Abstract
In a search for novel compounds with analgesic and anti-inflammatory activity, a series of regioisomeric 1-(3-pyridazinyl)-3-arylpyrazole (5a–f, 6a–f) and 1-(3-pyridazinyl)-5-arylpyrazole (7a–f, 8a–f) derivatives were synthesized. The structure of these regioisomers was confirmed by spectral techniques. The compounds were preliminarily screened at 8 μM concentration for their inhibitory activity against cyclooxygenase enzymes, COX-1 and COX-2, using a human whole blood test. The tested derivatives showed inhibitory activity for both enzymes and are worthy of further investigation for developing better leads.
Introduction
The discovery of two distinct cyclooxygenase (COX) isoforms, namely COX-1 and COX-2, made it possible to separate the pharmacological effects from the general side effects of traditional non-steroidal anti-inflammatory drugs (NSAIDs) [Citation1,Citation2]. Hence, this discovery suggested that the inhibition of COX-2, but not COX-1, was of importance for designing compounds that lack the gastrointestinal and renal side effects of currently used NSAIDs Citation2-4. Clinical data also indicated that selective inhibition of COX-2 produced efficacy equivalent to traditional NSAIDs in the treatment of inflammatory disorders with increased gastric safety profile Citation1-5. However, the occurrence of newer side-effects, most notably on the cardiovascular system, caused a hot debate on the usefulness of the selective COX-2 inhibition concept Citation6-10. During this time, the availability and widespread use of COX-2 selective inhibitors resulted in a better understanding of the roles of COX-1 and COX-2 in health and disease such as pain perception and the progression of cancers [Citation2,Citation11]. Apart from inflammation, a few COX-2 inhibitors are also being studied for treating other ailments like cancer Citation12-14 and Alzheimer's diseases [Citation15]. Although what is now known about the efficacy and safety of COX-2 inhibitors and their potential use in various pathologies including inflammation and cancer are under debate, we are still in need of better tolerated and different classes of COX-2 selective agents to extend our understanding on this drug class.
Pyridazine derivatives have recently received much attention due to their analgesic and anti-inflammatory activities Citation16-18. Various pyridazinone derivatives endowed with analgesic/anti-inflammatory effects have been reported and among them, emorfazone was launched in Japan as an anti-inflammatory drug [Citation19,Citation20]. In addition, 3-O-substituted benzyl pyridazinone derivatives [Citation21] and 4,5-diarylsubstituted pyridazinones [Citation22] were recently shown to exhibit in vivo potent anti-inflammatory activity using the carrageenan-induced rat paw oedema assay through the mechanism involving selective COX-2 inhibition. With the aim to discover new analgesic and anti-inflammatory drugs, our research group has long been interested in the chemistry and pharmacology of different lactamic heterocyclic systems and recently prepared distinct pyridazinone derivatives and found that they showed good analgesic and anti-inflammatory activities Citation23-28.
The objective of this study was the replacement of one of the aryl groups in the celecoxib template, a known COX-2 inhibitor drug, with pyridazine to evaluate the significance of the aryl substituents of the central pyrazole ring. For this purpose and based on the fact that pyridazinone derivatives bear potential analgesic/anti-inflammatory properties, we evaluated the biological consequences of incorporation of a pyridazine/pyridazinone ring as one of the aryl substituents, and also the effect of a 1,3- or 1,5-diarylsubstitution pattern around the pyrazole ring on the in vitro COX inhibitory potency of the resulting derivatives. Herein, we describe the synthesis and the in vitro preliminary screening of the first series of compounds to evaluate their COX-1/COX-2 inhibitory activity.
Materials and methods
Materials and instruments
3,6-Dichloropyridazine, hydrazine hydrate, ethyl trifluoroacetate, 4,4,4-trifluoro-1-phenyl-1,3-butanedione (2a) and substituted acetophenones were obtained from Sigma Chemicals. 3-Chloro-6-hydrazinopyridazine (3) was synthesized from the reaction of 3,6-dichloropyridazine with hydrazine hydrate in an ice bath according to the previously published procedure [Citation29] with modifications. Previously reported β-diketones [Citation30,Citation31] were synthesized by the condensation of the appropriate acetophenone or acetonaphthone derivatives with ethyl trifluoroacetate in the presence of NaH. All other chemicals were obtained from commercial sources. The yields reported are unoptimized. 1D NMR (1H: 500 MHz and 13C: 125 MHz) and 2D NMR spectra were measured with a JEOL JNM A500 and Bruker AVANCE500 NMR spectrometer at 30°C. All chemical shifts were reported as δ (ppm) values using TMS as an internal standard in DMSO-d6. Two dimensional phase-sensitive NOESY spectra were measured with field gradient mode in mixing time 1800 or 2000 msec. Inverse proton detected HMBC spectra were carried out with field gradient mode applying a delay optimized for long-range coupling constants of 8 Hz. Elemental analyses were performed with Leco-932 (C,H,N,S,O-Elemental Analyser) at the Scientific and Technical Research Council of Turkey, Instrumental Analysis Center (Ankara, Turkey). High resolution mass spectra (HRMS) was measured with a JEOL AX-505 spectrometer.
General procedure for the synthesis of 4,4,4-trifluoro-1-(4-substitutedphenyl)-1,3-butandiones (2b–d)
Appropriate p-substituted acetophenone (6′-Methoxy-2′-acetonaphtenone for 2f) (30 mmol) was dissolved in hexane (30 mL) and cooled to − 10°C in an ice–salt bath. To the cooled solution, a 60% suspension of NaH in parafin oil (30 mmol) was added and stirred for 10 min. Then, a solution of ethyl trifluoroacetate (33 mmol) in hexane (20 mL) was added dropwise over 30 min. After the addition was complete, the mixture was stirred at this temperature for 30 min and at room temperature for a further 1 h (in the case of 2e, it was refluxed for 6 h with vigorously stirring). The hexane layer was evaporated to half volume under reduced pressure, and the precipitate was suction-filtered, washed with cold hexane, dried and suspended in water (200 mL). The suspension was acidified with concentrated HCl and the precipitate was filtered off, dried and recrystallized from the appropriate solvent.
4,4,4-trifluoro-1-(4-chlorophenyl)-1,3-butandione (2b)
Recrystallized from petroleum ether (40–60°C). Yield: 69%, mp 63–64°C (lit. mp 61–62°C [Citation30,Citation31]). Anal. Calcd. for C10H6ClF3O2: C, 47.93; H 2.41. Found: C, 48.35; H 2.11%.
4,4,4-trifluoro-1-(4-methylphenyl)-1,3-butandione (2c)
Recrystallized from petroleum ether (40–60°C). Yield: 70%, mp 47–48°C (lit. mp 43–44°C [Citation30,Citation31]). Anal. Calcd. for C11H9F3O2: C, 57.40; H, 3.94. Found: C, 57.32; H, 3.54%.
4,4,4-trifluoro-1-(4-methoxyphenyl)-1,3-butandione (2d)
Recrystallized from petroleum ether (40–60°C). Yield: 67%, mp 58–59°C (lit. mp 57–58°C [Citation30,Citation31]). Anal. Calcd. for C11H9F3O3: C, 53.67; H, 3.68. Found: C, 53.50; H, 3.28%.
4,4,4-trifluoro-1-(6-methoxy-2-naphthyl)-1,3-butandione (2f)
Recrystallized from petreloum ether (40–60°C). Yield: 63%, mp 90–91°C. Anal. Calcd. for C15H11F3O3: C, 60.82; H, 3.74. Found: C, 60.90; H, 3.92%.
