Abstract
A series of pyrrolo[2,3-d]pyridazinones was synthesized and tested for their inhibitory activity on PDE4 subtypes A, B and D and selectivity toward Rolipram high affinity binding site (HARBS). New agents with interesting profile were reported; in particular compound 9e showed a good PDE4 subtype selectivity, being 8 times more potent (IC50 = 0.32 μM) for PDE4B (anti-inflammatory) than for PDE4D (IC50 = 2.5 μM), generally considered the subtype responsible for emesis. Moreover the ratio HARBS/PDE4B was particularly favourable for 9e (147), suggesting that the best arranged groups around the pyrrolopyridazinone core are an isopropyl at position-1, an ethoxycarbonyl at position-2, together with an ethyl group at position-6.
For compounds 8 and 15a the ability to inhibit TNFα production in PBMC was evaluated and the results are consistent with their PDE4 inhibitory activity.
Introduction
Phosphodiesterases (PDEs), are the enzymes responsible for the hydrolysis of cyclic adenosine monophosphate (c-AMP) and cyclic guanosine monophosphate (c-GMP) which play a key role as secondary messengers in cell functions. The PDE4 subfamily, specific for cAMP, is identified by four subtypes (PDE4A, PDE4B, PDE4C and PDE4D). Each subtype is differently expressed in various tissues, in particular PDE4A and PDE4B isoforms are ubiquitous, while PDE4C and PDE4D are essentially present in neuronal tissue and in inflammatory cells respectively[Citation1,Citation2]. In the last few years PDE4 has attracted considerable interest as a therapeutic target for a variety of pathologies, such as asthma, chronic obstructive pulmonary diseases (COPD) and inflammatory diseases[Citation3]. Since recent studies demonstrated the fundamental role of inflammation in asthma pathology[Citation4,Citation5], it is clear that selective PDE4 inhibitors which are able to both suppress inflammatory progression and to reduce the bronchial smooth muscle tone, represent a very attractive alternative to the classic combined therapy with corticosteroids and bronchodilators. New important applications of PDE4 inhibitors have been proposed, such as in rheumatoid arthritis and other important autoimmune diseases, this hypothesis being associated with the evidence that PDE4 selective inhibitors strongly reduce tumor necrosis factor α (TNF-α) production which plays an important role in these types of pathologies[Citation6]. Recent studies demonstrated an increase of PDE catalytic activity together with an increase of sensitivity to PDE4 inhibitors in atopic dermatitis, suggesting that this pathology could be an attractive target for PDE4 inhibitors[Citation7,Citation8]. Finally, the presence of differently located subtypes of PDE4 opens new opportunities for the development of selective inhibitors devoid of limiting side-effects[Citation9].
The archetypal PDE4 inhibitor Rolipram 1[Citation10] () showed very important unwanted side-effects, such as headache, nausea and vomiting, which are related to the binding of 1 with Rolipram high affinity binding site (HARBS)[Citation11]. This same effect was found in Rolipram analogues and in many other compounds structurally unrelated to Rolipram, such as quinazolinediones 2 and xanthine derivatives 3Citation12-14.
Figure 1 PDE4 inhibitors.
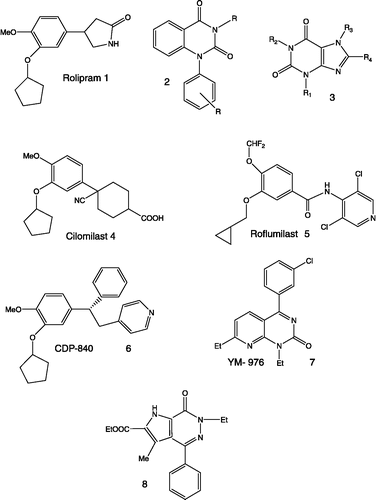
Thus it appears very important to dissociate the affinity for HARBS from PDE4 inhibitory activity, in order to obtain more selective antiasthmatic drugs, devoid of the above side-effects. In recent years, potent and selective PDE4 inhibitors with reduced affinity for HARBS have been described in the literature[Citation15] and a number of these are now in clinical trials. Compounds such as Cilomilast 4[Citation16,Citation17], Roflumilast 5[Citation18,Citation19], CDP-840 6[Citation20] or YM-976 7[Citation21] are well tolerated and are presently in phase III clinical trials for the treatment of asthma and COPD (compounds 4 and 5)Citation22-24 and in phase II clinical trials for asthmatic pathology (compounds 6 and 7)Citation25-27.
Roflumilast and CDP-840 didn't demonstrate PDE4 subtype selectivity[Citation7,Citation19], whereas Cilomilast is 10-fold more selective for PDE4D than for PDE4A and PDE4B[Citation28]. Presently some compounds with high selectivity for one PDE4 subtype (up to 300-fold) have been synthesized, and recent studies demonstrated a correlation between PDE4A/B inhibitory activity and TNFα release inhibition[Citation7]. Moreover some authors suggested that PDE4D is the subtype responsible for the emetic side-effects[Citation29]. These results and proposals seems to open new opportunities for the development of selective inhibitors with reduced adverse effects.
A systematic screening performed on our internal library, allowed us to identify compound 8 as an interesting lead. This compound exhibited IC50 in the same micromolar range as Rolipram (IC50 = 0.6 μM) and a good selectivity profile versus PDE4 (PDE3/PDE4 > 30). Moreover it displays low affinity for HARBS (IC50 = 2.0 μM). Thus it possesses a much better ratio of PDE4 inhibitory activity versus affinity for HARBS (HARBS/PDE4 = 3.33 for 9, and 0.02 for Rolipram).
