transformed cells was compared with that in cells transformed by other oncogenes. If FTase inhibitors were truly inhibiting the function of transforming Ras proteins, the cells transformed by an activated oncogene such as Raf, which does not require prenylation to achieve full biological activity, should be less sensitive to these inhibitors. The experimental basis for this hypothesis was based on work carried out by Stacey and coworkers, who showed that cells transformed by Raf were insensitive to the actions of a neutralizing Ras antibody or dormant negative forms of Ras [Citation75,Citation76]. In all reported biological assays, cells transformed by Raf have been resistant to the actions of FTase inhibitors at doses used to inhibit cells transformed by H-Ras, suggesting that the action of FTase inhibitors and their growth inhibitory profile is very different from the profiles observed with standard cytotoxic chemotherapeutic agents [Citation62,Citation69,Citation70].
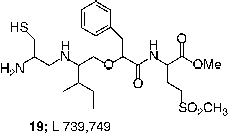
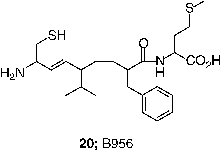
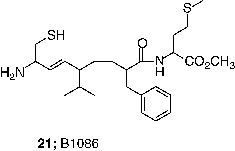
FTase inhibitors have also demonstrated antitumor efficacy in mice [Citation60,Citation44,Citation68,Citation77]. Initial studies utilized nude mice so that xenograft tumors could be produced using transformed rodent fibroblasts that were characterized for their sensitivity to FTase inhibitors in cell culture. The metabolic liability of the ester prodrug of FTIs became evident in this assay. In some cases, the free carboxylate forms of the FTIs were just as efficacious as the prodrug forms, FTI-276 (3) / FTI-277 (4) and B956 (20) /B1086 (21) [Citation68,Citation71]. This result may vary among compounds because the acid of L-739,749 had poorer activity than the prodrug. Nevertheless, given the high plasma esterase activity in rodents, this result is perhaps not so surprising and is probably a major reason why the doses required to achieve an antitumor effect (40–250 mg/Kg of body weight) were so high. However a clear dose-response relationship was observed between antitumor activity and exposure to active drug [Citation68,Citation71].
Human tumor cells have also shown sensitivity to FTIs in the nude mice xenograft model [Citation68,Citation71,Citation77]. However, not all tumor cells were equally sensitive. Researchers have suggested that the sensitivity of human tumor cells in this assay correlates with the Ras-mutational status [Citation68]. One of the more unexpected actions of FTIs in animals was seen in a transgenic mouse model of mammary cancer [Citation41]. In this model, viral Ha-Ras is expressed under the control of MMTV promoter, and mice develop mammary and salivary carcinomas [Citation78]. Thus it appears that a second genetic alteration must occur in these animals before they develop tumors.
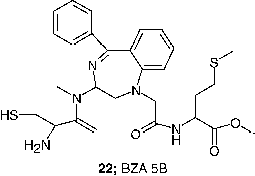
Although the biological results seen with FTIs are widely accepted, the question still to be convincingly addressed is ‘Why is there selectivity for tumor cells versus normal cells?’ An accurate explanation for this phenomenon has not been established, but several observations that address to this issue are noted. First, not all farnesylated proteins have the same sensitivity to FTase inhibition in cells. Ha-Ras appears to be one of the more sensitive polypeptides to the action of FTase inhibitors. Second, in the absence of functional FTase, some proteins may serve as substrates for GGPTase-I. The functional impact of geranylgeranylation on these proteins cannot be clearly anticipated, but in the case of Ki4B-Ras, which serves as a good substrate for GGPTase-I, the oncogenic forms of Ki4B-Ras that are geranylgeranylated can effectively transform cells [Citation79]. Geranylgeranylated wild type Ki4B-Ras may also provide critical biological functions to normal cells, although geranylgeranylated wild-type Ha-Ras has been reported to exhibit dominant negative properties [Citation80]. Third, it is possible that redundant pathways in normal cells compensate for the functional loss of proteins such as Ha-Ras. For example, MAPK activation by growth factors such as EFG is not sensitive to BZA-5B (22) [Citation81]. Fourth, it is unclear what degree of inhibition of farnesylation is required to block biological functions for any given protein.
Preclinical studies combining FTIs and chemotherapeutic agents
FTIs combined with other chemotherapeutic agents is an attractive approach for several reasons: FTIs have distinct spectra of activity compared with classical chemotherapeutic agents [Citation82]. Furthermore, the growth inhibition by FTIs can supplement cytotoxic effects of other drugs in an additive way under certain conditions. However, the cell cycle blocks induced by FTIs could inhibit the activity of certain cytotoxic agents. This is the reason why careful pre-clinical studies of FTI/drug interactions are essential. Several groups have reported the combination of FTIs with other drugs Citation83-85.