General procedure for the synthesis of 1-(6-chloropyridazine-3-yl)-3-aryl-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazoles (4a–f)
The solution of benzoylacetone or the appropriate 4,4,4-trifluoro-1-phenyl-1,3-butanedione derivative (15 mmol) and 6-chloro-3-hydrazinopyridazine (15 mmol) in absolute ethanol (30 mL) was refluxed for 8 h, and cooled to room temperature. After addition of a sufficient amount of water for complete precipitation, the precipitate was filtered off, dried and recrystallized from the appropriate solvent.
1-(6-chloropyridazine-3-yl)-3-phenyl-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole (4a)
Recrystallized from methanol. Yield, 66%; mp 142–143°C; 1H NMR (DMSO-d6) δ: 3.72(1H, d, J = 18.9 Hz, H-4α), 4.02(1H, d, J = 18.9 Hz, H-4β), 7.48(3H, m, H-3″, 4″, 5″), 7.79(1H, d, J = 9.5 Hz, H-5′), 7.84(2H, m, H-2″, 6″), 7.85 (1H, d, J = 9.5 Hz, H-4′), 8.14(1H, s, 5-OH). 13C NMR (DMSO-d6) δ: 44.81(C4), 92.93(C5, 2JF = 33 Hz), 120.08(C4′), 123.44 (CF3, 1JF = − 286 Hz), 126.38 (C2″, 6″), 128.75(C3″, 5″), 130.00(C5′), 130.28(C4″), 130.34(C1″), 149.62(C6′), 151.40(C3), 156.62 (C3′). Anal. Calcd. for C14H10ClF3N4O: C, 49.12; H, 2.95; N, 16.38. Found: C, 49.54; H, 2.7; N, 16.36%.
1-(6-chloropyridazine-3-yl)-3-(4-chlorophenyl)-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole (4b)
Recrystallized from acetonitrile. Yield, 64%; mp 207–208; 1H NMR (DMSO-d6) δ: 3.70(1H, d, J = 19.2 Hz, H-4α), 4.03(1H, d, J = 19.2 Hz, H-4β), 7.55(2H, dd, J = 8.9, 2.4 Hz, H-3″, 5″), 7.79(1H, d, J = 9.5 Hz, H-5′), 7.85(1H, d, J = 9.5 Hz, H-4′), 7.86(2H, dd, J = 8.9, 2.4 Hz, H-2″, 6″), 8.17(1H, s, 5-OH). 13C NMR (DMSO-d6) δ: 44.70(C4), 93.13(C5, 2JF = 33 Hz), 120.13(C4′), 123.38(CF3, 1JF = − 288 Hz), 128.14(C2″, 6″), 128.81(C3″, 5″), 129.27(C1″), 130.03(C5′), 134.85(C4″), 149.77(C6′), 150.43(C3), 156.53(C3′). Anal. Calcd. for C14H9Cl2F3N4O: C, 44.68; H, 2.41; N, 14.90. Found: C, 45.01; H, 2.05; N, 15.18%.
1-(6-chloropyridazine-3-yl)-3-(4-methylphenyl)-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole (4c)
Recrystallized from ethanol. Yield, 62%; mp 177–178°C; 1H NMR (DMSO-d6) δ: 2.36(3H, s, CH3), 3.68(1H, d, J = 18.9 Hz, H-4α), 3.98(1H, d, J = 18.9 Hz, H-4β), 7.29(2H, d, J = 8.6 Hz, H-3″, 5″), 7.73(2H, d, J = 8.6 Hz, H-2″, 6″), 7.77(1H, d, J = 9.5 Hz, H-5′), 7.83(1H, d, J = 9.5 Hz, H-4′), 8.1(1H, s, 5-OH). 13C NMR (DMSO-d6) δ: 20.96(CH3), 44.88(C4), 92.80(C5, 2JF = 33 Hz), 120.01(C4′), 123.47(CF3, 1JF = − 286 Hz), 126.36(C2″, 6″), 127.61(C1″), 129.32(C3″, 5″), 129.98(C5′), 140.17(C4″), 149.49(C6′), 151.42(C3), 156.63(C3′). Anal. Calcd. for C15H12ClF3N4O: C, 50.55; H, 3.40; N, 15.73. Found: C, 50.84; H, 3.02; N, 16.14%.
1-(6-chloropyridazine-3-yl)-3-(4-methoxyphenyl)-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole (4d)
Recrystallized from ethanol. Yield, 61%; mp 172–173°C; 1H NMR (DMSO-d6) δ: 3.68(1H, d, J = 18.9 Hz, H-4α), 3.81(3H, s, 4″-OCH3), 3.98(1H, d, J = 18.9 Hz, H-4β), 7.03(2H, d, J = 8.9 Hz, H-3″, 5″), 7.73(1H, d, J = 9.5 Hz, H-5′), 7.78(2H, d, J = 8.6 Hz, H-2″, 6″), 7.82(1H, d, J = 9.5 Hz, H-4′), 8.09(1H, s, 5-OH). 13C NMR (DMSO-d6) δ: 45.00(C4), 55.33(OCH3), 92.74(C5, 2JF = 33 Hz), 114.22(C3″. 5″), 119.95 (C4′), 122.86(C1″), 123.51(CF3, 1JF = − 288 Hz), 128.11(C2″, 6″), 129.97(C5′), 149.33(C6′), 151.25(C3), 156.68 (C3′), 160.94(C4″). Anal. Calcd. for C15H12ClF3N4O2: C, 48.38; H, 3.25; N, 15.05. Found: C, 47.99; H, 3.15; N, 14.9%.
1-(6-chloropyridazin-3-yl)-3-(4-fluorophenyl)-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole (4e)
Recrystallized from ethanol to give colorless prisms. Yield, 64%; mp 179–180°C; 1H NMR δ: 3.71(1H, d, J = 19.0 Hz, H-4α), 4.05(1H, d, J = 19.1 Hz, H-4β), 7.32(2H, dd, J = 8.9 Hz, H-3″, 5″), 7.78(1H, d, J = 9.4 Hz, H-4′), 7.85(1H, d, J = 9.4 Hz, H-5′), 7.90 (2H, dd, J = 8.9, 5.5 Hz, H-2″, 6″), 8.15(1H, s, 5-OH). 13C NMR δ: 44.90(C4), 93.05(q, 2JF = 33.2 Hz, C5), 115.84(d, 2JF = 22.0 Hz, C3″, 5″), 120.10(C5′), 123.44(q, 1JF = − 286.1 Hz, CF3), 127.01(d, 4JF = 2.8 Hz, C1″), 128.83(d, 3JF = 8.2 Hz, C2″, 6″), 130.04(C4′), 149.63(C3′), 150.58(C3), 156.62(C6′), 163.22(d, 1JF = − 248.4 Hz, C4″).Anal. Calcd. for C14H9ClF4N4O: C, 46.62; H, 2.52; N, 15.53. Found: C, 46.63; H, 2.74; N, 15.45%.