On this basis we report here some chemical modifications of lead compound 8 which afforded the new pyrrolo[2,3-d]pyridazines 9a–e and 15a–f and their PDE4 subtypes inhibitory activity.
Materials and methods
All melting points were determined on a Buchi apparatus and are uncorrected. 1H-NMR spectra were recorded with Varian Gemini 200 instruments. Chemical shifts are reported in ppm, using the solvent as internal standard. Extracts were dried over Na2SO4 and the solvents were removed under reduced pressure. Merck F-254 commercial plates were used for analytical TLC to follow the course of the reaction. Silica gel 60 (Merck 70–230 mesh) was used for column chromatography.
Chemistry
The synthesis of the novel pyrrolo[2,3-d]pyridazinones 9a–e and 15a–f is depicted in and respectively.
Figure 2 Synthesis of pyrrolo[2,3-d]pyridazinones 9a–e.
![Figure 2 Synthesis of pyrrolo[2,3-d]pyridazinones 9a–e.](/cms/asset/4bca8175-2350-4af4-bfd3-f243648ecb2a/ienz_a_211405_f0002_b.gif)
Figure 3 Synthesis of pyrrolo[2,3-d]pyridazinones 15a–f.
![Figure 3 Synthesis of pyrrolo[2,3-d]pyridazinones 15a–f.](/cms/asset/94f387e9-0b31-4048-8306-3be4e3a07680/ienz_a_211405_f0003_b.gif)
In , the procedure affording the N-1 substituted compounds 9a–e is reported. All final products were prepared starting from the lead compound 8 which was alkylated under standard conditions to afford the corresponding N-substituted derivatives 9a–e.
In , is depicted the synthetic route affording the pyrrolo[2,3-d]pyridazinones 15a–f in which modifications at positions- 3, -6 and on the phenyl ring of the bicyclic system were performed with respect to the lead 8.
Isoxazoles of type 10, which are the starting material for the final compounds 15 have been previously describedCitation30-32 with the exception of 10b. Treatment of isoxazoles 10 with hydrazine followed by alkylation with the appropriate alkyl halide afforded compounds 12, which were transformed into the 4-nitropyridazinone derivatives 13 through oxidative cleavage of the isoxazole ring[Citation30]. Displacement of the nitro group with glycine ethyl ester afforded the open intermediates 14 which led to the final pyrrolopyridazinones 15 by treatment with sodium ethoxide in absolute ethanol at room temperature (11d[Citation33], 12d–f [Citation34],13d–f [Citation33]).
General procedure for compounds 9a–e
A suspension of 8 (0.3 mmol), anhydrous K2CO3 (6 mmol) and the appropriate halide (0.5 mmol) in anhydrous DMF was heated for 30–60 min (16 h for 9e) at 80–110°C. After the mixture was cooled, ice cold water was added and the crude precipitate was recovered by suction, with the exception of 9d which was extracted with CH2Cl2 (3 × 15 mL).
Ethyl 6,7-dihydro-1,3-dimethyl-6-ethyl-7-oxo-4-phenyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 9a
Mp = 82–83°C; crystallization solvent = EtOH; yield = 45%.
1H-NMR (CDCl3), δ, ppm: 1.45 (m, 6H, NCH2CH3 and OCH2CH3), 2.10 (s, 3H, CCH3), 4.35 (m, 4H, NCH2CH3 and OCH2CH3), 4.50 (s, 3H, NCH3), 7.45 (s, 5H, Ar).
Anal. found: C, 67.40; H, 6.24; N, 12.34. C19H21N3O3 requires: C, 67.23; H, 6.25; N, 12.38%.
Ethyl 1,6-diethyl-6,7-dihydro-3-methyl-7-oxo-4-phenyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 9b
Mp = 77–78°C; crystallization solvent = EtOH; yield = 55%.
1H-NMR (CDCl3), δ, ppm: 1.40 (m, 9H, 2NCH2CH3 and OCH2CH3), 1.95 (s, 3H, CCH3), 4.35 (m, 4H, NCH2CH3 and OCH2CH3), 5.10 (q, 2H, NCH2CH3), 7.45 (s, 5H, Ar).
Anal. found: C, 68.03; H, 6.58; N, 11.92. C20H23N3O3 requires: C, 67.96; H, 6.57; N, 11.89%.
Ethyl 6,7-dihydro-6-ethyl-3-methyl-7-oxo-4-phenyl-1-propyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 9c
Mp = 77–78°C; crystallization solvent = EtOH; yield = 90%.
1H-NMR (CDCl3), δ, ppm: 0.95 (t, 3H, (CH2)2CH3), 1.40 (m, 6H, NCH2CH3 and OCH2CH3), 1.80 (m, 2H, CH2CH2CH3), 1.95 (s, 3H, CCH3), 4.35 (m, 4H, NCH2CH3 and OCH2CH3), 5.00 (t, 2H, NCH2CH2CH3), 7.45 (s, 5H, Ar).
Anal. found: C, 68.80; H, 6.89; N, 11.41. C21H25N3O3 requires: C, 68.63; H, 6.87; N, 11.44%.
Ethyl 1-benzyl-6,7-dihydro-6-ethyl-3-methyl-7-oxo-4-phenyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 9d
Mp = 105–106°C; crystallization solvent = EtOH; yield = 70%.
1H-NMR (CDCl3), δ, ppm: 1.25 (t, 3H, NCH2CH3), 1.40 (t, 3H, OCH2CH3), 2.00 (s, 3H, CCH3), 4.30 (m, 4H, NCH2CH3 and OCH2CH3), 6.40 (s, 2H, NCH2Ph), 7.10 (m, 2H, Ar), 7.25 (m, 3H, Ar), 7.50 (s, 5H, Ar).
