Abstract
The inhibition mechanism of electrophilic peptide-based protease inhibitors of full-length hepatitis C virus (HCV) NS3 has been investigated by determining the Ki-values for a series of compounds differing in the electrophilicity and acidity of the C-terminal residue at pH-values above and below the pKa of the catalytic histidine (6.85) and at two different ionic strengths. Electrophilic compounds with a pentafluoroethyl ketone group showed stronger inhibition at pH 8 than pH 6, as expected for a mechanism requiring an unprotonated catalytic histidine. However, the difference was only significant at high ionic strength. In contrast, electrophilic compounds with an acidic C-terminal group or a cyclic P1 residue showed a lower inhibitory effect at pH 8 than at pH 6, inconsistent with a mechanism-based inhibition. Moreover, all electrophilic compounds had an unexpectedly strong inhibition at pH 6, when mechanism-based inhibition is unlikely. The results suggest that for some of the electrophilic compounds the reactive group may not be properly positioned in the active site and that binding of these inhibitors is a result of non-covalent interactions. The nature of these interactions is discussed.
Introduction
Hepatitis C virus (HCV) is a global pathogen with a 3% average infection prevalence but with large geographic differences in the distribution of cases. The identification of the virus in 1989 has resulted in reduced transmission and improved therapeutic strategies for infected patients. But current therapy is inadequate and coming medication is predicted to be hampered by development of resistance (for a review, see [Citation1]). There is therefore a great need to discover and develop new therapeutic strategies for treatment of HCV infections.
One of the most actively pursued targets for HCV drug therapy is the non-structural protein 3 (NS3) of the virus. It is a bifunctional protease/helicase with the two different enzymatic activities situated in separate domains of the protein. The protease is a chymotrypsin-like serine protease, but with some distinct features of relevance for drug discovery. An important early observation was that N-terminal cleavage products of HCV NS3 protease are inhibitors of the enzyme itself [Citation2,Citation3]. This has been exploited for the creation of a variety of potent peptide-based inhibitors. Three compounds, which have been designed with such lead compounds as starting points, have shown proof-of-concept in clinical trials: BILN 2061, VX-950 and SCH 503034 Citation4-6, confirming that inhibition of NS3 protease is a viable strategy for HCV therapy. However, cell-based assays of viral replication in the presence of BILN 2061 [Citation7], VX-950 [Citation8] or SCH 503034 [Citation9] have confirmed that resistance against protease inhibitors arises as a result of single amino acid substitutions in the protease domain, and that cross-resistance between inhibitors can be expected [Citation10]. Furthermore, a cardiac histological toxicity towards BILN 2061 has been detected in rhesus monkeys [Citation11], indicating a potential, but severe, side effect of this compound. Hence, there are obviously still considerable challenges in the development of effective and safe drugs based on inhibition of HCV protease.
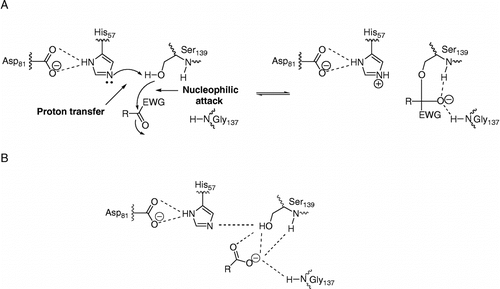
Optimization of peptide-based NS3 protease inhibitors includes the reduction of the peptidic nature of the inhibitors and the introduction of C-terminal carboxylic acid replacements. Potent peptide-based inhibitors of serine and cysteine proteases can be designed by replacing a C-terminal amide carbonyl by an electrophilic carbonyl group whereby a mechanism-based inhibition is achieved (for a review, see [Citation12]). This mechanism is illustrated in and resembles the first step of substrate hydrolysis. It involves a nucleophilic attack by the catalytic serine on the electrophilic carbonyl group of the inhibitor and the formation of a covalent, but reversible bond. The formed oxyanion is stabilized by interaction with two backbone amides in the oxyanion hole. Although the covalent complex is transient, it can be compared to the reversible non-covalent complex formed between product based inhibitors and the enzyme, as illustrated in . The stabilizing forces in this complex arises from interactions between the α-carboxylate oxygens of the inhibitor with the main chain amido groups of the residues forming the oxyanion hole, and possibly also with the Oγ of the catalytic serine residue [Citation2,Citation13,Citation14]. An interaction with the imidazole ring of the catalytic histidine has also been proposed [Citation13,Citation14], but this interaction is expected to be less significant at pH values above that of the catalytic histidine residue [Citation2]. Both types of inhibitors have succesfully been developed as far as clinical trials, with BILN 2061 representing an inhibitor with a C-terminal carboxylic acid [Citation4], and VX-950 and SCH 503034 representing inhibitors with a C-terminal electrophilic group (an α-ketoamide). Structural studies of VX-950 have confirmed that it acts according to the general serine protease inhibition mechanism [Citation15].