Several human cancer cell lines with K-Ras mutations were tested for sensitivity to FTI-2148 combined with 5-FU, melphalan and cisplatin in vitro [Citation86]. Combining FTI with 5-FU did not give a satisfactory result, whereas combinations with melphalan as well as gemcitabine were found to be synergistic. In A549 lung adenocarcinoma and T98G glioblastoma cells, SCH66336 (15) combined with cisplatin had an additive effect; while in the other cell lines, the combination did not have an effect [Citation84].
In contrast to the additive effects seen with most chemotherapeutic agents, the combination of FTI and taxol or desoxyepothilone was synergistic. Similar synergistic effects were reported for breast cancer and prostrate cancer cell lines T47D and DU-145. The fact that epothilones do not share structural similarity with taxol but also stabilize microtubules and exhibit synergistic effects in combination with FTI was interpreted as a mechanistic relationship between FTIs and microtubule-stabilizing agents. The effects of some FTIs on anchorage-independent growth of MIA-PaCa-2 cells in combination with taxol were supra-additive as determined using the Pearson correlation coefficient [Citation83]. Another FTI, SCH66336 (15), was also able to synergize with paclitaxel and to a somewhat lesser degree with docetaxel when 11 cell lines of different tumor entities were tested in vitro [Citation85]. In a study combining SCH66336 (15) with cyclophosphamide, 5-FU, and vincristine in a HTB177 human lung carcinoma xenographt (K-Ras mutant), an additive effect of combining FTI with these agents was reported [Citation87].
Clinical development of FTase inhibitors
Ambiguous results obtained in preclinical studies have complicated the selection of relevant end points in clinical trials [Citation88]. The traditional strategy for evaluation of cytotoxics in terms of their ability to shrink tumors may be sub-optimal if the primary effect of FTIs is growth inhibition cytostatis rather than cytotoxicity. Since the Ras hypothesis has fallen out of favor, the clinical studies have targeted a variety of cancers irrespective of their historical frequency of Ras mutations. There appears to be no relationship between clinical response and Ras status [Citation89,Citation90]. Pharmacodynamic effects of FTIs in patients have been monitored by quantitation of FTase substrates such as HDJ2 and prelamin A [Citation91,Citation92]. Summaries of only a few significant clinical findings are discussed here.
Therapy using single agent
In a Phase I clinical trial, 17 was administered orally twice daily to 28 patients with advanced solid tumors in a dose-escalating protocol [Citation89]. The dose limiting toxicities (DLTs) were myelosuppression and neurotoxicity and the maximum tolerated dose (MTD) was determined to be 300 mg twice daily. One patient with NSCLC showed a partial response (PR). In a Phase II breast cancer trial examining continuous and intermittent dosing schedules of 17 (300 mg twice daily), eight patients out of 76 (11%) showed a PR [Citation93]. However, 17 did not improve the survival time of patients with advanced colorectal cancer in a phase III trial [Citation92]. Perhaps the most impressive clinical efficacy reported for 17 as monotherapy has been in hematologic malignancies. A phase I dose-escalating trial in patients with refractory and relapsed acute leukemia's encountered DLT of central neurotoxicity at 1200 mg twice daily. Out of 34 patients, clinical responses occurred in 10 (29%), including 2 complete remissions (CRs) [Citation94]. Other clinical trials with 17 have shown objective responses in chronic myeloid leukemia (CML) [Citation95].
In a phase I trial, 15 was dosed orally twice a day in a dose escalating fashion to 20 patients with solid tumors. The DLTs were fatigue and gastrointestinal toxicity and the MTD was 350 mg twice daily. One PR was observed in a patient with NSCLC. In a phase II study of patients with pancreatic cancer, 15 was compared with standard gemcitabine therapy and two PRs were seen with 15 (200 mg twice daily) [Citation96].
A phase I study examined the administration of 8 as a continuous infusion to patients with advanced solid malignancies [Citation97]. The observed DLTs were myelosuppression, prolongation of the QTc interval and fatigue, and the MTD was 560 mg/m2 per day. No objective responses were seen, and the clinical development of this dual inhibitor has been discontinued. Compound 12 has been investigated clinically using both oral and intravenous delivery routes with later trials favoring infusion, in part to minimize gastrointestinal toxicity [Citation98]. In a phase I study, treatment with 12 led to objective responses in 24% of patients with advanced hematologic malignancies [Citation90] but the current development status of 12 is unclear.
Combination theraphy
Preclinical studies have revealed that FTIs can exhibit synergy when employed with other anticancer therapies [Citation99,Citation100]. FTIs have exhibited synergy with taxanes in particular and have been found to act as radiosensitizers [Citation98]. Recently 15 was shown to enhance the activity of imatinib against BCR-ABL-expressing cell lines [Citation101,Citation102], and evaluation of this combination in clinical trials was initiated. Other clinical studies have evaluated combinations of FTIs with a variety of current therapies including taxanes, gemcitabine, cisplatin, the antibody trastuzumab (Herceptin), and radiotherapy. Objective responses have been reported for 17, 15 and 12 in combination with paclitaxel and docetaxel [Citation98]. Compound 8 was evaluated in conjunction with standard radiotherapy in patients with either head and neck cancer or NSCLC suggesting that FTIs may be clinically useful as radiosensitizers [Citation103]. A phase I clinical trial examined the combination of 17 with gemcitabine and cisplatin in patients with advanced solid tumors and objective responses were seen in 33% of patients [Citation104].
