Abstract
A similarity-based algorithm based on a previously developed model is applied in the classification of two sets of anticonvulsant and non-anticonvulsant drugs. Each set is composed of a) anticonvulsant compounds that have shown moderate to high activity in the Maximal Electroshock Seizure (MES) test and b) drugs with other biological activities or poor activity in the MES test. The results from the analysis of variance (ANOVA) indicate that the proposed algorithm is able to differentiate anticonvulsant from non-anticonvulsant drugs. The proposed model may then be useful in the identification of new anticonvulsant agents through virtual screening of large virtual libraries of chemical structures.
Introduction
Despite the continuous development of 3D and 4D QSAR methodologies, 2D-descriptors still remain among the most widely used descriptors in pharmaceutical research today Citation1-5. This is, in part, because 3D descriptors depend on the conformation used to represent the molecule, which not necessarily corresponds to the conformation responsible for activity, which may be sometimes difficult to define Citation6-8. Furthermore, although their physicochemical meaning is not always clear, 2D descriptors do not depend on the molecule conformation, have a low computational expense and can be easily calculated for all the existing, new, and in-development chemical structures [Citation6,Citation9].
There are, essentially, two major types of molecular descriptors [Citation10]. The first of them has been defined as “holistic”. Descriptors of this class are numbers that usually represent some important physical property of the whole molecule. These descriptors are either empirical or theoretical. Examples that could be mentioned among many others are: octanol-water partition coefficient, [Citation11] Kier Shape Index and [Citation12] the shape indices of Hopfinger [Citation13] [Citation14].
The other descriptor category consists of the so-called chemical substructures [Citation1,Citation10]. They could be interpreted as subgraphs of the graph that is associated to a molecular structure. These subgraphs are labeled or colored in a way that reflects information such as: atomic species, bond types and connectivity of two or more atoms from the structure. Some examples of descriptors in this category are atom pairs (AP), [Citation1,Citation14,Citation15] topological torsions (TT), [Citation1,Citation10] atom sequences (AS) and the Index of Electrotopological state [Citation15,Citation16]. Chemical substructures are particularly useful in the generation of similarity algorithms for retrieval systems and compound classification [Citation17,Citation18], which are commonly used in virtual screening of large chemical structure libraries. One of the main advantages of similarity algorithms in pharmaceutical discovery is that little information is needed to formulate a reasonable query; similarity methods can be applied at the beginning of a drug discovery project when there is little or no information available on the molecular target [Citation6]. We present a similarity-based algorithm generated on the basis of the one previously developed by Bagchi and Maiti [Citation19]. We have applied the proposed model in the classification of two sets of molecules composed by both anticonvulsant and non-anticonvulsant drugs.
Motivation
Epilepsy is one of the most common neurological disorders that affect human condition. It is characterized by recurrent seizure attacks that range from a brief lapse of attention to severe, frequent convulsions due to synchronous neuronal firing. About 50 million people in the world suffer from epilepsy, especially in childhood, adolescence and old age [Citation20,Citation21]. Available treatment fails to control epilepsy in up to 30% of the patients. In non-developed countries about three-fourths of patients do not receive the treatment they need [Citation21]. Moreover, even the new generation of antiepileptic drugs (AEDs) causes important side effects, including headache, drowsiness, diplopia, ataxia, dizziness, nausea, allergies, blood dyscrasias and hepatotoxicity [Citation22]. Thus safer, more efficient, less toxic new AEDs are urgently needed.
Atom pair definition
In 1985 Carhart et al. [Citation15] defined an atom pair as a substructure composed of two non-hydrogen atoms and an interatomic separation.
“Separation” is measured as the number of atoms in the shortest bond-by-bond path that contains atoms 1 and 2. Since the “topological distance” is defined as the number of edges, by the shortest bond-by-bond path, between two atoms [Citation9], Carhart separation corresponds to the topological distance plus one. “Description” of each atom informs its chemical type, the number of non-hydrogen atoms attached to it and the number of bonding π electrons it bears. An asterisk or dot following an atom symbol represents the presence of a bonding π electron. Xn following an atom symbol indicates the presence of n non-hydrogen neighboring atoms.
For any given chemical structure denoted by j, the total number of APs d(j) that can be derived from it is obtained through the following expression:
n being the number of non-hydrogen atoms present in the compound j.
Pharmacophore definition
Pharmacophore models are hypotheses on the 3D arrangement of structural properties, such as hydrogen bond donor and acceptor properties, hydrophobic groups and aromatic rings of compounds that bind to a biological target [Citation23,Citation24].