Figure 1 A schematic representation of the active site of HCV NS3 and its interaction with non-electrophilic and electrophilic inhibitors, assuming typical serine protease inhibition mechanisms. (A) mechanism-based inhibition by compounds with a C-terminal electron withdrawing group (EWG), and (B) product-based inhibitors with C-terminal carboxyl groups.
We have also explored electrophilic groups in our attempts to design effective inhibitors of HCV NS3 protease. But we obtained some unexpected structure-activity relationships when comparing the inhibitory effect of electrophilic and non-electrophilic compounds. For example, peptide-based inhibitors with electrophilic C-terminals, e.g. pentafluoroethyl ketone, ketotetrazole or ketoacid, were not much better, or sometimes worse, than the corresponding inhibitors without the electrophilic group, for example carboxylates and acyl sulfonamides [Citation16]. The results supported an earlier exploration of different electrophilic groups which found that only α-ketoamides afforded an enhanced inhibitory effect [Citation17]. In addition, the characteristic slow-binding of many electrophilic inhibitors of serine and thiol proteases was absent in our studies of electrophilic NS3 protease inhibitors [Citation18], although it had been observed for α-ketoacid and α-ketoamide inhibitors [Citation15,Citation19]. Taken together, these observations have led us to question the inhibition mechanism for a number of electrophilic peptide-based inhibitors of NS3 protease.
The present experiments were designed to determine if a covalent bond was critical for the inhibition of HCV NS3 protease by electrophilic compounds and how much it contributed to the affinity of the interaction, as compared to alternative interactions in the active site, for example electrostatic interactions between an acidic C-terminal group and the catalytic histidine or the oxyanion hole. It was based on the two alternative mechanisms for inhibition of serine proteases illustrated in , and differences in the effects of varying the pH and ionic strength of the assay buffer. Electrophilic inhibitors are expected to be more efficient at pH values above the pKa of the catalytic histidine since a prerequisite for the formation of a covalent bond is the same as for catalysis. High pH thus ensures that the catalytic triad is functional so that the essential proton transfer from the catalytic serine to the catalytic histidine can occur. This mechanism should not be significantly influenced by changes in ionic strength. In contrast, the interaction with a non-electrophilic compound having a C-terminal carboxylate residue is expected to have higher affinity at pH values below the pKa of the catalytic histidine (but above the pKa of the C-terminal carboxylic acid group) and at low ionic strength conditions since these conditions favour the ionic interactions and hydrogen bonds that stabilize the enzyme-inhibitor complex. The strategy used to investigate the mechanism for inhibition of HCV NS3 was consequently based on a simple approach where the inhibition was studied at two pH values; one above and one below the pKa of the catalytic histidine residue. In addition, two ionic strengths were used for the measurements.