Anal. found: C, 72.09; H, 6.09; N, 10.08. C25H25N3O3 requires: C, 72.26; H, 6.08; N, 10.11%.
Ethyl 6,7-dihydro-6-ethyl-1-isopropyl-3-methyl-7-oxo-4-phenyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 9e
Mp = 99–101°C; crystallization solvent = EtOH; yield = 60%.
1H-NMR (CDCl3), δ, ppm: 1.40 (m, 6H, NCH2CH3 and OCH2CH3), 1.70 (d, 6H, CH(CH3)2), 1.85 (s, 3H, CCH3), 4.35 (m, 4H, NCH2CH3 and OCH2CH3), 5.85 (m, 1H, CH(CH3)2), 7.45 (s, 5H, Ar).
Anal. found: C, 68.79; H, 6.89; N, 11.42. C21H25N3O3 requires: C, 68.63; H, 6.87; N, 11.44%.
Ethyl-4-benzoyl-5-propylisoxazole-3-carboxylate 10b
To a cooled ( − 5–10°C) and stirred solution of sodium ethoxide, obtained from sodium (15 mmol) and anhydrous EtOH (30 mL), a solution of 1-phenylhexan-1,3-dione[Citation35] (15 mmol) in the same solvent (15 mL) was slowly added. A solution of ethyl chloro(hydroximino)acetate, commercially available (15 mmol) in anhydrous EtOH (10 mL) was added in a dropwise manner (over 1 h period). The mixture, neutralized with 6N HCl was evaporated to afford an oil which was washed with 0.5 N NaOH and water and then extracted with CH2Cl2 (3 × 25 mL). The organic layer was washed with water, dried with Na2SO4 and evaporated in vacuo to afford an oil, which was purified by column chromatography (cyclohexane/ethyl acetate 1:1). Yield = 57%.
1H-NMR (CDCl3), δ, ppm: 1.00 (t, 6H, 2CH3), 1.80 (m, 2H, CH2CH2CH3), 2.90 (t, 2H, CH2CH2CH3), 4.10 (q, 2H, OCH2CH3), 7.50 (s, 5H, Ar).
General procedure for compounds 11a–c
The appropriate isoxazole 10a–c (0.5 mmol) was dissolved in EtOH (5mL) and then hydrazine hydrate (0.8 mmol) was added at room temperature. After 10–60 min the crude product was collected by suction from the cooled mixture.
3-Ethyl-4-phenylisoxazolo[3,4-d]pyridazin-7-(6H)-one 11a
Mp = 204–206°C; crystallization solvent = EtOH; yield = 81%.
1H-NMR (CDCl3), δ, ppm: 1.20 (t, 3H, CH3), 2.80 (q, 2H, CH2), 7.50 (s, 5H, Ar).
4-Phenyl-3-n.propylisoxazolo[3,4-d]pyridazin-7-(6H)-one 11b
Mp = 146–148°C; crystallization solvent = EtOH; yield = 82%.
1H-NMR (CDCl3), δ, ppm: 0.90 (t, 3H, CH3), 1.80 (m, 2H, CH2CH2CH3), 2.80 (t, 2H, CH2CH2CH3), 7.50 (s, 5H, Ar), 10.00 (exch br s, 1H, NH).
4-(p-Chlorophenyl)-3-methylisoxazolo[3,4-d]pyridazin-7-(6H)-one 11c
Mp = 210–212°C; crystallization solvent = EtOH; yield = 74%.
1H-NMR (CDCl3), δ, ppm: 2.60 (s, 3H, CH3), 7.50 (m, 4H, Ar), 9.90 (exch br s, 1H, NH).
General procedure for compounds 12a–c
A mixture of the appropriate isoxazolo[3,4-d]pyridazinones 11 (1 mmol), anhydrous K2CO3 (5 mmol) and appropriate alkyl bromide (5 mmol) in anhydrous DMF (3–6 mL) was heated under stirring for 1 h at 110°C. After dilution with cold water (80–100 mL) the crude precipitate was recovered by suction.
3,6-Diethyl-4-phenylisoxazolo[3,4-d]pyridazin-7(6H)-one 12a
Mp = 67–70°C; crystallization solvent = EtOH; yield = 86%.
1H-NMR (CDCl3), δ, ppm: 1.20 (t, 3H, CCH2CH3), 1.40 (t, 3H, NCH2CH3), 2.85 (q, 2H, CCH2CH3), 4.30 (q, 2H, NCH2CH3), 7.55 (s, 5H, Ar).
6-Ethyl-4-phenyl-3-n.propylisoxazolo[3,4-d]pyridazin-7(6H)-one 12b
Mp = 70–72°C; crystallization solvent = EtOH; yield = 75%.
1H-NMR (CDCl3), δ, ppm: 0.90 (t, 3H, CH2CH2CH3), 1.50 (t, 3H, NCH2CH3), 1.80 (m, 2H, CH2CH2CH3), 2.80 (m, 2H, CH2CH2CH3), 4.25 (m, 2H, NCH2CH3), 7.50 (s, 5H, Ar).
4-(p-Chlorophenyl)-6-ethyl-3-methylisoxazolo[3,4-d]pyridazin-7(6H)-one 12c
Mp = 126–128°C; crystallization solvent = EtOH; yield = 70%.
1H-NMR (CDCl3), δ, ppm: 1.40 (t, 3H, NCH2CH3), 2.50 (s, 3H, CCH3), 4.20 (q, 2H, NCH2CH3), 7.50 (m, 4H, Ar).