Our group has identified a pharmacophore related to anticonvulsant activity mediated by sodium channels blockade [Citation25,Citation26] through fitting in SYBYL 6.6 [Citation27]. A model of the identified pharmacophore can be seen in .
Figure 1 Identified pharmacophore for anticonvulsant drugs acting through sodium channel blockade mechanism.
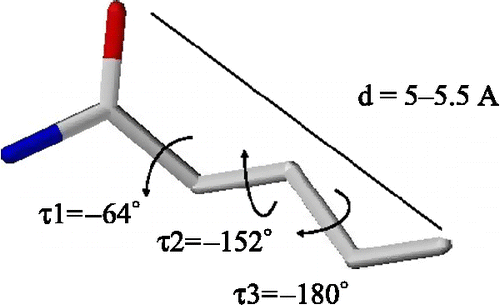
In the present work we have used information derived from this identified pharmacophore to generate a new mathematical model, specific for the search for potential new anticonvulsants, on the basis of the one from Bagchi and Maiti [Citation19].
On similarity determination
There are two major classes of similarity coefficients: association and distance coefficients. The essential difference between them is that the latter considers the common absence of certain structural features as the evidence of similarity between two chemical structures, whereas the former does not. Mentionable examples of association coefficients are Tanimoto, Dice and Cosine coefficients. Examples of distance coefficients are Hamming and Euclidean distances [Citation15,Citation18].
The work from Chen et al. [Citation15] indicates that the Tanimoto coefficient shows better performance than the Euclidean distance in 2D fragment-based similarity searching. Based on this information, we decided to use the Tanimoto coefficient as the similarity measure in the proposed model. We have tested the model with the Tanimoto coefficient in both its binary (Equation 2) and algebraic (Equation 3) forms:
where a is the number of types of fragments in compound A; b is the number of types of fragments in compound B and c is the number of types of fragments shared by compounds A and B.
where nai is the number of times that the ith AP appears in the AP set derived from compound A and nbi is the number of times that the ith AP appears in the AP set derived from compound B.
Materials and methods
Construction of the compounds sets
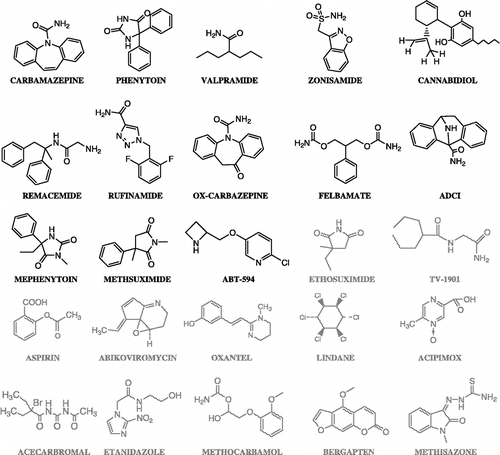
Two sets of compounds were constructed. Set A includes 12 compounds with high and moderate activity in the MES test (ED50 below 100 mg/kg, in mice) as well as 11 drugs with other therapeutic uses, extracted from Merck Index 13th [Citation28]: aspirin (analgesic), abikoviromycin (antibiotic), oxantel (antiparasitary), lindane (pediculicide), acipimox (antihyperlipoproteinemic), acecarbromal (hypnotic), etanidazole (antineoplasic), ABT-594 (analgesic), bergapten (antipsoriatic), methizasone (antiviral) and methocarbamol (muscle relaxant). Two anticonvulsants with poor activity in the MES test were also included (ethosuximide and TV-1901). The anticonvulsant agents included in Set A () are structurally diverse. Carbamazepine was chosen as the reference drug of this set, this is, the structure to which all the other molecules in the set will be compared through the proposed algorithm. Carbamazepine's measured activity in the MES test is the highest among set A (ED50 = 37 μmol/kg ip) [Citation25].
Figure 2 Structures of the compounds that compose set A. Anticonvulsant compounds are shown in black, while compounds with other therapeutic uses are presented in grey. Carbamazepine was chosen as the reference compound in this set.
Set B () is defined by 26 compounds. Besides the non-anticonvulsant drugs already used in set A, it includes 13 anticonvulsants: one benzoquinoxaline derivative (NBQX), nine benzodiazepine and tetrahydroisoquinoline derivatives selected from a set of drugs tested in the MES test [Citation29,Citation30], as well as carbamazepine and valpramide. THIQ – 10c, the compound with the highest measured activity in the MES test among set B (ED50 = 5.17 μmol/kg) was chosen as reference drug. Note that the anticonvulsants in set B are structurally less heterogeneous than those in set A.