For this study, a series of hexapeptide inhibitors differing both in the P1 side chain and the C-terminal group () was used. Three P1-side chains were selected as replacements for the P1 cysteine in the native substrate: L-aminobutyric acid (Abu), L-norvaline (Nva) and 1-aminocyclopropane-1-carboxylic acid (ACPC). These have all been used in product-based inhibitors with a C-terminal carboxylic acid [Citation3,Citation20]. The ACPC group has most successfully been exploited in BILN 2061 [Citation21]. In addition, four different C-terminal groups were evaluated in our study and these can be further divided into three groups on the basis of electrophilicity and acidity: 1) the electrophilic and non-acidic pentafluoroethyl ketone (PFK) group, 2) the electrophilic and acidic ketotetrazole (KT) and ketoacid (KA) groups and 3) the non-electrophilic and acidic carboxylic acid (COOH) group. This set of compounds was suitable for an initial exploration of the mechanism of electrophilic HCV NS3 inhibitors.
Table I. Inhibition of HCV NS3 by electrophilic and non-electrophilic inhibitors at different pH and ionic strengths†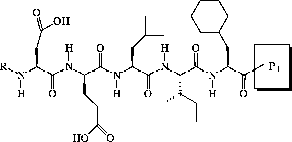 .
.
Materials and methods
Inhibitors
All inhibitors () except the L-aminobutyric acid peptide with a C-terminal carboxylic acid group (Abu COOH) have R = succinyl and have been described previously [Citation16]. Abu COOH had R = acetyl and has been described by [Citation22]. Compounds were dissolved in DMSO and stored at − 20°C between measurements.
Kinetic analysis
Measurements of the activity of full length HCV NS3 and the determination of kinetic constants have been carried out essentially as described earlier [Citation23]. Specifically, the enzyme was incubated in 25 mM MES, 25 mM MOPS, 25 mM BICINE, pH 6.0–9.0, 0–100 mM NaCl,10 mM DTT, 40% glycerol, 0.1% n-octyl-β-D-glucoside, 3.3% DMSO with 25 μM cofactor (KKGSVVIVGRIVLSGK) and inhibitor at 30°C for 10 min; the reaction was initiated by addition of substrate (Ac-DED(Edans)EEAbuψ[COO]ASK(Dabcyl)-NH2, AnaSpec, San Jose, CA, USA). All measurements were performed in triplicates. Kinetic constants (Vmax and KM) were determined with substrate concentrations from 0.25 to 4 μM and 1 nM enzyme. Inhibition measurements were performed with 1 nM enzyme, 0.5 μM substrate and different inhibitor concentrations.
Km- and Vmax-values were determined for each buffer separately by fitting the Michaelis-Menten equation to initial reaction rate velocities (v0) at different substrate concentrations by non-linear regression analysis (GraFit, Erithacus Software, Staines, MX, UK). Vmax was expressed in arbitrary units (AU), since the enzyme concentration was constant in all measurements, and there was no need to convert the relative fluorescence units into molar units. Ki-values were calculated from initial velocities at different inhibitor concentrations by non-linear regression analysis using the standard equation for competitive inhibition:
where Km was set to be constant.
Determination of pKa
The pKa of catalysis was determined by non-linear regression analysis, using:
and data of Vmax as a function of pH.
and
are the high and low limits of Vmax. The equation is a rearrangement of the equation for a singly ionizing system [Citation24] as implemented in GraFit.
Results
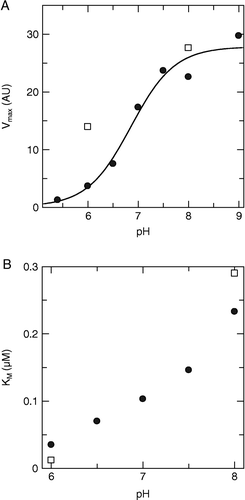
The activity of HCV NS3 protease as a function of pH was determined in order to identify the pH range for which the activity of the enzyme could be reliably measured, and to determine the pKa of the catalytic histidine residue. Therefore, Km and Vmax values were determined as a function of pH over range from 5.4 to 9.0 (). A full set of measurements was performed at low ionic strength, while measurements at high ionic strength (here “low” and “high” are relative terms for this assay, represented by 0 and 100 mM NaCl in the buffer, respectively) were limited to two pH values, as this was sufficient for verifying that the same trend was valid for both conditions. Both Km and Vmax were found to increase with pH, as illustrated in and . A wider pH range could be used for Vmax than for Km, (the graph only includes values that could be reliably determined). By fitting an equation representing a single ionic species to the plot of pH versus Vmax () the pKa of NS3 protease was estimated to be 6.85 ± 0.16, in accordance with that previously reported for the catalytic histidine of the truncated enzyme [Citation25]. The pH values used for inhibition analysis in the present study were selected to clearly span the pKa-value of the catalytic histidine and to represent the lowest pH where the enzyme was sufficiently active for accurate inhibition measurements, and the highest judged suitable for the current buffer (pH 6.0 and 8.0, respectively).