1-(6-chloropyridazine-3-yl)-3-(6-methoxynapht-2-yl)-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole (4f)
Recrystallized from ethanol. Yield, 71%; mp 169–170°C; 1H NMR (DMSO-d6) δ: 3.81(1H, d, J = 18.9 Hz, H-4α), 3.90(3H, s, 6″-OCH3), 4.11(1H, d, J = 18.9 Hz, H-4β), 7.23(1H, dd, J = 8.9, 2.7 Hz, H-7″), 7.38(1H, d, J = 2.4 Hz, H-5″), 7.79(1H, d, J = 9.5 Hz, H-5′), 7.88(1H, d, J = 8.6 Hz, H-4″), 7.89(1H, d, J = 9.5 Hz, H-4′), 7.90(2H, d, J = 8.9 Hz, H-8″), 8.03(1H, dd, J = 8.6, 1.8 Hz, H-3″), 8.16(1H, s, 5-OH), 8.21(1H, d, J = 1.8 Hz, H-1″). 13C NMR (DMSO-d6) δ: 44.88(C4), 55.28(OCH3), 92.90(C5, 2JF = 33 Hz), 106.33(C5″), 119.22(C7″), 120.03 (C5′), 123.29(C3″), 123.52(CF3, 1JF = − 286 Hz), 125.56(C2″), 127.09(C1″), 127.19(C4″), 127.94(C8a″), 130.00(C8″, 5′), 135.15(C4″a), 149.51(C6′), 151.58(C3), 156.63(C3′), 158.36(C6″). Anal. Calcd. for C19H14ClF3N4O2: C, 54.02; H, 3.34; N, 13.27. Found: C, 54.37; H, 3.13; N, 13.3%.
General procedure for the synthesis of 1-[3(2H)-pyridazinone-6-yl]-3-aryl-5-trifluoromethylpyrazoles (5a–f)
The appropriate 1-(6-chloropyridazine-3-yl)-3-aryl-5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole derivative (10 mmol) and sodium acetate trihydrate (15 mmol) in glacial acetic acid (30 mL) were refluxed for 15 h and poured into ice-water mixture (150 mL). The precipitate formed was filtered off, washed with sodium carbonate solution (2.5%, w/v) and then with water, dried and recrystallized from the appropriate solvent.
1-[3(2H)-pyridazinone-6-yl]-3-phenyl-5-trifluoromethylpyrazole (5a)
Recrystallized from toluene. Yield, 71.9%; mp 225–226°C; 1H NMR (DMSO-d6) δ: 7.16(1H, d, J = 10.1 Hz, H-4′), 7.43(1H, ddd, J = 7.3, 1.2 Hz, H-4″), 7.49(2H, ddd, J = 7.3, 1.2 Hz, H-3″, 5″), 7.78(1H, s, H-4), 7.96(2H, dd, J = 7.3, 1.2 Hz, H-2″, 6″), 7.98(1H, d, J = 10.1 Hz, H-5′), 13.29(1H, s, N2′–H). 13C NMR (DMSO-d6) δ: 108.79(C4), 119.35(CF3, 1JF = − 269 Hz), 125.77(C2″, 6″), 128.90(C3″, 5″), 129.19(C4″), 129.65(C5′), 130.52(C1″), 132.22(C5, 2JF = 39 Hz), 132.52(C4′), 140.20(C6′), 151.80(C3), 159.89(3′-CO). Anal. Calcd. for C14H9F3N4O: C, 54.91; H, 2.96; N, 18.29. Found: C, 54.87; H, 2.58; N, 18.08%.
1-[3(2H)-pyridazinone-6-yl]-3-(4-chlorophenyl)-5-trifluoromethylpyrazole (5b)
Recrystallized from ethanol. Yield, 79%; mp 247–248°C; 1H NMR (DMSO-d6) δ: 7.15(1H, d, J = 10.0 Hz, H-4′), 7.55(2H, dd, J = 8.6, 1.8 Hz, H-3″, 5″), 7.82(1H, s, H-4), 7.97(1H, d, J = 10.0 Hz, H-5′), 7.99(2H, dd, J = 8.6, 1.8 Hz, H-2″, 6″), 13.3(1H, s, N2′–H). 13C NMR (DMSO-d6) δ: 108.96(C4), 119.29(CF3, 1JF = − 269 Hz), 127.51(C2″, 6″), 128.96 (C3″, 5″), 129.42(C1″), 129.62(C5′), 132.34(C5, 2JF = 41 Hz), 132.53(C4′), 133.83(C4″), 140.13(C6′), 150.68(C3), 159.87(3′-CO). Anal. Calcd. for C14H8ClF3N4O: C, 49.36; H, 2.37; N, 16.45. Found: C, 49.53; H, 1.98; N, 16.48%.
1-[3(2H)-pyridazinone-6-yl]-3-(4-methylphenyl)-5-trifluoromethylpyrazole (5c)
Recrystallized from ethanol. Yield, 91%; mp 265–266°C; 1H NMR (DMSO-d6) δ: 2.35(3H, s, CH3), 7.14(1H, d, J = 10.0 Hz, H-4′), 7.29(2H, d, J = 8.2 Hz, H-3″, 5″), 7.73(1H, s, H-4), 7.85(2H, d, J = 8.2 Hz, H-2″, 6″), 7.96(1H, d, J = 10.0 Hz, H-5′), 13.3(1H, s, N2′-H). 13C NMR (DMSO-d6) δ: 20.81(4″-CH3), 119.37(CF3, 1JF = − 269 Hz), 125.69(C2″, 6″), 127.76(C1″), 129.44(C3″, 5″), 129.64(C5′), 132.11(C5, 2JF = 39 Hz), 132.58(C4′), 138.75(C4″), 140.23(C6′), 151.83(C3), 159.87(3′-CO). Anal. Calcd. for C15H11F3N4O: C, 56.25; H, 3.46; N, 17.49. Found: C, 56.62; H, 3.06; N, 17.63%.
1-[3(2H)-pyridazinone-6-yl]-3-(4-methoxyphenyl)-5-trifluoromethylpyrazole (5d)
Recrystallized from toluene. Yield, 84%; mp 233–234°C; 1H NMR (DMSO-d6) δ: 3.8(3H, s, OCH3), 7.03(2H, dd, J = 8.9, 2.4 Hz, H-3″, 5″), 7.14(1H, d, J = 10.0 Hz, H-4′), 7.69(1H, s, H-4), 7.89(2H, dd, J = 8.9, 2.4 Hz, H-2″, 6″), 7.96(1H, d, J = 10.0 Hz, H-5′), 13.26(1H, s, N2′-H). 13C NMR (DMSO-d6) δ: 55.19(4″-OCH3), 108.35(C4), 114.29(C3″, 5″), 119.40(CF3, 1JF = − 269 Hz), 123.06(C1″), 127.22(C2″, 6″), 129.64(C5′), 132.05(C5, 2JF = 39 Hz), 132.45(C4′), 140.25(C6′), 151.70(C3), 159.89(3′-CO), 160.04(C4″). Anal. Calcd. for C15H11F3N4O2: C, 53.58; H, 3.30; N, 16.66. Found: C, 53.98; H, 2.89; N, 16.59%.
1-(3(2H)-pyridazinon-6-yl)-3-(4-fluorophenyl)-5-trifluoromethylpyrazole (5e)
Recrystallized from ethanol to give colorless fine needles. Yield, 75%; mp 237.5–238°C; 1H NMR δ: 7.16(1H, d, J = 10.0 Hz, H-4′), 7.32(2H, dd, J = 8.8 Hz, H-3″, 5″), 7.79(1H, s, H-4), 7.97 (1H, d, J = 10.0 Hz, H-5′), 8.01(2H, dd, J = 8.8, 5.5 Hz, H-2″, 6″), 13.29(1H, s, N–H). 13C NMR δ: 108.81(C4), 115.90(d, 2JF = 22.0 Hz, C3″, 5″), 119.34(q, 1JF = − 268.9 Hz, CF3), 127.14(d, 4JF = 2.7 Hz, C1′), 127.14(d, 3JF = 8.2 Hz, C2″, 6″), 129.65(C-5′), 132.27(q, 2JF = 40.3 Hz, C5), 132.56(C4′), 140.19(C6′), 150.92(C3), 159.91(3-CO), 162.66(d, 1JF = − 246.5 Hz, C4″). Anal. Calcd. for C14H8F4N4O: C, 51.86; H, 2.49; N, 17.28. Found: C, 51.78; H, 2.70; N, 17.27%.