Recently, the pediatric oncology branch at the National Institute of Health, USA carried out a Phase I trial and pharmacokinetic study of the FTI tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. They concluded that oral tipifarnib is well tolerated in children receiving the drug twice daily for 21 days and a continuous dosing schedule at 200 mg/m2/dose, which is equivalent to the MTD in adults. The pharmacokinetic profile of tipifarnib in children is similar to that in adults [Citation105].
Clinicians at the Albert Einstein Cancer Center targeted inhibition of farnesyltransferase in locally advanced breast cancer by undertaking phase I and II trial of tipifarnib plus dose-dense doxorubicin and cyclophosphamide. They concluded that Tipifarnib may be safely combined with dose-dense cyclophosphamide using a dose and schedule that significantly inhibits FTase enzyme activity in human breast cancer in vivo and may enhance the pathologic complete response rate after four cycles of pre-operative dose-dense cyclophosphamide [Citation106].
Molecular modeling studies
Very few in silico attempts have been made to explore the structural requirements responsible for farnesyltransferase inhibition. Corey and co-workers had earlier reported the crystal structure of a farnesyl protein transferase complexed with a CaaX peptide and FPP analog [Citation107]. They reported that in the ternary complex, the bound substrates are within van der Waals contact of each other and the FTase enzyme. α-Hydroxyfarnesylphosphonic acid (α-HFP) binds in an extended conformation in the active site cavity where positively charged side chains and solvent molecules interact with the phosphate moiety and aromatic side chains pack adjacent to the isoprenoid chain. The backbone of the bound CaaX peptide adopts an extended conformation, and the side chains interact with both FTase and α-HFP. The cysteine sulfur of the bound peptide coordinates the active-site zinc. Pete and co-workers proposed a model for substrate binding [Citation108]. They concluded that both farnesyl diphosphate and peptide substrates can be accommodated in the hydrophobic active-site barrel, with the sole charged residue inside the barrel, Arg202 of the β-subunit, forming a salt bridge with the negatively charged carboxy terminus of the peptide substrates.
In order to clarify how FTase discriminates between FPP and larger prenyl diphosphates, Tammy and his co-workers have examined the interactions between the enzyme and several isoprenoid analogs, GGPP, and the farnesylated peptide product using a combination of biochemical and structural methods. Comparison of the GGPP binding mode with the binding mode of the farnesylated peptide product suggests that the bulkier isoprenoid cannot rearrange to convert to product without unfavorable steric interactions with the acceptor protein. They proposed a ‘second site exclusion model’ in which FTase binds larger isoprenoids in a fashion that prevents the subsequent productive binding of the acceptor protein [Citation109].
Markus and co-workers using benzophenone based FTIs reported two novel aryl binding sites. One is located in close proximity to the zinc ion and is defined by the aromatic side chains Try 300β, Trp 303β, Tyr 361β and Tyr 365β. The second aryl binding site is defined by the side chains of Try 300β, Leu 295β, Lys 294β, Lys 353β and Lys 356β. This second aryl binding site has been used for the design of non-thiol FTIs [Citation110].
Modeling of binding modes and inhibition mechanism of some natural ligands of FTase using molecular docking was carried out by Alessandro and co-workers. Their aim was to show how molecular docking analysis can be successfully used to underline the inhibition mechanism of the natural ligands. They have shown how the binding modes of the compounds can be related to lipophilicity values, which are appreciably less for peptidomimetics than for FPP mimetics and reveal a straightforward method to predict and understand the FTase inhibition mechanism [Citation111].
In an effort to develop isoprenoid diphosphate-based FTIs, striking variations have been observed in the ability of modified analogs to bind to the enzyme. For example 2Z-GGPP is an alternative substrate with high binding affinity, while GGPP is not an alternative substrate. In this view Brian and co-workers have used pharmacophore and docking studies to elucidate a new binding pocket for isoprenoid analogs. The unique conformations between the first two isoprene units of 2Z-GGPP, but not GGPP, allows 2Z-GGPP to exploit this new binding pocket. These studies suggest that ligand conformational flexibility may be an important design consideration for the development of both inhibitors and alternative substrates of FTase [Citation112]. Emanuele and co-workers have successfully applied virtual screening methodology for developing new leads as FTase inhibitors [Citation113].
We, in our molecular modeling laboratory have applied an analog-based drug design approach applying the Comparative Molecular Field Analysis (CoMFA) technique to a series of benzonitrile derivatives as FTIs in order to correlate the inhibitory activity with the structural features of the molecule. This study highlighted the importance of steric and electrostatic functional groups responsible for FTase inhibitory activity [Citation114]. Further, we were also successful in building predictive 3D-QSAR models for a tricyclic piperazinyl class of FTIs which guided our synthetic efforts [Citation115].