Figure 2 Dependence of the kinetic constants of HCV NS3 on pH. A) Vmax and B) Km as a function of pH, at low (•) and high (▪) ionic strength.
The inhibition constant for inhibition of HCV NS3 by a series of hexapeptide inhibitors, differing in the electrophilicity and acidity of the P1 residue was determined at pH-values above and below the pKa of the catalytic histidine and with or without 100 mM NaCl (). In general, the inhibition was significantly affected by changes in pH and ionic strengths, but the effect was characteristic for the different inhibitors and a general pattern could not easily be identified from the table. In order to reveal differences between the different compounds the data set was also visualized graphically ().
Figure 3 Effect of pH and NaCl concentrations on the inhibition constants for different inhibitors. A) Overview of Ki values for all compounds and conditions, see for details, B) Ratio of KipH 8.0/KipH 6.0 at 0 mM NaCl (grey) and 100 mM NaCl (black). C) Ratio of Ki100 mM NaCl/Ki0 mM NaCl at pH 6.0 (grey) and pH 8.0 (black).
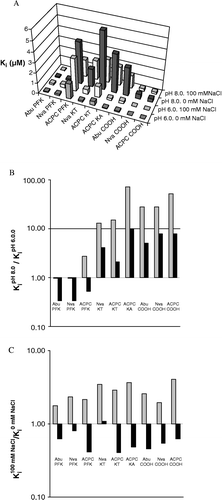
shows that the inhibitory effect was not simply correlated with either electrophilicity or acidity, irrespective of the conditions. The most potent inhibitors were either electrophilic and non-acidic (Abu PFK and Nva PFK) or non-electrophilic and acidic (Nva COOH and ACPC COOH), and significantly less effective compounds were found in these groups as well as in the group of electrophilic and acidic compounds. From a practical perspective, Abu PFK and Nva PFK are expected to be the most efficient inhibitors under physiological conditions which can be assumed to be best represented by pH > 6.85. This is independent on the ionic strength, even if 100 mM would be more realistic than 0 mM.
The inhibitory effects of the non-acidic electrophilic compounds (Abu PFK, Nva PFK and ACPC PFK) were not significantly different at pH 6 and pH 8 at low ionic strengths (). However, the inhibitory effects were significantly higher at pH 8 than at pH 6 when the ionic strength was “high” (here 100 mM), seen as values < 1 in the KipH 8.0/KipH 6.0 graph (). An exception was ACPC PFK which was more inhibitory at pH 6 than at pH 8 at low ionic strength. However, this compound had a relatively high Ki and other effects should perhaps be considered (see discussion). In contrast, all acidic compounds, irrespective of whether they were electrophilic or not (Abu COOH, Nva COOH, ACPC COOH, ACPC KT, Nva KT, ACPC KA), showed the opposite, i.e. had lower Ki at the lower pH (pH 6) at both high and low ionic strength (seen as values >1 in the KipH 8.0/KipH 6.0 graph, ).
The inhibitory potency of all compounds was significantly reduced (higher Ki-values) in the presence of salt at pH 6 (values > 1 in the Ki100 mM NaCl/Ki0 mM NaCl graph, ), while the Ki-values of all inhibitors (except Nva KT) were lower in the presence of salt at pH 8 (values < 1 in the Ki100 mM NaCl/Ki0 mM NaCl graph, ). The importance of electrostatic interactions was obviously different above and below the pKa of histidine, irrespective of whether compounds were electrophilic or acidic. But the influence of ionic strength on the potencies of the inhibitors was less pronounced as compared to the effect of changing the pH (note the differences in the y-axis scale in and 3C).