1-[3(2H)-pyridazinone-6-yl]-3-(6-methoxynapht-2-yl)-5-trifluoromethylpyrazole (5f)
Recrystallized from ethanol. Yield, 88%; mp 257–258°C; 1H NMR (DMSO-d6) δ: 3.89(3H, s, OCH3), 7.17(1H, d, J = 10.1 Hz, H-4′), 7.21(1H, dd, J = 8.9, 2.4 Hz, H-7″), 7.36(1H, d, J = 2.4 Hz, H-5″), 7.86(1H, s, H-4), 7.89(1H, d, J = 8.9 Hz, H-8″), 7.91(1H, d, J = 8.9 Hz, H-4″), 8.02(1H, d, J = 10.1 Hz, H-5′), 8.05(1H, dd, J = 8.9, 1.5 Hz, H-3″), 8.46(1H, d, J = 1.5 Hz, H-1″), 13,30(1H, s, N2′–H). 13C NMR (DMSO-d6) δ: 55.23(6″-OCH3), 106.05(C7″), 108.84(C4), 119.26(C7″), 119.40(CF3, 1JF = − 269 Hz), 123.83(C3″), 124.93(C1″), 125.69(C2″), 127.35(C4″), 128.29(C8a″), 129.67(C5′), 129.72(C8″), 132.22(C5, 2JF = 39 Hz), 132.52(C4′), 134.57(C4a″), 140.26(C6′), 151,98(C3), 157.90(C6″), 159.89(3′-CO). Anal. Calcd. for C19H13F3N4O2: C, 59.07; H, 3.39; N, 14.50. Found: C, 58.90; H, 3.32; N, 14.43%.
General procedure for the synthesis of 1-(6-chloropyridazine-3-yl)-3-aryl-5-trifluoromethylpyrazoles (6a–f)
The appropriate 1-[3(2H)-pyridazinone-6-yl]-3-aryl-5-trifluoromethylpyrazole (0.001 mol) was refluxed in POCl3 (10 mL) for 1 h, and poured into crushed-ice (150 g) and stirred for a further 30 min. The mixture was subsequently neutralized by K2CO3 and the precipitate was filtered off, washed with water, dried and recrystallized from the appropriate solvent.
1-(6-chloropyridazine-3-yl)-3-phenyl-5-trifluoromethylpyrazole (6a)
Recrystallized from isopropanol. Yield, 78%; mp 133–135°C; 1H NMR (DMSO-d6) δ: 7.51(3H, m, H-3″, 4″, 5″), 7.96(1H, s, H-4), 8.06(2H, d, J = 8.43, H-2″, 6″), 8.23(1H, d, J = 9.2 Hz, H-5′), 8.44(1H, d, J = 9.2 Hz, H-4′). Anal. Calcd. for C14H8ClF3N4: C, 51.79; H, 2.48; N, 17.26. Found: C, 51.94; H, 2.19; N, 16.93%.
1-(6-chloropyridazine-3-yl)-3-(4-chlorophenyl)-5-trifluoromethylpyrazole (6b)
Recrystallized from petroleum ether. Yield, 92%; mp 183–184°C; 1H NMR (DMSO-d6) δ: 7.56(2H, dd, J = 8.6, 2.1 Hz, H-3″, 5″), 7.95(1H, s, H-4), 8.04(2H, dd, J = 8.6, 2.1 Hz, H-2″, 6″), 8.20(1H, d, J = 9.2 Hz, H-5′), 8.39(1H, d, J = 9.2 Hz, H-4′). 13C NMR (DMSO-d6) δ: 110.90(C4), 119.40(CF3, 1JF = − 269 Hz), 124.42(C4′), 127.70(C2″, 6″), 129.01(C3″, 5″), 129.14(C1″), 132.00(C5′), 132.95(C5, 2JF = 39 Hz), 134.18(C4″), 151.73(C3), 153.93(C6′), 155.70(C3′). Anal. Calcd. for C14H7Cl2F3N4: C, 46.82; H, 1.96; N, 15.60. Found: C, 47.20; H, 2.30; N, 15.60%.
1-(6-chloropyridazine-3-yl)-3-(4-methylphenyl)-5-trifluoromethylpyrazole (6c)
Recrystallized from isopropanol. Yield, 95%; mp 169–171°C; 1H NMR (DMSO-d6) δ: 7.34(2H, d, J = 8.04 Hz, H-3″, 5″), 7.91(1H, s, H-4), 7.94(2H, d, J = 8.14 Hz, H-2″, 6″), 8.22(1H, d, J = 9.2 Hz, H-5′), 8.43(1H, d, J = 9.2 Hz, H-4′), 2.36 (3H, s, CH3). Anal. Calcd. for C15H10ClF3N4: C, 53.19; H, 2.98; N, 16.54. Found: C, 53.28; H, 2.27; N, 16.14%.
1-(6-chloropyridazine-3-yl)-3-(6-methoxyphenyl)-5-trifluoromethylpyrazole (6d)
Recrystallized from isopropanol. Yield, 94%; mp 159–160°C; 1H NMR (DMSO-d6) δ: 7.08(2H, d, J = 8.8 Hz, H-3″, 5″), 7.87(1H, s, H-4), 7.96(2H, d, J = 8.8 Hz, H-2″, 6″), 8.22(1H, d, J = 9.2 Hz, H-4′), 8.41(1H, d, J = 9.2 Hz, H-5′), 3.83 (3H, s, OCH3). Anal. Calcd. for C15H10ClF3N4O: C, 50.79; H, 2.84; N, 15.79. Found: C, 50.93; H, 2.39; N, 15.47%.
1-(6-chloropyridazin-3-yl)-3-(4-fluorophenyl)-5-trifluoromethylpyrazole (6e)
Recrystallized from n-hexane to give colorless fine bar. Yield, 90%; mp 176.5–177°C; 1H NMR δ: 7.35(2H, dd, J = 8.9 Hz, H-3″, 5″), 7.93(1H, s, H-4), 8.08(2H, d, J = 8.9, 5.5 Hz, H-2″, 6″), 8.22(1H, d, J = 9.2 Hz, H-5′), 8.40(1H, d, J = 9.2 Hz, H-4′). 13C NMR δ: 110.79(C4), 115.96(d, 2JF = 22.0 Hz, C3″, 5″), 119.46(q, 1JF = − 268.6 Hz, CF3), 124.39(C5′), 126.85(d, 4JF = 2.8 Hz C1″), 128.24(d, 2JF = 8.2 Hz, C2″, 6″), 132.02 (C4′), 132.89(q, 2JF = 40.7 Hz, C3), 151.95(C5), 153.96(C6′), 155.64(C3′), 162.84(d, 1JF = − 246.6 Hz, C4″). Anal. Calcd. for C14H7ClF4N4: C, 49.07; H, 2.06; N, 16.35. Found: C, 49.10; H, 2.25; N, 16.30%.
1-(6-chloropyridazin-3-yl)-3-(6-methoxynapht-2-yl)-5-trifluoromethylpyrazole (6f)
Recrystallized from isopropanol. Yield, 95%; mp 185–187°C; 1H NMR (DMSO-d6) δ: 3.91(3H, s, OCH3), 7.24(1H, dd, J = 8.9, 2.5 Hz, H-7″), 7.4(1H, dd, J = 8.5, 1.6 Hz, H-3″), 7.94(2H, m, H-4″, 5″), 8.04(1H, s, H-4), 8.14(1H, d, J = 10 Hz, H-8″), 8.24 (1H, d, J = 9.1 Hz, H-5′), 8.46(1H, d, J = 9.1 Hz, H-4′) 8.55(1H, s, H-1″). Anal. Calcd. for C19H12ClF3N4O: C, 56.38; H, 2.99; N, 13.84. Found: C, 56.91; H, 2.55; N, 13.55%.