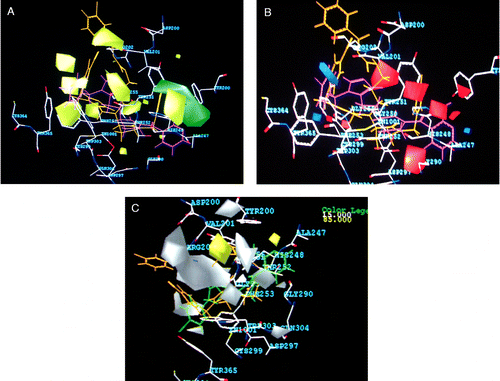
3D-QSAR analysis of antimalarial farnesyltransferase inhibitors based on a 2,5-diaminobenzophenone scaffold was performed by Doerkson and group in order to optimize FTase inhibitors potentially active against malaria [Citation116]. In continuation with our efforts to identify the pharmacophoric requirements of optimum FTase inhibitory activity we also carried out molecular modeling studies of 3-aminopyridones, imidazolymethyl ethers and 2-amino nicotinonitriles. The superimposition of contour maps on the FTase active site are depicted in . These results are yet to be published. Researchers from Cuba carried out modeling of FTase inhibition by some thiol and non-thiol peptidomimetic inhibitors using genetic neural networks and RDF approaches. Descriptors in the developed model suggested the occurrence of a strong dependence of FTase inhibition on the molecular shape and size rather than on electronegativity or polarizability characteristics of the studied compounds [Citation117]. Baldwin and co-workers illustrated how a large ECLiPS library of 26,908 compounds, based on a tricyclic core structure, was used to define a multitude of SARs for the oncogenic target, FTase [Citation118]. Further in silico studies will not only help us to address the selectivity issue of structurally diverse FTIs but also assist in the identification of lead compounds that can be used in cancer therapy.
Figure 4 STDDEV*COEFF contour plots for 3-aminopyrrolidinones. (A) CoMFA steric (B) CoMFA electrostatic (C) CoMSIA hydrophobic maps superimposed on FTase active site.
Conclusion and future directions
In this review, we have tried to summarize the current knowledge concerning the development of FTIs, their status in pre-clinical and clinical trials and some in silico attempts made to develop FTIs. Despite encouraging preclinical observations, FTIs have not succeeded as single-agent anticancer drugs for most solid cancers. Identifying the tumor molecular signatures that predict response to FTIs and GGTIs is a critical issue to be addressed in future hypothesis-driven mechanistic studies. Proteomics and the elucidation of oncogenic and survival pathways are potential avenues by which these questions could be answered.
| Abbreviations | ||
| FTase: | = | farnesyltransferase |
| FTIs: | = | farnesyl tranferase inhibitors |
| FPP/FDP: | = | farnesyl diphosphate |
| GGPP: | = | geranylgeranyl diphosphate |
| GAPs: | = | GTPase activating proteins |
| GEFs: | = | guanine nucleotide exchange factors |
| MTD: | = | maximum tolerated dose |
| AML: | = | acute myeloid leukemia |
| SCLC: | = | small cell lung cancer |
| GGTIs: | = | geranylgeranyl transferase inhibitors |
| NSCLC: | = | non small cell lung cancer |
| GGTIs: | = | geranylgeranyl transferase inhibitors |
| α-HFP: | = | α-hydroxyfarnesylphosphonic acid |
| DLT: | = | dose limited toxicity |
| CoMFA: | = | comparative molecular field analysis |
| CML: | = | chronic myeloid leukemia |
| ECLiPS: | = | encoded combinatorial library on polymeric support |
Acknowledgements
The work was supported by the All India Council for Technical Education, New Delhi, India, Grant No: 8021/RID/NPROJ/TAP-201/2002-03). One of the authors (DSP) would like to thank the University Grants Commission for the award of a junior research fellowship.