Figure 3 Schematic representation of compounds that define set B. Anticonvulsant compounds are shown in black, while compounds with other therapeutic uses are presented in grey. It can be appreciated that many of the anticonvulsants of this set are structurally closely-related. THIQ-10c was chosen as the reference compound in Set B.
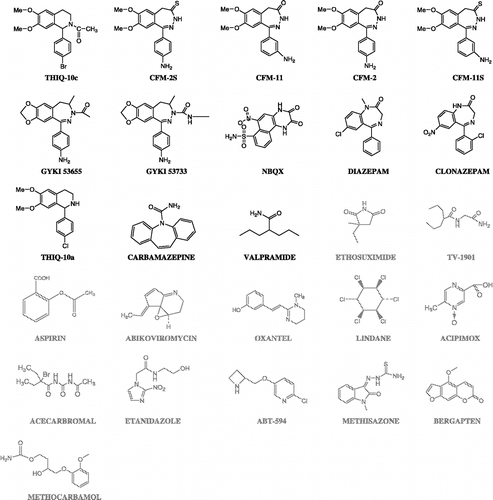
The structures of each set with high and moderate anticonvulsant activity in the MES test will be referred to as active compounds from now on. This category is presented in black in and . On the other hand, the compounds with other therapeutic uses or poor anticonvulsant activity in the MES test are presented in grey and will be referred to as inactive compounds.
Construction of the new model
We propose the following model:
A is a measure of the expected relative activity to a reference drug; this is chosen as the most active structure of the working set (carbamazepine for set A and THIQ-10c for set B). S is the binary form of the Tanimoto similarity coefficient. ni represents the number of ith AP in the reference drug; Δni stands for the difference of ith AP between the reference drug and the tested drug; λi is the separation in the ith AP and α is the separation in the longest AP that can be derived from the identified pharmacophore (or, in other words, α represents the maximal topological distance in the pharmacophore plus one). For the term between brackets we consider only those types of AP that contain heteroatoms, based on the common knowledge that bioactive compounds tend to present heteroatoms and heterocycles and that functional groups involved in non-covalent short-distance interactions are essential for the binding of the drug to its site of action. b and c are constants which moderate the influence over the predicted relative activity of the similarity coefficient and the second term between brackets.
The value of each one of the terms in the sum between brackets can be interpreted as the contribution of the ith type of AP to the activity of the structure. Whether these terms are positive or negative (adding to or reducing the relative activity of the molecule that is being compared) is defined by the Δni factor, which is the only one among the factors involved in the model that can take values either greater or smaller than zero. Since the reference drug of each set is chosen as the most active structure in it, the types of AP that are present in the reference drugs can be thought of as desired features involved in the interaction of the molecule and its site of action. This was one of the fundamental hypotheses in the development of our model. Therefore, if the number of APs of the ith type is greater in the reference drug than in the tested compound, then the tested molecule is lacking a desired feature and the fraction of the activity that can be explained, through that particular element will be smaller in the tested compound than in the reference one. If the number of ith type elements is greater in the tested compound than in the reference drug, then the tested compound has more of a desired characteristic and a greater probability of this feature being expressed at the time of interacting with the site of action.
However, are all the types of atom pairs equally significant to the activity of a particular structure? Since the reference is the drug with the highest activity among the available structures, we believe that the more times a particular type of AP appears in the reference structure, the more important is the contribution of that type of AP to the activity. Thus, our model includes the factor ni in the numerator of the second term between brackets. We also believe that, having indentified a specific pharmacophore, we should give more attention to the types of AP that somehow accomplish the characteristics of the pharmacophoric pattern. The topological distances are one of the features of the pharmacophore that can be considered. We decided to give prominence to those types of AP that include heteroatoms and a separation of six, since six is the maximum number of atoms comprised in the possible AP derived from the identified pharmacophore for anticonvulsant drugs with activity in the MES test.
Although the pharmacophore itself could be used as a 3D-search query in virtual screening for new anticonvulsant agents through compound databases, this requires the previous generation of reasonable, low-energy conformers of the molecules in the database to be screened. Our model presents the advantage that, since only topological, 2D-features of the pharmacophore are considered, the results of the query do not depend on the conformation of the molecules from the database being screened.