General procedure for the synthesis of 1-(6-chloropyridazin-3-yl)-5-aryl-3-trifluoromethylpyrazoles (7a–f)
The solution of the appropriate 4,4,4-trifluoro-1-phenyl-1,3-butanedione derivative (0.001 mol) and the hydrochloride salt of 6-chloro-3-hydrazinopyridazine (0.001 mol) in diglyme (30 mL) was refluxed for 5 h, and cooled to room temperature. Concentrated hydrochloric acid was dropped over concentrated sulfuric acid for liberation of gaseous HCl which was passed through the reaction medium to prepare the salt of 6-chloro-3-hydrazinopyridazine. After addition of a sufficient amount of water for complete precipitation, the precipitate was filtered off, dried and recrystallized from the appropriate solvent.
1-(6-chloropyridazin-3-yl)-5-phenyl-3-trifluoromethylpyrazole (7a)
Recrystallized from n-hexane. Yield, 65%; mp 131–133°C; 1H NMR (DMSO-d6) δ: 7.34(1H, s, H-4), 7.35–7.41(5H, m, H–Ph), 8.22(2H, s, H-4′, 5′). 13C NMR δ: 107.28(C4), 120.94(q, 1JF = − 269.2 Hz, CF3), 127.31(C5′), 128.43(C1″), 128.56(C2″, 6″), 128.70(C3″, 5″), 129.22(C4″), 131.85(C4′), 143.44(q, 2JF = 38.3 Hz, C3), 146.35(C5), 154.89(C6′), 156.11(C3′). EI-HRMS: 324.0388, Calcd. for 324.0396.
1-(6-chloropyridazin-3-yl)-5-(4-chlorophenyl)-3-trifluoromethylpyrazole (7b)
Recrystallized from n-hexane. Yield, 56%; mp 143–146°C; 1H NMR (DMSO-d6) δ: 7.38(1H, s, H-4), 7.42(2H, d, J = 8.4 Hz, H-2″, 6″), 7.48(2H, d, J = 8.4 Hz, H-3″, 5″), 8.22(1H, d, J = 9.1 Hz, H-5′), 8.24(1H, d, J = 9.1 Hz, H-4′). 13C NMR δ: 107.75(C4), 120.87(q, 1JF = − 269.3 Hz, CF3), 127.10(C5′), 127.49(C1″), 128.58(C3″, 5″), 130.59(C2″, 6″), 131.92(C4′), 134.11(C4″), 143.48(q, 2JF = 38.2 Hz, C3), 145.07(C5), 154.75(C6′), 156.08(C3′). EI-HRMS: 357.9972, Calcd. for 358.0000.
1-(6-chloropyridazin-3-yl)-5-(4-methoxyphenyl)-3-trifluoromethylpyrazole (7c)
Recrystallized from n-hexane. Yield, 48%; mp 159–160°C; 1H NMR (DMSO-d6) δ: 3.77(3H, s, OCH3), 6.95(2H, d, J = 8.8 Hz, H-3″, 5″), 7.25(1H, s, H-4), 7.28(2H, d, J = 8.8 Hz, H-2″, 6″), 8.18(1H, s, H-5′), 8.20(1H, s, H-4′). 13C NMR δ: 55.22(OCH3), 106.57(C4), 114.06(C3″, 5″), 120.55(C1″), 121.01(q, 1JF = − 268.3 Hz, CF3), 127.47(C5′), 130.19(C2″, 6″), 131.79(C4′), 143.36(q, 2JF = 36.2 Hz, C3), 146.34(C5), 155.06(C6′), 156.09(C3′), 159.92(C4″). EI-HRMS: 354.0485, Calcd. for 354.0495.
1-(6-chloropyridazin-3-yl)-5-(4-methylphenyl)-3-trifluoromethylpyrazole (7d)
Recrystallized from cyclohexane. Yield, 50%; mp 136–137°C; 1H NMR (DMSO-d6) δ: 2.31(3H, s, CH3), 7.20(2H, d, J = 8.1 Hz, H-3″, 5″), 7.24(2H, d, J = 8.1 Hz, H-2″, 6″), 7.29(1H, s, H-4), 8.20(2H, s, H-4′, 5′). 13C NMR δ: 20.75(CH3), 106.88(C4), 120.97(q, 1JF = − 269.2 Hz, CF3), 125.50(C1″), 127.40(C5′), 128.57(C2″, 6″), 129.15(C3″, 5″), 131.82(C4′), 138.93(C4″), 143.40(q, 2JF = 38.1 Hz, C3), 146.46(C5), 154.96(C6′), 156.10(C3″). EI-HRMS: 338.0554, Calcd. for 338.0546.
1-(6-chloropyridazin-3-yl)-5-(4-fluorophenyl)-3-trifluoromethylpyrazole (7e)
Recrystallized from n-hexane to give colorless needles. Yield, 51%; mp 136–137°C; 1H NMR (DMSO-d6) δ: 7.25(2H, d, J = 8.8 Hz, H-3″, 5″), 7.34(1H, s, H-4), 7.44(2H, d, J = 8.7, 5.4 Hz, H-2″, 6″), 8.22(1H, d, J = 9.1 Hz, H-4′), 8.22(1H, d, J = 9.1 Hz, H-5′). 13C NMR δ: 108.06(C4), 116.10(d, 2JF = 22.0 Hz, C3″, 5″), 121.44(q, 1JF = − 269.3 Hz, CF3), 125.59(d, 4JF = 3.2 Hz, C1″), 127.72(C5′), 131.70(d, 2JF = 8.2 Hz, C2″, 6″), 132.40(C4′), 143.93(q, 2JF = 38.3 Hz, C3), 145.84(C5), 155.34(C6′), 156.58(C3′), 162.99(d, 1JF = − 247.5 Hz, C4″). Anal. Calcd. for C14H7ClF4N4: C, 49.07; H, 2.06; N, 16.35. Found: C, 49.15; H, 2.29; N, 16.28%.
1-(6-chloropyridazin-3-yl)-5-(6-methoxy-2-naphthyl)-3-trifluoromethylpyrazole (7f)
Recrystallized from cyclohexane. Yield, 67%; mp 152.5–154°C; 1H NMR (DMSO-d6) δ: 3.88(3H, s, OCH3), 7.21(1H, dd, J = 8.9, 2.5 Hz, H-7″), 7.31(1H, dd, J = 8.5, 1.6 Hz, H-3″), 7.34(1H, d, J = 2.4 Hz, H-5″), 7.40(1H, s, H-4), 7.78(1H, d, J = 8.6 Hz, H-4″), 7.82(1H, d, J = 9.0 Hz, H-8″), 7.96(1H, s, H-1″), 8.21 (1H, d, J = 9.1 Hz, H-4′), 8.24(1H, d, J = 9.1 Hz, H-5′). 13C NMR δ: 55.27(OCH3), 105.89(C5″), 107.27(C4), 119.33(C7″), 120.99(q, 1JF = − 269.1 Hz, CF3), 123.52(C2″), 126.38(C3″), 126.81(C4″), 127.14 (C5′), 127.83(C8a″), 128.06(C1″), 129.76(C8″), 131.82(C4′), 134.24(C4a″), 143.52(q, 2JF = 37.8 Hz, C3), 146.60(C5), 154.94(C6′), 156.00(C3′), 158.21(C6″). EI-HRMS: 404.0652, Calcd. for 404.0652.