General procedure for compounds 13a–c
To a stirred suspension of the appropriate compounds 12 (2.5 mmol) in AcOH (50% w/v; 20 mL) and HNO3 (65% w/v; 2.5 mL), CAN (15 mmol) was added in a portionwise manner at 55°C over 30–45 min period. Then ice-cold water (100 mL) was added and compounds 13a and 13c were recovered by suction; to obtain 13b the mixture was extracted with CH2Cl2 (3 × 20 mL). Evaporation of the solvent afforded an oil which was purified by column chromatography using toluene/ethyl acetate (8:2) as eluent.
2-Ethyl-4-nitro-6-phenyl-5-propionylpyridazin-3(2H)-one 13a
Mp = 97–100°C; crystallization solvent = EtOH; yield = 65%.
1H-NMR (CDCl3), δ, ppm: 0.95 (t, 3H, COCH2CH3), 1.50 (t, 3H, NCH2CH3), 2.35 (q, 2H, COCH2CH3), 4.40 (q, 2H, NCH2CH3), 7.50 (s, 5H, Ar).
5-Butyryl-2-ethyl-6-phenyl-4-nitropyridazin-3(2H)-one 13b
oil; yield = 52%.
1H-NMR (CDCl3), δ, ppm: 0.90 (t, 3H, COCH2CH2CH3), 1.50 (m, 5H, NCH2CH3 and COCH2CH2CH3), 2.35 (m, 2H, COCH2CH2CH3), 4.40 (q, 2H, NCH2CH3), 7.50 (s, 5H, Ar).
5-Acetyl-6-(p-chlorophenyl)-2-ethyl-4-nitropyridazin-3(2H)-one 13c
Mp = 105–107°C; crystallization solvent = EtOH; yield = 59%.
1H-NMR (CDCl3), δ, ppm: 1.50 (t, 3H, NCH2CH3), 2.20 (s, 3H, COCH3), 4.40 (q, 2H, NCH2CH3), 7.50 (m, 4H, Ar).
General procedure for compounds 15a–f
A suspension of the appropriate nitroderivative 13 (0.3 mmol) and glycine ethyl ester (0.6 mmol) in EtOH (3 mL) was heated at 40–50°C for 20–160 min. After cooling, the intermediate 14 was recovered by suction. The crude 14, completely dried, was treated with EtONa (0.5 mmol) in absolute. EtOH (3 mL) at room temperature for 20–30 min. After dilution with water (10 mL), the mixture was acidified with 6N HCl to afford compounds of type 15 which were recovered by suction.
Ethyl 3,6-diethyl-6,7-dihydro-4-phenyl-7-oxo-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 15a
Mp = 127–131°C; crystallization solvent = EtOH; yield = 68%.
1H-NMR (CDCl3), δ, ppm: 0.80 (t, 3H, CCH2CH3), 1.40 (m, 6H, NCH2CH3 and OCH2CH3), 2.60 (q, 2H, CCH2CH3), 4.35 (m, 4H, NCH2CH3 and OCH2CH3), 7.50 (m, 4H, Ar).
Anal. found: C, 67.40; H, 6.24; N, 12.40. C19H21N3O3 requires: C, 67.23; H, 6.25; N, 12.38%.
Ethyl 6,7-dihydro-6-ethyl-4-phenyl-3-n.propyl-7-oxo-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 15b
Mp = 98–100°C; crystallization solvent = EtOH; yield = 52%.
1H-NMR (CDCl3), δ, ppm: 0.50 (t, 3H, CH2CH2CH3), 1.35 (m, 8H, NCH2CH3, CH2CH2CH3 and OCH2CH3), 2.50 (t, 2H, CH2CH2CH3), 4.40 (m, 4H, NCH2CH3 and OCH2CH3), 7.50 (m, 5H, Ar), 10.0 (exch. br. s. 1H, NH).
Anal. found: C, 67.79; H, 6.58; N, 11.91. C20H23N3O3 requires: C, 67.96; H, 6.57; N, 11.89%.
Ethyl 4-(p-chlorophenyl)-6,7-dihydro-6-ethyl-3-methyl-7-oxo-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 15c
Mp = 206–208°C; crystallization solvent = EtOH; yield = 82%.
1H-NMR (CDCl3), δ, ppm: 1.40 (m, 6H, NCH2CH3 and OCH2CH3), 2.10 (s, 3H, CCH3), 4.40 (m, 4H, NCH2CH3 and OCH2CH3), 7.50 (m, 4H, Ar).
Anal. found: C, 60.23; H, 5.06; N, 11.65. C18H18N3O3Cl requires: C, 60.08; H, 5.05; N, 11.68%.
Ethyl 6,7-dihydro-3-methyl-7-oxo-4-phenyl-6-n.propyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 15d
Mp = 175–178°C; crystallization solvent = EtOH; yield = 75%.
1H-NMR (CDCl3), δ, ppm: 1.00 (t, 3H, NCH2CH2CH3), 1.45 (t, 3H, OCH2CH3), 1.95 (m, 2H, NCH2CH2CH3), 2.10 (s, 3H, CCH3), 4.35 (m, 4H, NCH2CH2CH3 and OCH2CH3), 7.50 (m, 5H, Ar), 11.50 (exch. br. s. 1H, NH).
Anal. found: C, 67.05; H, 6.26; N, 12.37. C19H21N3O3 requires: C, 67.23; H, 6.25; N, 12.38%.
Ethyl 6,7-dihydro-6-isopropyl-3-methyl-7-oxo-4-phenyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 15e
Mp = 145–147°C; crystallization solvent = EtOH; yield = 74%.