Application of the models
The AP set derived of each of the compounds considered for this study was generated. shows the complete sets of AP for carbamazepine and THIQ-10c, the reference compounds of set A and set B, in that order. Once we had knowledge of all the AP sets the similarity coefficients of Tanimoto were calculated as specified in Equations (2) and (3); the values of these coefficients for each structure are shown in. In order to determine the common heteroatom APs, the APs of each structure was compared with the APs of the corresponding reference drug. Different values for b and c were tested for each set of compounds. b was given values of 0.5, 0.75 and 1. c was given values of 50 and 75. As an example, shows the types of heteroatom AP common to carbamazepine and phenytoin, and the occurrence of each type in each one of these structures. The calculated value for the negative term between brackets in the proposed model is also shown; c was chosen as equal to 75 for the example presented in . Once we calculated the value of A for the different forms of model (4), analysis of variance (ANOVA) was performed in order to determine if the proposed model could be used to classify anticonvulsant and non-anticonvulsant agents. In other words, it was checked if there was a statistically significant difference between the mean values of A for the active and the inactive categories of both set A and B. It was also verified if that difference was either similar or higher, in terms of statistical significance, than the difference between the mean values of the Tanimoto coefficients for both groups (active and inactive) and both sets.
Table I. AP sets derived from carbamazepine and THIQ-10c structures.
Table II. Heteroatom APs common to carbamazepine and phenytoin, and the value of the negative term between brackets in expression (3) for each type of AP.
Table III. Value of the Tanimoto coefficient (in its binary and algebraic form) for every compound tested. Compounds which compose set A were compared to carbamazepine. Compounds which composed set B were compared to THIQ-10c. Inactive compounds are presented in grey.
Scatter plots of log ED50 in the MES test versus A (obtained through Equation (5) using Tanimoto coefficient in its algebraic form) were drawn in order to determine if there was correlation between the activity predicted with our model and the experimental activity measured in the MES test, for both sets A and B. For comparison purposes, the scatter plots of log ED50 versus the Tanimoto coefficient in its algebraic form were also drawn.
The model was finally validated with a set of ten compounds with bioactivities different from anticonvulsant activity ().
Results and discussion
shows the values of A for one of the forms of the proposed model that showed good performance in both sets A and B, both with the binary and algebraic forms of the Tanimoto coefficient:
Table IV. Value of A for the compounds of Set A and B, obtained with the form of the proposed model specified in expression (5).
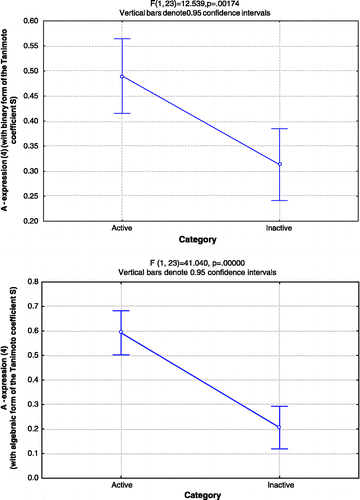
shows the results from the ANOVA test for the different forms of the proposed model that were evaluated and for the bare Tanimoto coefficient (in its binary and algebraic form). We present the value of the F Statistical (which represents the ratio between the variance between-groups and the variance within-group) and the p-value (which indicates the level of significance of the difference between the mean values of the considered categories). It can be observed that the performance of various forms of the proposed model in the classification of compounds from set A is similar to that of the Tanimoto coefficients, while in the case of set B the difference between the mean value of A is more significant than the difference between the mean values of the Tanimoto coefficients for each considered category. Taking into account that most of the anticonvulsants which compose set B are structurally related to THIQ 10c, results seem to indicate that the proposed model is effective in the identification of anticonvulsant compounds whose structure is close-related to the structure of the compound chosen as a reference (). also shows that the difference between the categories is statistically much more significant when using in the model the Tanimoto coefficient in its algebraic form. This reflects the fact that the differences between the mean values of the Tanimoto coefficient for the active and inactive categories is also much greater for Set B for the algebraic than for the binary form.
Table V. Values of the statisticals F and p for the different forms of the proposed model that were tested. Each form of the model corresponds to different values of b and c in (3). Values of F and p are also presented for the Tanimoto coefficient. The first set of data corresponds to the models which incorporate the binary form of the Tanimoto coefficient; the second set of data, to the models which incorporate the algebraic form of the Tanimoto coefficient. Note that difference between the mean values of the algorithm for both considered categories is statistically more significant when the algebraic form of Tanimoto coefficient is used.