Discussion
The present study confirms our previous observation that the structure-activity relationship of electrophilic inhibitors of HCV NS3 protease is unexpected and that the formation of a covalent bond can be questioned [Citation16]. The data for this series of inhibitors only support a mechanism-based inhibition of HCV NS3 protease by the pentafluoroethyl ketones, i.e. the non-acidic electrophilic compounds tested. This is concluded from the fact that they are the only compounds that show a more effective inhibition at pH 8 than at pH 6, but only at high ionic strength when electrostatic interactions are less pronounced. The weaker effect for the ACPC PFK compound is in accordance with the sterically unfavourable formation of the tetrahedral intermediate when the P1 side chain is α,α-disubstituted [Citation26], as for ACPC compounds.
So how do the remaining electrophilic compounds become potent inhibitors of the enzyme without exploiting the reactivity of the active site? An obvious alternative is that inhibition occurs by a mechanism similar to that for non-electrophilic compounds. In this study we have therefore used a series of non-electrophilic analogues with a C-terminal carboxylate for comparison. The acidic group of these compounds interacts with the oxyanion hole of the enzyme, forming a non-covalent but tight enzyme-inhibitor complex (). We propose that the acidic electrophilic compunds interact in a similar way via the acidic residue (i.e. the carboxylate or the tetrazole). Although this does not rule out the formation of a covalent bond, it appears that electrostatic interactions are more important and that they interfere with the proper positioning of the electrophilic carbonyl group relative to the catalytic residues and that they thereby hinder the nucleophilic attack. The effect is probably accentuated for the sterically hindered ACPC-compounds (see above).
However, this does not explain why the non-acidic electrophilic compounds are such potent inhibitors at pH 6, when the covalent bond is unlikely to form, or why the acidic electrophilic compounds are more inhibitory at pH 6 than at pH 8, a pH range where the interactions with the oxyanion hole should be unaffected. The most obvious residue that may be involved is the catalytic histidine, a residue that is protonated and positively charged at pH 6, but not at pH 8. Thus, the present data can be rationalized by assuming that the electrophilic carbonyl is oriented towards the catalytic histidine, where it can form a hydrogen bond with the imidazole ring, instead of towards the oxyanion hole as is typical for serine proteases. This carbonyl orientation has been found for the binding of α-ketoacid inhibitors to HCV NS3 protease [Citation19,Citation27] and a related orientation for α-ketoamides [Citation28]. Another residue that has a similar pH dependence is His528, located in the helicase domain of NS3. It may form a hydrogen bond with the carbonyl oxygen of the P4 residue [Citation13,Citation29]. Note that this interaction is only possible in the full-length HCV NS3, and may be overlooked in studies based on the truncated enzyme. The non-electrophilic acidic compounds show a more enhanced inhibition at pH 6, suggesting that two histidine residues may be involved, supporting a role for His528.
In addition to the specific interactions between the active site and the C-terminal moiety of the inhibitors, there are other interactions of importance for the binding of these compounds. They include interactions between the positively charged arginine and lysine residues in the S5/S6-sites (Arg161 and Lys165) and the negative P5/P6 residues [Citation30]. There are also hydrophobic interactions with the P1–P4 side chains. These non-active site interactions are not expected to be significantly influenced by the changes in pH in the range from 6 to 8, but may be affected by the changes in ionic strength. The somewhat enhanced affinity of non-acidic electrophilic compounds at higher ionic strength at pH 8 suggests that electrostatic interactions are of minor importance under these conditions. Whether the increase is simply due to the formation of a covalent bond or if hydrophobic interactions contribute significantly cannot be discerned.
The present data suggest that the orientation of the carbonyl oxygen of electrophilic compounds is critical for the ability of the catalytic serine to perform a nucleophilic attack, and thus for the inhibitory effect. Thus, both the electrophilic group and the groups adjacent to the electrophilic carbon have an effect. For example, the ACPC group was not favourable for the formation of the covalent bond. Likewise, the ketoacid group did not result in a mechanism where the formation of a covalent group contributed significantly to the inhibitory effect. This indicates that the nature of the peptide scaffold also has an indirect effect since the ketoacid group has been found to result in mechanism-based inhibition when incorporated into two other peptide scaffolds [Citation27].