1H-NMR (CDCl3), δ, ppm: 1.45 (m, 9H, CH(CH3)2 and OCH2CH3), 2.20 (s, 3H, CCH3), 4.45 (q, 2H, OCH2CH3), 5.50 (m, 1H, CH(CH3)2), 7.60 (m, 5H, Ar), 11.60 (exch. br. s. 1H, NH).
Anal. found: C, 67.08; H, 6.24; N, 12.40. C19H21N3O3 requires: C, 67.23; H, 6.25; N, 12.38%.
Ethyl 6-n.butyl-6,7-dihydro-3-methyl-7-oxo-4-phenyl-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate 15f
Mp = 136–138°C; crystallization solvent = EtOH; yield = 64%.
1H-NMR (CDCl3), δ, ppm: 0.95 (t, 3H, NCH2CH2CH2CH3), 1.40 (t, 3H, OCH2CH3), 1.80 (m, 4H, NCH2CH2CH2CH3), 2.10 (s, 3H, CCH3), 4.40 (m, 4H, NCH2CH2CH2CH3 and OCH2CH3), 7.50 (m, 5H, Ar), 10.80 (exch. br.s. 1H, NH).
Anal. found: C, 68.09; H, 6.58; N, 11.92. C20H23N3O3 requires: C, 67.96; H, 6.57; N, 11.89%.
Pharmacology
Purification of phosphodiesterase 3 (PDE3)
Cyclic nucleotide phosphodiesterases 3 were obtained from guinea pig ventricular tissue following the procedure described by Gristwood et al.[Citation36]. Briefly, the tissue was homogenized in 20 mM Bis-Tris pH 6.5 buffer, containing 50 mM sodium acetate, 2 mM benzamidine, 2 mM EDTA, 5 mM β-mercapto-ethanol and 50 μM PMSF using an Ultraturrax homogenizer. The sample was centrifuged at 40000 × g for 20 min and the supernatant was filtered through a 0.22 μm filter. The clean sample was chromatographed on a 1 mL ion-exchange MONO-Q column equilibrated with the same buffer using a FPLC system. The column was developed at a flow rate of 1 mL/min using a linear gradient of sodium acetate from 50 to 1000 mM in a total volume of 25 mL. Fractions of 500 μL were collected. The isoenzymes were characterized prior to use in terms of substrate selectivity and affinity and by the effect of calcium ions (10 μM) plus calmodulin (1.2 μM) and the selective inhibitors Rolipram and SK& 94120. Active fractions were pooled and kept frozen at − 20°C in the presence of g/L bovine serum albumin until used.
PDE3 activity determination
Cyclic nucleotide phosphodiesterase activities were measured using a two step procedure according to Thompson and Strada[Citation37]. Inhibition assays were run in duplicate at a substrate concentration of 0.25 μM. Substrate was cAMP for PDE3. IC50 values were obtained by nonlinear regression using the program InPlot from GraphPad Software. Drugs were dissolved in DMSO, and the effects of this solvent were taken into consideration in the calculations.
Purification of phosphodiesterase 4 (PDE4)
Yeast strains overexpressing PDE4 were inoculated into 500 ml flasks with 100 ml YPD (1 g yeast extract, 2 g peptone, 95 ml H2O, and 5 ml 40% glucose) and grown at 30°C under orbital shaking (150 rpm) for 48 h to ensure reaching one near OD600 = 2.0. Then cells were harvested by centrifugation at 1500 rpm for 10 min at 20°C. All supernatants were removed and pellets resuspended in the appropriate volume of SD (1.7 g yeast nitrogen base w/o aminoacids and sulphate, 5 g (NH4)2SO4, 30% galactose, 0.5% adenine sulphate and 0.50 mg/mL L-lysine-HCl) to obtain an OD600 between 0.4 and 0.6 and then incubated with shaking at 30°C until an OD600 of 2.0 was reached (usually 48 h). Then cells were centrifuged at 4000 rpm for 20 min at 4°C and the supernatants discarded. Pellets were resuspended in 100 ml of ice-cold sterile water and centrifuged at 4000 rpm for 20 min at 4°C. The supernatants were discarded and the wet weight of pellets was measured and 1.3 volume/weight of YHB (2 M KCl, 0.5 M EDTA, 1 M HEPES) supplemented with DTT and protease inhibitors added and then resuspended. Another centrifugation at 4000 rpm for 20 min at 4°C was carried out. The supernatants were discarded and using a spatula the yeast paste was transferred into a 10-mL syringe and yeast “noodles” were made and placed into a small plastic beaker filled with liquid nitrogen. The frozen yeast “noodles” can be stored at − 80°C until required for extract preparation.
Preparation of whole-cell extract
The yeast “noodles” were poured into a cooled porcelain mortar filled with liquid nitrogen and crushed under liquid nitrogen until the yeast showed a powdery smooth consistency. Then yeast/liquid nitrogen suspension was poured into a plastic beaker and the remaining yeast powder scraped into the beaker with a spatula. Dialysis buffer (1.4 volume/ weight), YDB (2 M KCl, 0.5 M EDTA, 1 M HEPES, 100% glycerol, 1 M DTT, 0.25 M PMSF, 1 mg/mL leupeptin and supplemented with DTT and protease inhibitors) was then added to resuspend the yeast powder and then centrifugated at 25000 rpm for 2 h at 4°C. The lipid layer was removed, supernatants recovered and transferred to dialysis tubing and dialyzed overnight against 500 ml YDB. Then dialysis supernatants were transferred to microtubes in small aliquots (100 μl) and quick-frozen and stored at − 80°C; The protein concentration was determined by the method of Bradford with the Bio-Rad protein assay kit using bovine serum albumin (BSA) as a standard.