Figure 4 Results from ANOVA test on the values of A calculated from expression (5) for set B, for both considered categories. The upper graph corresponds to the model that incorporates the binary form of the Tanimoto coefficient, while the lower corresponds to the model that incorporates the algebraic form.
shows that there are strong correlations between the measured log ED50 in the MES test and the values of A. The correlation coefficients r for both sets A and B (0.8149 and 0.9055, in that order) are higher than those obtained when plotting log ED50 versus the algebraic form of the Tanimoto coefficient (0.7962 and 0.8568).
Figure 5 On the left, scatter plots of log10 ED50 versus A (calculated through expression 5) for the anticonvulsant compounds of both sets A and B. On the right, log10 ED50 versus algebraic form of the Tanimoto coefficient.
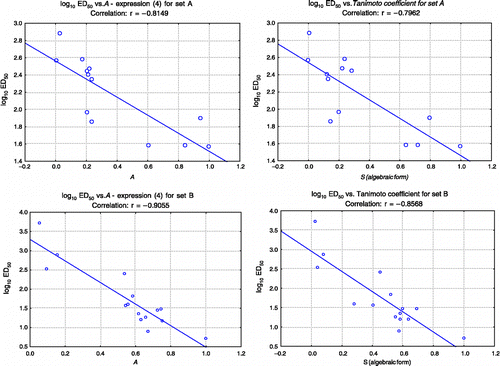
Figure 6 Structures of the 10 non-anticonvulsant drugs used for validation purposes.
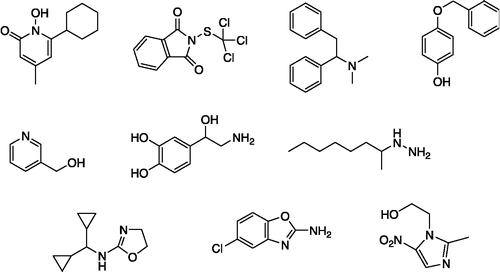
shows the results from the validation of model (5) (using the algebraic form of the Tanimoto coefficient and taking THIQ10c as the reference compound). Nine of the ten inactive compounds have low A values that fall inside or below the 0.95 confidence interval for the inactive category (see ). The A value of the remaining compound (lefetamine), although higher, falls below the 0.95 confidence interval for the active category.
Table VI. Results from the validation of the model with 10 non-anticonvulsant compounds. The second column shows the use of each drug according to Merck Index 13th Edition. For determination of A with expression (5) Tanimoto coefficient is used in its algebraic form and THIQ10c is used as reference drug.
Conclusions
A new similarity-based algorithm for the classification of anticonvulsant and non-anticonvulsant drugs is introduced and applied in the classification of two sets of compounds, both composed of anticonvulsant and non-anticonvulsant agents in the MES test. The ANOVA test showed statistically significant differences between the mean values of the algorithm for each considered category. Results indicate that the proposed algorithm performs better in the identification of anticonvulsant compounds structurally close-related to the reference compound chosen for the query. The algorithm could be applied in the search for new anticonvulsant agents through virtual screening.
Acknowledgements
Eduardo A. Castro is a member of the Consejo Nacional de Investigaciones Científicas y Técnicas de la República Argentina (CONICET). Luis E. Bruno-Blanch is researcher of the Facultad de Ciencias Exactas, Universidad Nacional de La Plata. Alan Talevi thanks CONICET for his postgraduate fellowship. This work was supported in part through grants from Agencia de Promoción Científica y Tecnológica (PICT BID 1728/06–11985) and Universidad Nacional de La Plata, Argentina.