Related Research Data
References
- Levitzki A, Gazit A. Science 1995; 276: 1782–1788
- Gibbs JB, Oliff A. Cell 1994; 79: 193–198
- Clark GJ, Der CJ. Ras prota-oncogene activation in human malignancy. Cellular markers, CT Garnett, S Sell, 1995; 17–52
- Lowry DR, Willumsen BM. Annu Rev Biochem 1993; 62: 851–891
- Bernards R. Biochim Biophys Acta 1995; 1242: 43–59
- Largacspada DA, Branan CI, Jenkins NA, Copeland NG. Nat Genet 1996; 12: 137–143
- Gibbs JB. Semin Cancer Biol 1992; 3: 383–390
- Spaargaren M, Bischoff JR, McCornick F. Gene Exp 1995; 4: 345–356
- Manne V, Ricca CS, Brown JG, Tuomari AV, Yan N. Drug Dev Res 1995; 34: 121–137
- Willumsen BM, Norris K, Papageorge AG, Hubbert NL, Lowrey DR. EMBO J 1984; 3: 2581–2585
- Casey PJ, Solski PA, Der C, Bus JE. Proc Natl Acad Sci USA 1989; 86: 8323–8327
- Zhang FL, Casey PJ. Annu Rev Biochem 1996; 65: 241–269
- Leonard DM. J Med Chem 1997; 40: 2971–2990
- Fu HW, Casey PJ. Recent Prog Horm Res 1999; 54: 315–343
- Long SB, Casey PJ, Beese LS. Nature 2002; 419: 645–650
- Hightower KE, Huang C, Casey PJ, Fierke CA. Biochemistry 1998; 37: 15555–15562
- Rozema DB, Poulter CD. Biochemistry 1999; 38: 13138–13146
- Tschantz WR, Furfine ES, Casey PJ. J Biol Chem 1997; 272: 9989–9993
- Adjei A. J Natl Cancer Inst. 1062–1074, 932001a
- Cox D. Drugs 2001; 61: 723–732
- Hill TH, Hunt JT, Ding CZ, Batorsky R, Bednarz M, Bhide R, Cho Y, Chong S, Cho S, Gullo-Brown J, Guo P, Kim SH, Lee FY, Leftheris K, Miller A, Mitt T, Patel M, Penhallow BA, Ricca C, Rose WC, Schmidt R, Slusarchyk WA, Vite G, Perrin D, Kruczynski L. Crit Rev Oncol Hematol 2000; 33: 7–23
- Hahn SM, Bernhard E, McKenna WG. Semi Oncol 2001; 28: 86–93
- Karp JE, Kauffman SH, Adjei AA, Lancet JE, Wright JJ. Curr Opin Oncol 2001a; 13: 470–476
- Katz ME, McCormick F. Curr Opin Genet Dev 1997; 7: 75–79
- Boettner B, Van Aelst L. Genes Dev 2002; 16: 2033–2038
- Olson MF, Marais R. Semin Immunol 2000; 12: 63–73
- Hamad NE, Elconin JE, Karnoub AE, Bai W, Rich NJ, Abraham NT, Der CJ, Counter CM. Genes Dev 2002; 16: 2045–2057
- Bos JL. Cancer Res 1989; 49: 4682–4689
- Takai Y, Sasaki T, Matozaki T. Physiol Rev 2001; 81: 153–208
- Purcell WT, Donehower RC. Curr Oncol Rep 2002; 4: 29–36
- Falugi C, Trombino S, Granote P, Margaritota S, Russo P. Curr Cancer Drug Targets 2003; 3: 109–118
- Sebti SM, Hamilton AD. Oncogene 2000; 19: 6584–6593
- Crespo NC, Ohkanda J, Yen TJ, Hamilton AD, Sebti SM. J Biol Chem 2001; 276: 16161–16167
- Crespo NC, Delarve F, Ohkanda J, Carrico D, Hamilton AD, Sebti SM. Cell Death Differ 2002; 9: 702–709
- Ashar HR, James L, Gray K, Carr D, Black S, Armstrong L, Bishop WR, Kirschmeier P. J Biol Chem 2000; 275: 30451–30457
- Ashar HR, James L, Gray K, Carr D, McGuirk M, Maxwell E, Black S, Armstrong L, Doll RJ, Taveras AG, Bishop WR, Kirschmeier P. Exp Cell Res 2001; 262: 17–27
- Sebti SM, Hamilton AD. Drug Discov Today 1998; 3: 26–33
- Williams TM. Expert Opin Ther Patents 1999; 9: 1263–1280
- Rowinsky EK, Patnaik A. Emerg Drugs 2000; 5: 161–199
- Taveras G, Kirschneier P, Baum CM. Curr Top Med Chem 2003; 3: 1103–1114
- Kohl N, Omer C, Conner M, Anthony N, Davide J, Desolms S, Giuliani E, Gomez R, Graham S, Hamilton K, Handt L, Hartman G, Koblan K, Kral K, Miller P, Mosser S, O'Neil T, Rands T, Schabler M, Gibbs J, Oliff A. Nature Med 1995; 1: 792–797
- Lerner LC, Qian Y, Blaskowich MA, Ossum RD, Vogt A. J Biol Chem 1995; 270: 26802–26806