PDE4 activity determination
PDE4 activity from various subtypes (PDE4B1, PDE4A4 and PDE4D3) was monitored by measuring the hydrolysis of [3H]-AMP using a PDE-SPA kit (Amersham International) as reported in the literature[Citation38]. Extracted Enzyme (4 μg of protein) was incubated in “low binding” plates (Costar 3604) for 60 min at room temperature. The assay mixture (80 μL) contained 15 nM [3H]-cAMP (1 μC/mL) in the assy buffer (50 mM Tris pH 7.5, 8.3 mM MgCl2, 1.7 mM EGTA) and 10 μL of test compound. These compounds were resuspended in DMSO at a stock concentration of 1 mM. The compounds were tested at different concentrations varying fron 10 μM to 10 pM to calculate the ED50.Hydrolysis of [3H]-cAMP was initiated by adding 10 μL of a solution containing enzyme extract, and the plate was then incubated under agitation at room temperature. The reaction was stopped after 60 min (with 10–20% substrate conversion) by addition of 50 μL Phosphodiesterase Scintillation Proximity Assay (SPA) Beads and the reaction was incubated for another 20 min. [3H]-AMP, captured by the SPA beads, was determined by counting the plates in a Wallac-Microbeta Trilux scintillation counter.
[3H]Rolipram displacement
The binding of [3H]Rolipram to rat brain membranes was performed according to Schneider et al.[Citation39] At least six drug concentrations were assayed in duplicate to generate individual displacement curves. IC50 values were calculated from those curves by nonlinear regression using the program Inplot, from GraphPad Sofware. The effect of drug vehicle was taken into account in the calculation.
PBMC assay
Human whole blood of healthy donors was collected in 50 ml Falcon tubes with heparin (20 units/mL). The isolation of PBMCs by centrifugation on a Ficoll-Plaque and hypotonic lysis of remaining red blood cells was performed essentially as described previously by Gantner and coworkers[Citation40]. The purity of PBMCs was >90% and the viability, as measured by trypan blue exclusion, as >95%. Cells were incubated in 96-well plates at a density of 2 × 105 cells/well in a total assay volume of 200 μL (RPMI medium with 2 mM L-Gln, 20 mM Hepes and 10%FBS). A 1 μg/mL of LPS (from Escherichia coli, Sigma, St. Louis, MO) together with vehicle or test compound was incubated in a bath at 37°C for 24 h. Each drug was evaluated at six different concentrations in duplicate determinations. After the 24 h period, the reaction was stopped and media obtained by centrifugation of PBMCs at 13000 rpm at 4°C for 10 min was then subjected to ELISA (R&D Systems) or kept at − 80°C until use.
IC50 values were obtained by non-linear regression using InPlot, GraphPad. The results were expressed as the mean IC50 ± SEM value obtained in 4 independent experiments. Results related to this experiment are reported in .
Results and discussion
All the final compounds were tested for their ability to inhibit PDE4 (PDE4A, PDE4B and PDE4D) and PDE3 isoenzymes, as well as for their affinity for HARBS ().
SAR studies were performed in comparison with the lead 8 which did not demonstrate subtype selectivity showing IC50s of 0.11, 0.067 and 0.049 μM for PDE4A, PDE4B and PDE4D respectively, but a significant selectivity versus HARBS (HARBS/PDE4A-C = 20–40) was observed.
Starting from 15a–b, where the methyl group at position- 3 was elongated with respect to lead 8, an IC50 in the nanomolar range can be observed, but unfortunately these agents were completely devoid of selectivity versus PDE4 subtypes, as well as versus HARBS. These results clearly indicated that elongation of the carbon chain at position-3 did not modify the inhibitory activity but it led to a decrease in selectivity versus HARBS.
Replacement of the hydrogen at position-1 (compounds 9a–e) by alkyl groups was associated with significant effects on the selectivity. In fact, even though 9a–c and 9e showed less potent PDE4 inhibitory activity with respect to the previous compounds, the selectivity versus HARBS (HARBS/PDE4B) was significantly increased, in particular for 9b (ratio = 67) and 9e (147). This compound was also the best one in the present series as regards the PDE4 subtypes selectivity profile, being 8 times more potent for PDE4B, the subtype responsible for the anti-inflammatory effect (IC50 = 0.32 μM) than for PDE4D (IC50 = 2.5 μM), which is generally considered the subtype responsible for central side-effects.
Introduction of a benzyl group at position-1 (compound 9d) was found very detrimental to activity with respect to 9a–c and 9e, where the substituent is a small alkyl group. This result seems to indicate that there is a small liphophilic pocket in the catalytic site of the enzyme which is able to receive linear or branched low molecular weight alkyls groups.
Introduction of a chlorine in the para position of the phenyl group (15c) was associated with a moderate activity for PDE4B and the loss of the selectivity versus HARBS.
Compounds 15d–f, where the carbon chain at position-6 was elongated and branched in comparison to 8, showed a reduced potency towards the three isoenzymes. Thus in this group of differently 6-substituted derivatives, a two carbon chain was found to be optimal.
Finally, all synthesized compounds showed a good PDE3/PDE4 selectivity profile, as reported in .
Table I. Effects of compounds 9 and 15 on PDE4 isoenzymes, PDE3 and displacement of [3H]Rolipram from its binding site (HARBS).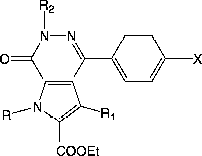 .
.