General procedure for the synthesis of 1-[3(2H)-pyridazinone-6-yl]-5-aryl-3-trifluoromethylpyrazoles (8a–f)
The appropriate 1-(6-chloropyridazine-3-yl)-5-aryl-3-trifluoromethylpyrazole derivative (10 mmol) and sodium acetate trihydrate (15 mmol) in glacial acetic acid (30 mL) were refluxed for 15 h and poured into an ice-water mixture (150 mL). The precipitate formed was filtered off, washed with sodium carbonate solution (2.5%, w/v) and then with water, dried and recrystallized from the appropriate solvent.
1-(3(2H)-pyridazinon-6-yl)-5-phenyl-3-trifluoromethylpyrazole (8a)
Recrystallized from toluene. Yield, 74%; mp 189–190°C; 1H NMR (DMSO-d6) δ: 7.10(1H, d, J = 9.9 Hz, H-4′), 7.25(1H, s, H-4), 7.40–7.45(5H, m, H–Ph), 7.76(1H, d, J = 9.9 Hz, H-5′), 13.12(1H, brs, N–H). 13C NMR δ: 106.07(C4), 121.02(q, 1JF = − 268.9 Hz, CF3), 128.08(C1″), 128.49(C2″, 6″), 128.76(C3″, 5″), 129.28(C4″), 131.30(C5′), 132.18(C4′), 140.47(C6′), 142.47(q, 2JF = 36.8 Hz, C3), 145.73(C5), 159.86(C3′). EI-HRMS: 306.0709 Calcd. For 306.0728.
1-(3(2H)-pyridazinon-6-yl)-5-(4-chlorophenyl)-3-trifluoromethylpyrazole (8b)
Crude product washed with hot cyclohexane. Yield, 71%; mp 209–210°C; 1H NMR (DMSO-d6) δ: 7.11(1H, d, J = 9.9 Hz, H-4′), 7.30(1H, s, H-4), 7.45(2H, d, J = 8.6 Hz, H-2″, 6″), 7.53(2H, d, J = 8.5 Hz, H-3″, 5″), 7.78(1H, d, J = 10.0 Hz, H-5′), 13.10(1H, s, N–H). 13C NMR δ: 106.52(C4), 120.93(q, 1JF = − 269.9 Hz, CF3), 127.08(C1″), 128.78(C3″, 5″), 130.37(C2″, 6″), 131.04(C5′), 132.28(C4′), 134.13(C4″), 140.32(C6′), 142.46(q, 2JF = 37.9 Hz, C3), 144.41(C5), 159.81(C3′). EI-HRMS: 340.0331, Calcd. for 340.0339.
1-(3(2H)-pyridazinon-6-yl)-5-(4-methoxyphenyl)-3-trifluoromethylpyrazole (8c)
Crude product washed with hot cyclohexane. Yield, 94%; mp 169–170°C; 1H NMR (DMSO-d6) δ: 3.77(3H, s, OCH3), 6.99(2H, d, J = 8.7 Hz, H-3″, 5″), 7.03(1H, d, J = 9.9 Hz, H-4′), 7.15(1H, s, H-4), 7.32(2H, d, J = 8.7 Hz, H-2″, 6″), 7.67(1H, d, J = 9.9 Hz, H-5′), 13.11(1H, br, N–H). 13C NMR δ: 55.23(OCH3), 105.22(C4), 114.21(C3″, 5″), 120.30(C1″), 121.09(q, 1JF = − 269.2 Hz, CF3), 129.92(C2″, 6″), 131.08(C5′), 131.41(C4′), 140.64(C6″), 142.26(q, 2JF = 37.7 Hz, C3), 145.64(C5), 159.90(C4″), 160.53(C3′). EI-HRMS: 336.0837, Calcd. for 336.0834.
1-(3(2H)-pyridazinon-6-yl)-5-(4-methylphenyl)-3-trifluoromethylpyrazole (8d)
Crude product washed with hot cyclohexane. Yield, 91%; mp 214.5–216.5°C; 1H NMR (DMSO-d6) δ: 2.31(3H, s, CH3), 7.08(1H, d, J = 9.9 Hz, H-4′), 7.20(1H, s, H-4), 7.24(2H, d, J = 8.2 Hz, H-3″, 5″), 7.28(2H, d, J = 8.2 Hz, H-2″, 6″), 7.21(1H, d, J = 9.9 Hz, H-5′), 13.11(1H, brs, N–H). 13C NMR δ: 20.74(CH3), 105.67(C4), 121.04(q, 1JF = − 269.1 Hz, CF3), 125.20(C1″), 128.36(C2″, 6″), 129.33(C3″, 5″), 131.31(C5′), 132.03(C4′), 138.99(C4″), 140.53(C6′), 142.42(q, 2JF = 37.8 Hz, C3), 145.82(C5), 159.95(C3′). EI-HRMS: 320.0887, Calcd. for 320.0885.
1-(3(2H)-pyridazinon-6-yl)-5- (4-fluorophenyl)-3-trifluoromethylpyrazole (8e)
Recrystallized from ethanol to give colorless fibers. Yield, 90%; mp 197–198°C; 1H NMR (DMSO-d6) δ: 7.20(1H, d, J = 9.9 Hz, H-4′), 7.25(1H, s, H-4), 7.30 (2H, dd, J = 8.9 Hz, H-3″, 5″), 7.48(2H, d, J = 8.8, 5.4 Hz, H-2″, 6″), 7.76(1H, d, J = 9.9 Hz, H-5′), 13.10(1H, s, N–H). 13C NMR δ: 106.30(C4), 115.80(d, 2JF = 22.0 Hz, C3″, 5″), 121.00(q, 1JF = − 268.9 Hz, CF3), 124.71(d, 4JF = 3.9 Hz, C1″), 130.97(d, 3JF = 8.2 Hz, C2″, 6″), 131.19(C5′), 132.23(C4′), 140.40(C6′), 142.41(q, 2JF = 37.9 Hz, C3), 144.69(C5), 159.86(3-CO), 162.49(d, 1JF = − 248.0 Hz, C4″). Anal. Calcd. for C14H8F4N4O: C, 51.86; H, 2.49; N, 17.28. Found: C, 51.75; H, 2.64; N, 17.23%.
1-(3(2H)-pyridazinon-6-yl)-5-(6-methoxy-2-naphthyl)3-trifluoromethylpyrazole (8f)
Crude product washed with hot cyclohexane. Yield, 93%; mp 158–159°C; 1H NMR (DMSO-d6) δ: 3.87(3H, s, OCH3), 6.70 (1H, d, J = 9.7 Hz, H-4′), 7.19(1H, dd, J = 8.9, 2.3 Hz, H-7″), 7.25(1H, s, H-4), 7.33(1H, s, H-5″), 7.34(1H, dd, J = 10.2, 1.3 Hz, H-3″), 7.41(1H, d, J = 9.7 Hz, H-5′), 7.80(2H, d, J = 8.7 Hz, H-4″, 8″), 7.93(1H, s, H-5″), N–H was not observed. 13C NMR δ: 55.27(OCH3), 105.30(C4), 105.84(C5″), 119.37(C7″), 121.28(q, 1JF = − 268.5 Hz, CF3), 123.53(C2″), 126.07(C3″), 126.92(C1″, 4″), 127.79(C4′), 127.89(C8a″), 128.72 (C5′), 129.73(C8″), 129.89(C1″), 134.09(C4a″), 141.02(C6′), 141.78(q, 2JF = 37.6 Hz, C3), 145.61(C5), 158.12(C6″), 164.82(C3′). EI-HRMS: 386.0996, Calcd. for 386.0991.