- Stephens TC, Wardleworth MJ, Matuziak MZ, Ashton SE, Hancox UJ, Bate M, Ferguson R, Boyle T. Proc Am Assoc Cancer Res 2003; 44: R4870
- Williams TM, Giccarone TM, MacTough SC, Bock RL, Conner MW, Davide JP, Hamilton K, Koblan KS, Kohl NE, Kral AM, Mosser SD, Omer CA, Pompliano DE, Rands E, Schaber MD, Shah D, Wilson FR, Gibbs JB, Graham SL, Hartman GD, OLiff AI, Smith RL. J Med Chem 1996; 39: 1345–1348
- Anthony NJ, Gomez RP, Schaber MD, Mosser SD, Hamilton KA, O'Neill TJ, Koblan KS, Graham SL, Hartman GD, Shah D, Rands E, Kohl NE, Gibbs JB, Oliff AI. J Med Chem 1999; 42: 3356–3368
- Williams TM, Bergman JM, Braschear K, Breslin MJ, Dinsmore CJ, Hutchinson JH, MacTough SC, Stump CA, Wei DD, Zartmen CB, Bogusky MJ, Culberson JC, Buser-Doepner C, Davide J, Greenberg IB, Hamilton KA, Koblan KS, Kohl NE, Liu D, Lobell RB, Mosser SD, O'Neill TJ, Rands E, Schabler MD, Wilson F, Senderak E, Motzel SL, Gibbs JB, Graham SL, Heimbook DC, Hartman GD, Oliff AI, Huff JR. J Med Chem 1999a; 42: 3779–3784
- Dinsmore CJ, Bogusky MJ, Culberson JC, Bergman JM, Homnick CF, Zartman CB, Mosser SD, Schaber MD, Robinson RG, Koblan KS, Huber HE, Graham SL, Hartman GD, Huff JR, Williams TM. J Am Chem Soc 2001; 123: 2107–2108
- Bell M, Gallicchio SN, Abrams M, Beese LS, Bishore DC, Bhimnathwala H, Bogusky MJ, Buser CA, Culberson JC, Divide J, Ellis-Hutchings M, Fernandes C, Gibbs JB, Graham SL, Hartman GD, Heimbrook DC, Homnick CF, Huber HE, Huff JR, Kassahun K, Koblan KS, Kohl NE, Lobell RB, Lynch JJ, Robinson R, Rodrigues AD, Taylor JS, Walsh ES, Williams TM, Zartmen CB. J Med Chem 2002; 45: 2388–2409
- Hunt JT, Lee VJ, Leftheris K, Seizinger B, Carboni J, Mabus J, Ricca C, Yan N, Manne V. J Med Chem 1996; 39: 353–358
- Ding CZ, Batorsky R, Bhide R, Chao HJ, Cho Y, Chong S, Gullu-Brown J, Guo P, Kim SH, Lee F, Leftheris K, Miller A, Mitt T, Patel M, Penhallow BA, Ricca C, Rose WC, Schmidt R, Slusarchyk WA, Vite G, Yan N, Manne V, Hunt JT. J Med Chem 1999; 42: 5241–5253
- Njoroge FG, Taveras AG, Kelly J, Remiszewiski S, Mallams AK, Wolin R, Afonso A, Cooper AB, Rane DF, Liu YT, Wong J, Vibhulban B, Pinto P, Deskus J, Alvarez CS, Del Rosario J, Connoly M, Wang J, Desai J, Rossman RR, Venet M, End D, Anggibaud P. Curr Top Med Chem 2003; 3: 1095–1102
- End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, Venet M, Sanz G, Poignet H, Skrzat S, Devine A, Wouters W, Bowden C. Cancer Res 2001; 61: 131–137
- Esteva MI, Kettler K, Maidana C, Fichera L, Ruiz AM, Bontempi EJ, Anderson B, Dahse H-M, Haebel P, Ortmann R, Klebe G, Schlitzer M. J Med Chem 2005; 48: 7186–7191
- Huckeo O, Gelb MH, Verlinde CLMJ, Buckner FS. J Med Chem 2005; 48: 5415–5421
- Huang C-Y, Stauffer TM, Strickland CL, Reader JC, Huang H, Li G, Cooper AB, Doll RJ, Ganguly AK, Baldwin JJ, Rokosz LL. Bioorg Med Chem Lett 2006; 16: 507–511
- Li Q, Li T, Woods KW, Gu WZ, Cohen J, Stoll VS, Galicia T, Hutchins C, Frost D, Rosenberg SH, Sham HL. Bioorg Med Chem Lett 2005; 15: 2918–2922
- Kettler K, Wiesner J, Sibler K, Haebel P, Ortmann R, Sattler I, Dahse H-M, Jomaa H, Klebe G, Schlitzer M. Eur J Med Chem 2005; 40: 93–101
- Guida WC, Hamilton AD, Crotty JW, Sebti SM. J Comput Aided Mol Des 2006; 19: 871–885
- Graham SL. Expert Opin Ther Patents 1995; 5: 1269–1285
- Hara M, Akasaka K, Akinaga S, Okabe M, Nakano H. Proc Natl Acad Sci USA 1993; 90: 2281–2285
- Gibbs JB, Pompliano DL, Mosser SD, Rands E, Lingham RB. J Biol Chem 1993; 268: 7617–7620
- Kohl NE, Mosser SD, dsolms SJ, Giuliani EA, Pomliano DL. Science 1993; 260: 1934–1937