In conclusion we synthesized novel PDE4 inhibitors endowed with IC50 in the nanomolar range. Selectivity versus HARBS was strongly improved, both in comparison with Rolipram 1 and our lead 8.
The best profile was found for 9e, which showed a high potency for PDE4B and a very high selectivity (147) versus HARBS.
These results suggest that the best arranged groups around the pyrrolopyridazinone core are an isopropyl at position-1, an ethoxycarbonyl at position-2 together with an ethyl group at position-6.
Finally, we evaluated the ability of the most potent compounds (8 and 15a) and of the less active in this series (15f) to reduce TNFα production in human peripheral blood mononuclear cells (PBMC), using Cilomilast as reference drug (). The obtained results really demonstrated a correlation between PDE4 inhibitory activity and TNFα release being the IC50s = 0.198 and 0.192 μM for the most active molecules respectively 4.9 μM for compound 15f.
Table II. TNFα inhibition values for compounds 8, 15a and 15f. .
.
Further studies are in progress to optimize the profile of the above reported compounds.
References
- Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med 1998; 157: 351–370
- Houslay MD. PDE4 cAMP-specific phosphodiesterases. Prog Nucleic Acid Res Mol Biol 2001; 69: 249–315
- Dal Piaz V, Giovannomi MP. Phosphodiesterase 4 inhibitors, structurally unrelated to rolipram, as promising agents for the treatment of asthma and other pathologies. Eur J Med Chem 2000; 35: 463–480
- Bousquet J, Chanez P, Vignola AM, Lacoste JY, Michel FB. Eosinophil inflammation in asthma. Am J Respir Crit Care Med 1994; 150: S33–S38
- Whelan CJ. Inhibition of PAF-, LPS-, and cytokine-induced granulocyte accumulation in guinea pig lung by dexamethasone: Evidence that inhibition of IL-5 release is responsible for the selective inhibition of eosinophilia by glucocorticoids in guinea-pigs. Inflamm Res 1996; 45(4)166–170, Apr;
- Nyman U, Mussener A, Larsson E, Lorentzen J, Klareskog L. Amelioration of collagen II-induced arthritis in rats by the type IV phosphodiesterase inhibitor. Rolipram Clin Exp Immunol 1997; 108: 415–419
- Spina D. Phosphodiesterase-4 inhibitors in the treatment of inflammatory lung disease. Drugs 2003; 63: 2575–2594
- Hanifin JM, Chan SC, Cheng JB, Tofte SJ, Henderson WRJr, Kirby DS, Weiner ES. Type 4 phosphodiesterase inhibitors have clinical and in vitro anti-inflammatory effects in atopic dermatitis. J Invest Dermatol 1996; 107: 51–56
- Muller T, Engels P, Fozard JR. Subtypes of the type 4 cAMP phosphodiesterases: Structure, regulation and selective inhibition. TIPS 1996; 17: 294–298
- Schneider HH, Schmiechen R, Brezinski M, Seidler J. Stereospecific binding of the antidepressant rolipram to brain protein structures. Eur J Pharmacol 1986; 127: 105–115
- Souness JE, Rao S. Proposal for pharmacologically distinct conformers of PDE4 cyclic AMP phosphodiesterases. Cell Signal 1997; 9: 227–236
- Kaulen P, Bruning G, Schneider HH, Sarter N, Baumgarten HG. Autoradiographic mapping of a selective cyclic adenosine monophosphate phosphodiesterase in rat brain with the antidepressant [3H]rolipram. Brain Res 1989; 503: 229–245
- Buckle DR, Arch JR, Connolly BJ, Fenwick AE, Foster KA, Murray KJ, Readshaw SA, Smallridge M, Smith DG. Inhibition of cyclic nucleotide phosphodiesterase by derivatives of 1,3-bis(cyclopropylmethyl)xanthine. J Med Chem 1994; 37(4)476–485
- Larson LJ, Pino MV, Geiger LE, Simeone CR. The toxicity of repeated exposures to rolipram, a type IV phosphodiesterase inhibitor, in rats. Pharmacol Toxicol 1996; 78: 44–49
- Burnouf C, Pruniax MP. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs. Curr Pharm Des 2002; 8: 1255–1296
- Christensen SB, Guider AM, Forster CJ, Gleason JG, Bender PE, Karpinski JM, DeWolf WE, Barnette MS, Underwood DC, Griswold DE, Cielinski LB, Burman M, Bochnowicz S, Osborn RR, Manning CD, Grous M, Hillegas LM, Bartus JO, Ryan MD, Eggleston DS, Haltiwanger RC, Torphy TJ. 1,4-Cyclohexanecarboxylates: Potent and selective inhibitors of phosophodiesterase 4 for the treatment of asthma. J Med Chem 1998; 41: 821–835
- Barnette MS, Christensen SB, Essayan DM, Grous M, Prabhakar U, Rush JA, Kagey-Sobotka A, Torphy TJ. SB 207499 (Ariflo), a potent and selective second-generation phosphodiesterase 4 inhibitor: In vitro anti-inflammatory actions. J Pharmacol Exp Ther 1998; 284: 420–426
- Karish SB, Gagnon JM. The potential role of roflumilast: The new phosphodiesterase-4 inhibitor. Ann Pharmacother 2006; 40(6)1096–1104