Effect of compounds on human whole blood COX-1 and COX-2 activities
COX-1 assay
Fresh blood from healthy volunteers who had not taken NSAIDs in the previous week was collected into vacutainers containing no anticoagulants. Aliquots of 0.5 mL were immediately transferred into tubes containing vehicle or the test compound. Each drug was evaluated at a final 8 μM concentration in duplicate determinations. The samples were incubated at 37°C with gentle shaking for 60 min to allow the blood to clot. At the end of incubation, the reaction was stopped by submerging the tubes in a cold bath and centrifuging at 13 000 rpm for 10 min at 4 °C. Levels of TXB2 in the serum were determined by using an enzyme immunoassay kit (Amersham, TXB2 Biotrak Assay, #RPN 220).
COX-2 assay
The blood was collected into heparinized (20 U/mL) tubes and distributed in 0.5 aliquots in tubes containing 10 μg/mL of LPS (Sigma, St. Louis, MO, #L-2630 from E. coli serotype 0111:B4, 100 mg/mL final concentration, diluted in phosphate-buffered saline) together with vehicle or test compound at a final concentration of 8 μM. Each drug was evaluated in duplicate determinations. The samples were incubated in a bath at 37 °C for 24 h; during this time COX-2 was induced in mononuclear cells. The reaction was stopped by submerging the tubes in a cold bath and centrifuging at 13,000 rpm for 10 min at 4 °C. Levels of PGE2 in the supernatant were determined by using an enzyme immunoassay kit (Amersham, PGE2 Biotrak Assay, #RPN 222).
Results
The general method employed for the preparation of regioisomeric 1-(3-pyridazinyl)-5-arylpyrazole and 1-(3-pyridazinyl)-3-arylpyrazole derivatives is illustrated in Scheme . As a starting point, Claisen condensation of an appropriate acetophenone derivative (1a–f) with ethyl trifluoroacetate provided the expected 1,3-dicarbonyl adduct (2a–f) in good yield. As might be expected that the reaction of these unsymmetrical β-diketones with a monoaryl-substituted hydrazine derivative might lead to the formation of a mixture of pyrazole isomers, depending on the reaction conditions which affect the site of initial nucleophilic attack in a regioselective process. As seen in Scheme , when we condensed trifluoromethyl-substituted β-diketones (2a–f) with 3-chloro-6-hydrazinopyridazine (3) in refluxing ethanol without any acid or base catalyst, stable 1,3-diaryl-5-hydroxy-4,5-dihydropyrazole derivatives (4a–f) were isolated while we were expecting ring closure followed by dehydration to obtain the pyrazole. The 1,3-diaryl-5-hydroxy-4,5-dihydropyrazoles were only converted to the aromatic pyrazoles on treatment with acetic acid at elevated temperature and regioisomeric 1-(pyridazin-3(2H)-on-6-yl)-3-arylpyrazole derivatives (5a–f) were obtained, which were subsequently converted to 1-(6-chloro-3-pyridazinyl)-3-arylpyrazole derivatives (6a–f) by reaction with phosphorus oxychloride. The other regioisomers, 1-(3-pyridazinyl)-5-arylpyrazoles (7a–f), were generated almost exclusively by carrying out the condensation in the presence of the hydrochloride salt of the 3-chloro-6-hydrazinopyridazine in diglyme. These were subsequently converted to the corresponding 3(2H)-pyridazinone derivatives (8a–f) by refluxing in glacial acetic acid.
Scheme 1 The synthetic methodology used in the preparation of title compounds.
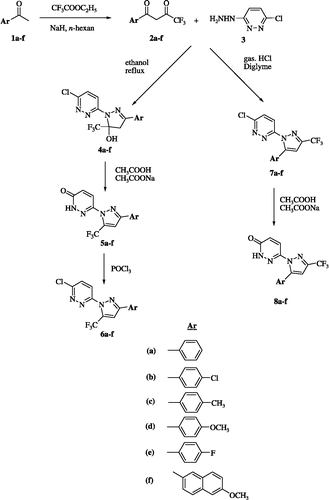
The structures of synthesized 1,3- and 1,5-isomers were fully elucidated using 1H- and 13C-NMR, DEPT and HMBC experiments. To avoid repetition in describing the structural verification between the 1,3- and 1,5-regioisomers, only the structure elucidations of 6e and 7e as representative examples will be discussed here.
For both compounds 6e and 7e, elemental analysis and mass spectral data (m/z 342 M+) established their molecular formula as C14H7ClF4N4, suggesting that they are possibly the 1,3- and 1,5-diarypyrazole regioisomers. The summary of spectral properties of the two regioisomeric structures is shown in . First of all, the 1H-NMR spectrum of compound 6e indicated very simple analysis of their structures showing a singlet proton signal at δ 7.93 (H-4) and two sets of aromatic proton signals attached to the phenyl and pyridazine rings, respectively. 13C-NMR and its DEPT experiment indicated the presence of a quaternary sp2 carbon signal at 151.95 ppm which showed a remarkable 2J-correlation with two proton signals at δ 7.93 ppm (s, pyrazole H-4) and δ 8.07 ppm (dd, H-2″, 6″). Moreover, two quartet C signals at δ 119.4 ppm (CF3, J = 269 Hz) and δ 132.9 ppm (pyrazole C-5, J = 40 Hz) showing JCH long-range correlation with the proton signal at δ 7.93 ppm (pyrazole-H4) were observed. All of the obtained correlations unambiguously confirmed that the 13C signal at δ 151.95 ppm belongs to the imino carbon (C-3) indicating that ring closure had occurred to put the phenyl substituent at 3-position of the central pyrazole. In the case of the 1,5-isomer (7e), the olefinic carbon signal (C-5) was observed at about δ 145.8 ppm as a singlet, and C-3 was observed at 143.9 as a quartet (J = 38.3 Hz). In conclusion, the structures of compound 6e and 7e were unambiguously elucidated as 1,3- and 1,5-diaryl substituted regioisomers, respectively, as shown in .
Figure 1 Summary of spectral differences between compound 6e and 7e.
After comparing the physical and spectral data of the 1,3- and 1,5-regioisomers, it was discovered that melting points of all the 1,5-regioisomers (i.e. 7e, mp 136°C) were relatively lower than those of the corresponding 1,3-regioisomers (i.e. 6e, mp 176°C), and that the similar characteristic spectral patterns were observed for the same type of regioisomers. Firstly, the 1H-NMR spectra of all the 1,5-regioisomers showed an overlapping couple of doublet signals at about 8.2 and 8.22 ppm belonging to pyridazine-H4′ and H5′ protons, respectively. In contrast, these proton peaks were observed as clearly separated two doublets at about 8.4 ppm (H4′) and 8.2 ppm (H5′) in the 1,3-regioisomers indicating a chemical shift of pyridazine-H4′ to lower field. Secondly, in the 1H NMR spectra of all the 1,3-regioisomers, the signal of phenyl-H2″ and 6″ appeared at low field (8.07 ppm in 6e) as compared to the signal of these phenyl protons in the 1,5-regioisomer which occurred at higher field (7.44 ppm in 7e). Therefore, we can indicate another slight distinction between 1,3- and 1,5-regioisomers in the aromatic region indicating that any phenyl proton signals lower than 7.5 ppm did not appear in the 1,5-type derivatives, which were characteristic low field phenyl proton signals corresponding to the 1,3-type (). It is worthwhile mentioning that our observations of the difference in mp and NMR spectra between the above regioisomers can be used as a valuable reference when dealing with structure verification of related compounds.