Despite the rational strategy for the insertion of an electrophilic group in peptide-based inhibitors of NS3 protease, the benefit of forming a covalent bond can be questioned since high affinity can obviously be achieved even without the electrophilic group (e.g. Nva COOH and ACPC COOH). The effective binding of non-electrophilic acidic compounds is afforded both by the general electrostatic and hydrophobic interactions mentioned above, and more specifically by electrostatic interactions with the oxyanion hole and the histidine. Although a second electrostatic interaction may also be involved (as discussed above), it cannot be distinguished from that with the catalytic histidine since these two interactions have the same pH and ionic strength dependencies. Even if the acidic electrophilic compounds contain an electrophilic group, it does not appear to be responsible for the inhibitory effect since these compounds interact in the same mode as the non-electrophilic compounds and show the same pH- and ionic strength-dependency.
Nevertheless, electrophilic groups have successfully been incorporated into HCV NS3 protease inhibitors, with VX-950 being the most successful so far. A practical problem with electrophilic inhibitors is that they have been found to have a lower selectivity compared to their non-electrophilic counterparts [Citation17,Citation18]. VX-950 is an interesting exception to this principle [Citation15], but the clinical potential of electrophilic compounds as drugs for HCV therapy remains to be established.
In addition to the information about the interaction between the enzyme and inhibitors, this study has revealed the importance of an electrostatic interaction between the enzyme and the C-terminal moiety of the substrate. It comes from the observation that the pH-dependency of the Km determined with the substrate was different in this study compared to that found in a previous study [Citation2]. Here it was similar to that for inhibitors with C-terminal carboxylate groups, indicating that substrate and inhibitor interactions were similar. However, Steinkühler et al. found that Km was greatly increased as pH was reduced below the pKa expected for a histidine residue. There appears to be electrostatic repulsion between the substrate and the enzyme at pKa-values below that for a histidine, consistent with that expected from the histidine residue in the P3′-position of the substrate used for their studies (Ac-DEMEECASHLPYK-NH2). In contrast, the substrate used in this study (DED(Edans)EEAbuψ[COO]ASK(Dabcyl)-NH2) contained a lysine in the P3′-position and the Km for this substrate showed less pH-dependency and in the opposite direction, not at all characteristic of a histidine. The P3′residue in the natural HCV NS3 protease substrates varies, and includes both histidine and lysine. (Note that the Steinkühler et al. study cannot show effects of an interaction with His528 since this residue is located in the helicase domain, not present in the truncated form of NS3 used in their study.)
Conclusions
The present study has shed light on the mechanism of electrophilic peptide-based inhibitors of HCV NS3 protease. Importantly, it has revealed that inhibition of HCV NS3 protease by electrophilic compounds via a mechanism-based formation of a covalent bond is only significant if they are non-acidic and if the ionic strength is high. In contrast, inhibition at low ionic strength is dominated by electrostatic interactions, particularly if the compounds contain an acidic C-terminal group. Moreover, at least two types of interactions are responsible for the high affinity of the non-acidic but electrophilic compounds, where the covalent interaction dominates at high pH and electrostatic interactions dominate at low pH. The acidic electrophilic and non-electrophilic compounds achieve their inhibitory effect by electrostatic interactions which are stronger at pH 6 than pH 8. The findings are consistent with the unconventional orientation of the carbonyl oxygen in electrophilic inhibitors towards the catalytic histidine in HCV NS3 protease, and/or the involvement of an additional interaction between a histidine residue and the N-terminal moiety of the inhibitor.