- James GL, Goldstein JL, Brown MS, Rawson TE, Sommers TC. Science 1993; 260: 1937–1942
- Dalton MB, Fantle KS, Beehtold HE, DeMaio L, Evans RM. Cancer Res 1995; 55: 3295–3304
- James GL, Goldstein JL, Pathak RK, Anderson RJW, Brown MS. J Biol Chem 1994; 269: 14182–14190
- Vogt A, Qian Y, Blaskowich MA, Fossum RD, Hamilton AD. J Biol Chem 1995; 270: 660–664
- Lerner EC, Qian Y, Blaskowich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM. J Biol Chem 1995; 270: 26802–26806
- Nagasu T, Yoshimatsu K, Rowel C, Lewis MD, Garcin AM. Cancer Res 1995; 55: 5310–5314
- Cox D, Garcia AM, Westwick JK, Kowalczyk JJ, Lewis MD. J Biol Chem 1994; 269: 19203–19206
- Prendergast GC, Davide JP, deSolms SJ, Giulani EA, Graham SL. Mol Cell Biol 1994; 14: 4193–4202
- Sun J, Qian Y, Hamilton AD, Sebti SM. Cancer Res 1995; 55: 4243–4247
- Pai JJ-K, Bishop WR, Catino J, Kirschemeier P, Whyte D. Proc Am Assoc Cancer Res 1996; 37: 503–510
- Yan N, Ricca C, Fletcher J, Glover T, Scizinger BR. Cancer Res 1995; 55: 3569–3575
- Sepp-Lorenzino L, Ma Z, Rands E, Kohl NE, Gibbs JB. Cancer Res 1995; 55: 5302–5309
- Smith MR, DeGudiebus SJ, Staccay DW. Nature 1986; 320: 540–543
- Staccy DW, Roudebush M, Day R, Mosser SD, Gibbs JB. Oncogene 1991; 6: 2297–2304
- Ito T, Kawata S, Tamura S, Igura T, Nagase T. Jpn J Cancer Res 1996; 87: 113–116
- Sinn E, Muller W, Pattengale P, Tepler I, Wallace R. Cell 1987; 49: 465–475
- Hancock JF, Kadwalleder K, Patterson H, Marshall CJ. EMBO J 1991; 10: 4033–4039
- Cox D, Hisaka MM, Buss JE, Der CJ. Mol Cell Biol 1992; 12: 2606–2615
- James GL, Brown MS, Cobb MH, Goldstein JH. J Biol Chem 1994; 269: 27705–27714
- Prevost GP, Pradines A, Viossat I, Brezak MC, Miquek K, Lonchampt MO, Kaspryzk P, Favre G, Pignol B, Le Breton C, Dong J, Morgan B. Int J Cancer 1999; 83: 283–287
- Prevost GP, Pradines A, Brezak MC, Lonchampt MO, Viossat I, Ader I, Toulas C, Kaspryzk P, Gordon T, Favre G, Morgan B. Int J Cancer 2001; 91: 718–722
- Adjei A, Davis JN, Bruzek LM, Erlichman C, Kauffman SH. Clin Cancer Res 2001; 7: 1438–1445
- Shi B, Yaremko B, Hajian G, Terracina G, Bishop WR, Liu M, Nielson LL. Cancer Chemother Pharmacol 2000; 46: 387–393
- Mangues R, Corral T, Kohl NE, Symanns WF, Lu S, Malumbres M, Gibbs JB, Oliff A. Cancer Res 1998; 58: 1253–1259
- Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Lipari P, Malkowiski M, Ferrari E, Nielson L, Prioli N, Dell J, Sinha D, Syed J, Korfmacher WA, Nomier AA, Lin CC, Wang L, Taveras AG, Doll RJ, Njoroge FG, Mallams AK, Remiszewiski S, Catino JJ, Girijavallabhan VM, Bishop WR. Cancer Res 1998; 58: 4947–4956
- Dinsmore CJ, Bell IM. Curr Top Med Chem 2003; 3: 1075–1093
- Crul M, de Klerk GJ, Swart M, van't Veer LJ, de Jong D, Boerrigter L, Palmer PA, Bol CJ, Tan H, de Gast GC, Beijnen JH, Schellens JH. J Clin Oncol 2002; 20: 2726–2735
- Kantarjian HM, Cortes JE, Rowe JM, Kurzrock R. Semin Hematol 2002; 39: 36–38
- Lobell RB, Liu D, Buser CA, Davide JP, DePuy E, Hamilton K, Koblan KS, Lee Y, Mosser S, Motzel SL, Abruzzesse JL, Fuchs CS, Rowinsky EK, Rubin EH, Sharma S, Deustch PJ, Mazina KE, Morrison BW, Wildonger L, Yao SL, Kohl NE. Mol Cancer Ther 2002; 1: 747–758
- Adjei A, Davis JN, Erlichman C, Svigen PA, Kaufman SH. Clin Cancer Res 2000; 6: 2318–2325
- , Johnston HRD, Hickish T, Houston S, Ellis PA, Howes AJ, Thibault A. Presented at the 38th Annual meeting of the American Society of Clinical Oncology 2002. Abstract 138.