The title compounds at a concentration of 8 μM were tested for their inhibitory potency against COX-1 and COX-2 in a human whole blood assay which is considered to be the more biologically relevant way to assess the inhibition of the cyclooxygenase isoenzymes, COX-1 and COX-2, by a test compound [Citation32,Citation33]. The results of the % inhibitory activity on COX-1 and COX-2 enzymes are shown in .
Table I. % Inhibition of compounds against COX-1 and COX-2 at 8 μM concentration
Discussion
The in vitro potencies of regioisomeric 1-pyridazinyl-3(5)-aryl-5(3)-trifuoromethylpyrazoles are shown in . The compounds illustrated here were tested at an initial dose of 8 μM for preliminary screening of inhibitory potency. As is evident from , all compounds with both a 1,3- and 1,5-diarylsubstitution pattern did not show an appreciable selectivity on the inhibition of COX enzymes as expected. The inhibitory activities at this high dose level were not very marked except for compounds 7a–f, 8c and 8d, which showed comparable activity to the reference compound SC-560, giving 100% inhibition toward COX-1, but with low selectivity (selectivity ratios of 0.58–0.96). Most of the compounds showed preference for the COX-1 isoform to some extent, and this was particularly noted when the phenyl and pyridazine groups were adjacent to each other about a central pyrazole template as in compounds 7a–f (1,5-diaryl substitution pattern). The replacement of pyridazine with pyridazinone in this particular series resulted in compounds (8a–f) with reduced inhibitory potency and selectivity against COX-1. As also seen from , a range of substituents on the phenyl is tolerated to the same extent and did not contribute to the selectivity as much at this high test concentration. The replacement of the phenyl substituent with a larger naphthalene moiety usually caused a small decrease in the inhibitory potency in the series.
In conclusion, the results of our preliminary screening of the novel compounds incorporating the pyridazine nucleus as one of the aryl substituents about the central pyrazole ring showed promising leads for developing newer COX inhibitors. Although the spatial orientation of the two aromatic rings of the tricyclic inhibitor class (coxibs) was known to be critical for COX inhibition, our initial results indicated that the synthesized 1-pyridazinyl-3-phenyl-5-trifuoromethylpyrazoles, showing the 1,3-diarylsubstitution pattern, also resulted in inhibition of both COX isoforms, although the inhibitory potency was not very pronounced at the 8 μM screening dose used.
In addition, the possibility of easy functionalization(s) at various ring positions of pyridazinones, the electron withdrawing ability of the pyridazinone moiety, their stability and moderate solubility in organic solvents make this ring system interesting for synthetic applications to obtain biologically active distinct derivatives [Citation34]. Further studies for developing better candidates are under ongoing investigation in our laboratory.
Acknowledgements
The investigation was supported by a research grant from the SPO (2003K120470-01).
References
- Brooks P, Emery P, Evans JF, Fener H, Hawkey CJ, Patrono C, Smolen J, Breedveld F, Day R, Dougados M, Ehrich EW, Gijon-Banos J, Kvien TK, Van Rijswijk MH, Warner T, Zeidler H. Rheumatology 1999; 38: 779–788
- Van Ryn J, Trummlitz G, Pairet M. Curr Med Chem 2000; 7: 1145–1161
- Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Proc Natl Acad Sci USA 1994; 91: 3228–3232
- Meyer-Kirchrath J, Schrör K. Curr Med Chem 2000; 7: 1121–1129
- Gans KR, Galbraith W, Roman RJ, Haber SB, Kerr JS, Schmidt WK, Smith C, Hewes WE, Ackerman NR. J Pharmacol Exp Ther 1990; 254: 180–187
- Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan H, Gertz BJ, FitzGerald GA. J Pharmacol Exp Ther 1999; 289: 735–741
- Dannhardt G, Kiefer W. Eur J Med Chem 2001; 36: 109–126
- Sciulli MG, Capone ML, Tacconelli S, Patrignani P. Pharmacol Res 2005; 57: 66–85
- Wallace JL. Trends Pharmacol Sci 1999; 20: 4–6
- Pham K, Hirschberg R. Curr Top Med Chem 2005; 5: 465–473
- Masferrer J. Adv Exp Med Biol 2003; 532: 209–213
- Amir M, Agarwal HK. Pharmazie 2005; 60: 563–570
- Gee J, Lee IL, Jendiroba D, Fischer SM, Grossman HB, Sabichi AL. Oncol Rep 2006; 15: 471–478
- Klenke FM, Gebhard MM, Ewerbeck V, Abdollahi A, Huber PE, Sckell A. BMC Cancer 2006; 6: 1–8
- Hoozemans JJ, O'Banion MK. Curr Drug Tar CNS Neurol Disord 2005; 4: 307–315
- Coudert P, Rubat C, Rohet F, Leal F, Fialip J, Couquelet J. Pharm Pharmacol Commun 2000; 6: 387–396
- Dal Piaz V, Giovannoni MP, Ciciani G, Barlocco D, Giardina G, Petrone G, Clarke GD. Eur J Med Chem 1996; 31: 65–70
- Moreau S, Coudert P, Rubat C, Albuisson E, Couquelet J. Arzneim-Forsch Drug Res 1996; 46: 800–805
- Sato M, Ishizuka Y, Yamaguchi A. Arzneimittelforschung 1981; 31: 1738–1745
- Takaya M, Sato M, Terashima K, Tanizawa H. J Med Chem 1979; 22: 53–58
- Chintakunta VK, Akella V, Vedula MS, Mamnoor PK, Mishra P, Casturi SR, Vangoori A, Rajagopalan R. Eur J Med Chem 2002; 37: 339–347
- Li CS, Brideau C, Chan CC, Savoie C, Claveau D, Charleson S, Gordon R, Greig G, Gauthier JY, Lau CK, Riendeau D, Therien M, Wong E, Prasit P. Bioorg Med Chem Lett 2003; 13: 597–600
- Banoglu E, Akoglu Ç, Ünlü S, Küpeli E, Yesilada E, Sahin MF. Arch Pharm Pharm Med Chem 2004; 337: 7–14
- Banoglu E, Akoğlu C, Ünlü S, Ergün BC, Küpeli E, Yeşilada E, Şahin MF. Arzneim-Forsch Drug Res 2005; 55: 520–527
- Dogruer DS, Sahin MF, Ünlü S, Shigeru I. Arch Pharm Pharm Med Chem 2000; 333: 79–86
- Gökçe M, Dogruer D, Şahin MF. Farmaco 2001; 56: 233–237
- Okçelik B, Ünlü S, Banoglu E, Küpeli E, Yesilada E, Sahin MF. Arch Pharm Pharm Med Chem 2003; 336: 406–412
- Süküroglu M, Ergün BC, Ünlü S, Şahin MF, Küpeli E, Yeşilada E, Banoglu E. Arch Pharm Res 2005; 28: 509–517
- Druey J, Meier K, Eichenberger K. Helv Chim Acta 1954; 37: 121–133
- Lee SG, Kim JJ, Kim HK, Kweon DH, Kang YJ, Cho SD, Kim SK, Yoon YJ. Curr Org Chem 2004; 8: 1463–1480
- Uekawa M, Miyamoto Y, Ikeda H, Kaifu K, Nakaya T. Synt Metals 1997; 91: 259–262
- Brideau C, Kargman S, Liu S, Dallob AL, Ehrich EW, Rodger IW, Chan CC. Inflamm Res 1996; 45: 68–74
- Patrignani P, Panara MR, Greco A, Fusco O, Natoli C, Iacobelli S, Cipollone F, Ganci A, Creminon C, Maclouf J, Patrono C. J Pharmacol Exp Ther 1994; 271: 1705–1712
- Matyus P. J Heterocyclic Chem 1998; 35: 1075–1089