References
- Merlot C, Domine D, Cleva C, Church DJ. Chemical substructures in drug discovery. Drug Discov Today 2003; 8: 594–602
- Merlot C, Domine D, Church DJ. Fragment analysis in small molecule discovery. Curr Opin Drug Discov Devel 2002; 5: 391–399
- Bajaj S, Sambia SS, Madanb AK. Topological models for prediction of Anti-HIV activity of acylthiocarbamates. Bioorg Med Chem 2005; 13: 3263–3268
- Duart MJ, Antón-Fos GM, de Julian-Ortiz JV, Gozalbes R, Gálvez J, García-Domenech R. Use of molecular topology for the prediction of physicochemical, pharmacokinetic and toxicological properties of a group of antihistaminic drugs. Int J Pharm 2002; 246: 111–119
- García-García A, Gálvez J, de Julián-Ortiz JV, García-Domenech R, Muñoz C, Guna1 R, Borrás R. New agents active against mycobacterium avium complex selected by molecular topology: A virtual screening method. J Antimicrob Chemother 2004; 53: 65–73
- Sheridan RP, Kearsley SK. Why do we need so many chemical similarity search methods?. Drug Discov Today 2002; 7: 903–911
- Matter H, Pötter T. Comparing 3D pharmacophore triplets and 2D fingerprints for selecting diverse compound subsets. J Chem Inf Comput Sci 1999; 39: 1211–1225
- Schuffenhauer A, Gillet VJ, Willett P. Similarity searching in files of three-dimensional chemical structures: analysis of the bioster database using two-dimensional fingerprints and molecular field descriptors. J Chem Inf Comput Sci 2000; 40: 295–307
- Devillers J, Balaban AT. Topological indices and related descriptors in QSAR and QSPR. Gordon and Breach Science Publishers. 1999; 1–17
- Nilakantan R, Bauman N, Dixon S, Venkataraghavan R. Topological torsión: A new molecular descriptor for SAR applications. comparison with other descriptors. J Chem Inf Comput Sci 1987; 27: 82–85
- Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chem Rev 1971; 71: 525–616
- Kier L. A shape index from molecular graphs. Quant Struct-Act Relat Pharmacol Chem Biol 1985; 4: 109–116
- Walters DE, Hopfinger AJ. Case studies of the application of molecular shape analyis to elucidate drug action. J Mol Struct Theochem 1986; 134: 317–323
- Carhart RE, Smith DH, Venkataraghavan R. Atom pairs as molecular features in structure-activity studies: definitions and applications. J Chem Inf Comput Sci 1985; 25: 64–73
- Chen X, Reynolds CH. Performance of similarity measures in 2D fragment – based similarity searching: comparison of structural descriptors and similarity coefficients. J Chem Inf Comput Sci 2002; 42: 1407–1414
- Kier LB, Hall LH. The electrotopological state. Molecular structure description. Academic Press, New York 1999; 1
- Brown RD, Martin YC. Use of structure-activity data to compare structure-based clustering methods and descriptors for use in compound selection. J Chem Inf Comput Sci 1996; 36: 572–584
- Wilett P. Chemical similarity searching. J Chem Inf Comput Sci 1998; 38: 983–996
- Bagchi MC, Maiti BC. On application of atom pairs in drug design. J Mol Struct Theochem 2003; 623: 31–37
- http://www.who.int/. World Health Organization Fact sheet n° 265. Mental and neurological disorders 2001.
- http:// www.who.int/. World Health Organization Fact sheet n° 165. Epilepsy: aetiogy, epidemiology and prognosis. Revised on February 2001.
- Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, Loiseau P, Perucca E. Progress report on new antiepileptic drugs: A summary of the sixth Eilat conference (EILAT VI). Epilepsy Res 2002; 51: 31–71
- Langer T, Wolber G. Pharmacophore definition and 3D searches. Drug Discov Today Technol 2004; 1: 203–207
- Bywater RP, Poulsen TA, Røgen P, Hjorth PG. De novo generation of molecular structures using optimization to select graphs on a given lattice. J Chem Inf Comput Sci 2004; 44: 856–861
- Tasso SM, Moon SCh, Bruno-Blanch LE, Estiú GL. Characterization of the anticonvulsant profile of valpromide derivatives. Bioorg Med Chem 2004; 12: 3857–3869
- Gavernet L, Dominguez-Cabrera MJ, Bruno-Blanch LE, Estiú G. 3D- QSAR Design of novel antiepileptic sulfamides. Bioorg Med Chem 2007; 15: 1556–1567
- SYBYL is a product by Tripos Inc., San Diego CA.
- The Merck Index. Encyclopedia of Chemical, Drugs and Biologicals13th edition. Merck and co, Withehouse Station, NJ 2001
- De Sarro G, De Sarro A, Gareri P, Gitto R, Russo E, Chimirri A, Ferreri G. Comparative anticonvulsant activity of N-acetyl-1-aryl-6,7-dimethoxy-1,2,3,4- tetrahydroisoquinoline derivatives in rodents. Pharmacol Biochem Behav 2004; 77: 85–94
- De Sarro G, Ferreri G, Gareri P, Russo E, De Sarro A, Gitto R, Chimirri A. Comparative anticonvulsant activity of some 2,3-benzodiazepine derivatives in rodents. Pharmacol Biochem Behav 2003; 74: 595–602