- Karp JE, Lancet JE, Kauffman SH, End DW, Wright JJ, Bol K, Horak I, Tidwell ML, Liesweld J, Kottke TJ, Ange D, Buddharaju L, Gojo I, Highsmith WE, Belly RT, Hohl RJ, Rybak ME, Thibault A, Rossenbat J. Blood 2001; 97: 3361–3369
- Cortes J, Albitar M, Thomas D, Giles F, Kurzrock R, Thibault A, Rackoff W, Koller C, O'Brien S, Garcia-Manero G, Talpaz M, Kantarjian H. Blood 2003; 101: 1692–1697
- , Lersch C, van Custem E, Amado R, Ehninger G, Heike M, Kerr D, Rothenberg ML, Baum CM, Zakneon SL. Presented at the 37th Annual meeting of the American Society of Clinical Oncology 2001. Abstract 608.
- Britten CD, Rowinsky EK, Soignet S, Patnaik A, Yao SL, Deutsch P, Lee Y, Lobell RB, Mazina KE, McCreery H, Pezulli S, Spriggs DA. Clin Cancer Res 2001; 7: 3894–3903
- Haluska P, Dy GK, Adjei AA. Eur J Cancer 2002; 38: 1685–1700
- Moasser MM, Sepp-Lorenzino L, Kohl NE, Oliff A, Su DS, Danishefsky SJ, Rosen N. Proc Natl Acad Sci USA 1998; 95: 1369–1374
- Sun J, Blaskowich MA, Knowles D, Qian Y, Ohka J, Bailey RD, Hamilton AD, Sebti SM. Cancer Res 1999; 59: 4919–4926
- Hoover RR, Mahon FX, Melo JV, Daley GQ. Blood 2002; 100: 1068–1071
- Nakajima A, Tauchi T, Sumi M, Bishop WR, Ohyashik K. Mol Cancer Ther 2003; 2: 219–224
- Hahn SM, Bernhard EJ, Regine W, Mohiuddin M, Haller DG, Stevenson JP, Smith D, Pramanik B, Tepper J, DeLaney TF, Kiel KD, Morrison B, Deutsh P, Muschel RJ, McKenna WG. Clin Cancer Res 2002; 8: 1065–1072
- Adjei A, Croghan GA, Erlichman C, Marks RS, Reid JM, Sloan JA, Pitot HC, Alberts SR, Goldberg RM, Hanson LJ, Bruzek LM, Atherton P, Thibault A, Palmer PA, Kauffman SH. Clin Cancer Res 2003; 9: 2520–2526
- Widemann BC, Salzer WL, Arceci RJ, Blaney SM, Fox E, End D, Gillespie A, Whitcomb P, Palumbo JS, Pitney A, Jayaprakash N, Zannikos P, Balis FM. J Clin Oncol 2006; 24(3)507–516
- Sparano JA, Moulder S, Kazi A, Vahdat L, Li T, Pellegrino C, Munster P, Malafa M, Lee D, Hoschander S, Hopkins U, Hershman D, Wright JJ, Sebti SM. J Clin Oncol 2006; 24(19)3013–3018
- Corey LS, William TW, Rosalinda S, Lyan W, Richard B, Zhen W, Jeffry S, Hung VL, Lorena SB, Patricia CW. Biochemistry 1998; 37: 16601–16611
- Pete D, Ursula K, Robert C, David W, Robert P, Jens B. Biochemistry 1998; 37: 7907–7912
- Tammy CT, Corey LS, Mark DD. Biochemistry 2003; 42: 3716–3724
- Markus B, Andreas M, Pia W, Isbel S, Martin S. J Med Chem 2001; 44: 3317–3324
- Alessandro P, Luigi V, Giulio V. J Med Chem 2002; 45: 1460–1465
- Brian SH, Todd JZ, Jrffery DE, Steven MF, Richard AG. J Chem Inf Model 2005; 45: 1047–1052
- Perola E, Xu K, Kollmeyer TM, Kauffmann SH, Prendergast FG, Pang YP. J Med Chem 2000; 43: 401–408
- Puntambekar D, Giridhar R, Yadav MR. Bioorg Med Chem Lett 2006; 167: 1821–1827
- Puntambekar DS, Giridhar R, Yadav MR. Eu Med Chem 2006, In Press
- Xie A, Sivprakasan P, Doerkson R. Bioorg Med Chem 2006, Jul 10: In Press
- Gonzalez MP, Caballero J, Tundidor-Camba A, Helguera AM, Fernandez M. Bioorg Med Chem 2006; 14(1)200–213
- Rokosz LL, Huang CY, Reader JC, Stauffer TM, Southwick EC, Li G, Chelsky D, Baldwin JJ. Comb Chem High Throughput Screen 2006; 9(7)545–548