- Hatzeman A, Schudt C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther 2001; 297: 267–279
- Perry MJ, O'Connell J, Walker C, Crabbe T, Baldock D, Russell A, Lumb S, Huang Z, Howat D, Alle R, Merriman M, Walls J, Daniel T, Hughes B, Laliberte F, Higgs GA, Owens RJ. CDP840: A novel inhibitor of PDE-4. Cell Biochem Biophys 1998; 29: 113–132
- Aoki M, Fukunaga M, Sugimoto T, Hirano Y, Kobayashi M, Honda K, Yamada T. Studies on mechanisms of low emetogenicity of YM976, a novel phosphodiesterase type 4 inhibitor. J Pharmacol Exp Ther 2001; 298: 1142–1149
- Compton CH, Gubb J, Nieman RB, Edelson J, Amit O, Bakst A, Ayres JG, Creemers JPHH, Schultze-Werninghaus G, Brambilla C, Barnes NC. Cilomilast, a selective phosphodiesterase-4 inhibitor for treatment of patients with chronic obstructive pulmonary disease: A randomised, dose-ranging study. Lancet 2001; 358: 265–270
- Bateman ED, Izquierdio JL, Harnest U, Hofbauer P, Magvar P, Schmid-Wirlitsch C, Leichtl S, Bredenbroker D. Efficacy and safety of roflumilast in the treatment of asthma. Ann Allergy Asthma Immunol 2006; 96: 679–686
- Lipworth BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 2005; 365: 167–175
- Harbinson PL, MacLeod D, Hawksworth R, O'Toole S, Sullivan PJ, Kilfeather S, Page CP, Costello J, Holgate ST, Lee TH. The effect of a novel orally active selective PDE4 isoenzyme inhibitor (CDP840) on allergen-induced responses in asthmatic subjects. Eur Respir J 1997; 10: 1008–1014
- Alexander RP, Warrellow GJ, Eaton MAW, Boyd EC, Head JC, Porter JR, Brown JA, Reuberson JT, Hutchinson B, Turner P, Boyce B, Barnes D, Mason B, Cannell A, Taylor JR, Zomaya A, Millican A, Leonard J, Morphy R, Wales M, Allen RA, Gozzard N, Hughes B. CDP840. A prototype of a novel class of orally active anti-inflammatory phosphodiesterase 4 inhibitors. Bioorg Med Chem Lett 2002; 12: 1451–1456
- Aoki M, Kobayashi M, Ishikawa J, Saita Y, Terai Y, Takayama K, Miyata K, Yamada T. A novel phosphodiesterase type 4 inhibitor, YM976 (4-(3-chlorophenyl)-1,7-diethylpyrido[2,3-d]pyrimidin-2(1H)-one), with little emetogenic activity. J Pharmacol Exp Ther 2000; 295: 255–260
- Hersperger R, Bray-French K, Mazzoni L, Muller T. Palladium-catalyzed cross-coupling reactions for the synthesis of 6, 8-disubstituted 1,7-naphthyridines: a novel class of potent and selective phosphodiesterase type 4D inhibitors. J Med Chem 2000; 43: 675–682
- Robichaud A, Stamatiou PB, Jin SLC, Lachance N, MacDonald D, Lalibertè F, Liu S, Huang Z, Conti M, Chan CC. Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest 2002; 110: 1045–1052
- Dal Piaz V, Ciciani G, Turco G. Oxidative cleavage of 7-oxo-6,7-dihydroisoxazolo[3,4-d]pyridazines by cerium(IV) ammonium nitrate. A synthetic approach to new 5-acyl-4-nitro-3-oxo-2,3-dihydropyridazines. Synthesis 1989; 3: 213–214
- Dal Piaz V, Ciciani G, Giovannoni MP. Synthesis of 4,5-functionalized-2-methyl-6-(substituted aryl)-3 (2H)-pyridazinones: A new group of potent platelet aggregation inhibitors. Il Farmaco 1997; 52: 173–178
- Renzi G, Dal Piaz V, Musante C. Condensation between b-oxobenzoyl compounds and hydroximic chloride. Gazz Chim Ital 1968; 98: 656–666
- Renzi G, Pinzauti S. New derivatives of isoxazolo[3,4-d]pyridazin-7(6H)-one. Il Farmaco Ed Sci 1969; 24: 885–889
- Dal Piaz V, Ciciani G, Giovannoni MP, Franconi F. Synthesis and evaluation as platelet aggregation inhibitors of 6-phenyl-2,4-substituted-3(2H)-pyridazinones and their rigid analogues benzo[h]cinnolin-3,5-diones. Drug Des Disc 1996; 14: 53–75
- Adams J, Hauser CR. Condensations. XXIX. Acylation of ketones with aliphatic anhydrides by means of BF3. Synthesis of β-diketones. J Am Chem Soc 1945; 67: 284–286
- Gristwood RW, Beleta J, Bou J, Cardelus I, Fernandez AG, Lenas J, Berga P. Studies on the cardiac actions of flosequinam in vitro. Br J Pharmacol 1992; 105: 985–991
- Thompson WJ, Strada SJ. Method of Enzymatic Analysis3th ed., HU Bergmayer. Verlag-Chemie, Weinheim 1984; IV: 127–134
- Percival MD, Yeh B, Falgueyret JP. Zinc dependent activation of cAMP-specific phosphodiesterase (PDE4A). Biochem. Biophys Res Commun 1997; 241: 175–180
- Schneider HH, Schmiechen R, Brezinski M, Seidler J. Stereospecific binding of the antidepressant rolipram to brain protein structures. Eur J Pharmacol 1986; 127: 105–115
- Gantner F, Kupferschmitd R, Schudt C, Wendel A, Hatzelmann A. In vitro differentiation of human monocytes to macrophages: Change of PDE profile and its relationship to suppression of tumour necrosis factor-alpha release by PDE inhibitors. Br J Pharmacol 1997; 121: 221–231