References
- De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature 2005; 436(7053)953–960
- Steinkühler C, Biasiol G, Brunetti M, Urbani A, Koch U, Cortese R, Pessi A, De Francesco R. Product inhibition of the hepatitis C virus NS3 protease. Biochemistry 1998; 37(25)8899–8905
- Llinàs-Brunet M, Bailey M, Fazal G, Goulet S, Halmos T, Laplante S, Maurice R, Poirier M, Poupart MA, Thibeault D, Wernic D, Lamarre D. Peptide-based inhibitors of the hepatitis C virus serine protease. Bioorg Med Chem Lett 1998; 8(13)1713–1718
- Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bös M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagacé L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinàs-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003; 426(6963)186–189
- Reesink HW, Zeuzem S, Weegink CJ, Forestier N, van Vliet A, van de Wetering de Rooij J, McNair LA, Purdy S, Chu H-M, Jansen PLM. Final results of a phase 1B, multiple-dose study of VX-950, a hepatitis C virus protease inhibitor. Hepatology (New York) 2005; 42(233A), abst 96
- Zeuzem S, Sarrazin C, Rouzier R, Tarral A, Brion N, Forestier N, Gupta S, Deckman D, Fellows K, Hussain M, Cutler DL, Zhang J. Anti-viral activity of SCH 503034, a HCV protease inhibitor, administered as monotherapy in hepatitis C genotype-1 (HCV-1) patients refractory to pegylated interferon (Peg-IFN-a). Hepatology 2005; 42(233A), abst 94
- Lu L, Pilot-Matias TJ, Stewart KD, Randolph JT, Pithawalla R, He W, Huang PP, Klein LL, Mo H, Molla A. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob Agents Chemother 2004; 48(6)2260–2266
- Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL, Fulghum JR, Hsiao HM, Ma S, Maxwell JP, Cottrell KM, Perni RB, Gates CA, Kwong AD. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: Structural analysis indicates different resistance mechanisms. J Biol Chem 2004; 279(17)17508–17514
- Tong X, Chase R, Skelton A, Chen T, Wright-Minogue J, Malcolm BA. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res 2006; 70(2)28–38
- Lin C, Gates CA, Rao BG, Brennan DL, Fulghum JR, Luong YP, Frantz JD, Lin K, Ma S, Wei YY, Perni RB, Kwong AD. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J Biol Chem 2005; 280(44)36784–36791
- Reiser M, Hinrichsen H, Benhamou Y, Reesink HW, Wedemeyer H, Avendano C, Riba N, Yong CL, Nehmiz G, Steinmann GG. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology 2005; 41(4)832–835
- Mehdi S. Synthetic and naturally occurring protease inhibitors containing an electrophilic carbonyl group. Bioorg chem 1993; 21: 249–259
- Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 1997; 7(11)1353–1363
- Tsantrizos YS, Bolger G, Bonneau P, Cameron DR, Goudreau N, Kukolj G, LaPlante SR, Llinàs-Brunet M, Nar H, Lamarre D. Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection. Angew Chem Int Ed Engl 2003; 42(12)1356–1360
- Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob Agents Chemother 2006; 50(3)899–909
- Johansson A, Poliakov A, Åkerblom E, Wiklund K, Lindeberg G, Winiwarter S, Danielson UH, Samuelsson B, Hallberg A. Acyl sulfonamides as potent protease inhibitors of the hepatitis C virus full-length NS3 (protease-helicase/NTPase). A comparative study of different C-terminals. Bioorg Med Chem 2003; 11: 2551–2568
- Llinàs-Brunet M, Bailey M, Déziel R, Fazal G, Gorys V, Goulet S, Halmos T, Maurice R, Poirier M, Poupart MA, Rancourt J, Thibeault D, Wernic D, Lamarre D. Studies on the C-terminal of hexapeptide inhibitors of the hepatitis C virus serine protease. Bioorg Med Chem Lett 1998; 8(19)2719–2724
- Poliakov A, Johansson A, Åkerblom E, Oscarsson K, Samuelsson B, Hallberg A, Danielson UH. Structure-activity relationships for the selectivity of hepatitis C virus NS3 protease inhibitors. Biochim Biophys Acta 2004; 1672(1)51–59
- Narjes F, Brunetti M, Colarusso S, Gerlach B, Koch U, Biasiol G, Fattori D, De Francesco R, Matassa VG, Steinkühler C. Alpha-ketoacids are potent slow binding inhibitors of the hepatitis C virus NS3 protease. Biochemistry 2000; 39(7)1849–1861
- Llinàs-Brunet M, Bailey M, Fazal G, Ghiro E, Gorys V, Goulet S, Halmos T, Maurice R, Poirier M, Poupart MA, Rancourt J, Thibeault D, Wernic D, Lamarre D. Highly potent and selective peptide-based inhibitors of the hepatitis C virus serine protease: towards smaller inhibitors. Bioorg Med Chem Lett 2000; 10(20)2267–2270
- Llinàs-Brunet M, Bailey MD, Bolger G, Brochu C, Faucher AM, Ferland JM, Garneau M, Ghiro E, Gorys V, Grand-Maître C, Halmos T, Lapeyre-Paquette N, Liard F, Poirier M, Rhéaume M, Tsantrizos YS, Lamarre D. Structure-activity study on a novel series of macrocyclic inhibitors of the hepatitis C virus NS3 protease leading to the discovery of BILN 2061. J Med Chem 2004; 47(7)1605–1608
- Oscarsson K, Poliakov A, Oscarson S, Danielson UH, Hallberg A, Samuelsson B. Peptide-based inhibitors of hepatitis C virus full-length NS3 (protease-helicase/NTPase): Model compounds towards small molecule inhibitors. Bioorg Med Chem 2003; 11(13)2955–2963
- Poliakov A, Hubatsch I, Shuman CF, Stenberg G, Danielson UH. Expression and purification of recombinant full-length NS3 protease-helicase from a new variant of hepatitis C virus. Protein Expr Purif 2002; 25: 363–371
- Fersht A. Structure and mechanism in protein science. A guide to enzyme catalysis and protein folding. W.H. Freeman and Company, New York 1999
- Urbani A, Bazzo R, Nardi MC, Cicero DO, De Francesco R, Steinkühler C, Barbato G. The metal binding site of the hepatitis C virus NS3 protease. A spectroscopic investigation. J Biol Chem 1998; 273(30)18760–18769
- Perni RB, Britt SD, Court JC, Courtney LF, Deininger DD, Farmer LJ, Gates CA, Harbeson SL, Kim JL, Landro JA, Levin RB, Luong YP, O'Malley ET, Pitlik J, Rao BG, Schairer WC, Thomson JA, Tung RD, Van Drie JH, Wei Y. Inhibitors of hepatitis C virus NS3.4A protease 1. non-charged tetrapeptide variants. Bioorg Med Chem Lett 2003; 13(22)4059–4063
- Di Marco S, Rizzi M, Volpari C, Walsh MA, Narjes F, Colarusso S, De Francesco R, Matassa VG, Sollazzo M. Inhibition of the hepatitis C virus NS3/4A protease. The crystal structures of two protease-inhibitor complexes. J Biol Chem 2000; 275(10)7152–7157
- Perni RB, Pitlik J, Britt SD, Court JJ, Courtney LF, Deininger DD, Farmer LJ, Gates CA, Harbeson SL, Levin RB, Lin C, Lin K, Moon YC, Luong YP, O'Malley ET, Rao BG, Thomson JA, Tung RD, Van Drie JH, Wei Y. Inhibitors of hepatitis C virus NS3.4A protease 2. warhead SAR and optimization. Bioorg Med Chem Lett 2004; 14(6)1441–1446
- Johansson A, Poliakov A, Åkerblom E, Lindeberg G, Winiwarter S, Samuelsson B, Danielson UH, Hallberg A. Tetrapeptides as potent protease inhibitors of Hepatitis C Virus full-length NS3 (protease-helicase/NTPase). Bioorg Med Chem 2002; 10(12)3915–3922
- Koch U, Biasiol G, Brunetti M, Fattori D, Pallaoro M, Steinkühler C. Role of charged residues in the catalytic mechanism of hepatitis C virus NS3 protease: Electrostatic precollision guidance and transition-state stabilization. Biochemistry 2001; 40(3)631–640