Abstract
Based on the prodrug concept as well as the combination of two different classes of anti-HIV agents, we have designed and synthesized a series of anti-HIV double-drugs consisting of a nucleoside reverse transcriptase inhibitor (NRTI) conjugated with an integrase inhibitor (INI) through a spontaneously cleavable linker in an effort to enhance the antiviral activity. These conjugates combined in their structure a dideoxy-didehydro-nucleoside (ddN) such as d4T and an INI such as α,γ-diketo acid (DKA) analogues of L-708,906 and L-731,988 linked through an appropriate self-immolative spacer. Among these novel bis-substrate inhibitors, several conjugates exhibited antiviral activity but this effect was accompanied for some of them by an increased cytotoxicity by comparison to d4T, DKA or even some precursors. These compounds are nevertheless interesting candidates for further investigations.
Introduction
Human immunodeficiency virus type 1 (HIV-1) is the etiological agent of acquired immunodeficiency syndrome (AIDS) [Citation1]. The pol gene of HIV encodes for three enzymes, reverse transcriptase (RT), protease (PR) and integrase (IN). Drug discovery directed at inhibitors of RT and PR has produced clinically useful compounds for the treatment of AIDS Citation2-3. Currently, the standard treatment strategy for AIDS is referred to the highly active antiretroviral therapy (HAART) which is a combination of three or more anti-HIV drugs inhibiting two viral enzymes (reverse transcriptase and protease) and virus fusion (interaction with glycoprotein 41) Citation4-6. However, HAART strategy still suffers from issues of patient compliance, cost, deliterious side effects, and the ever-increasing emerging drug resistance Citation7-9.
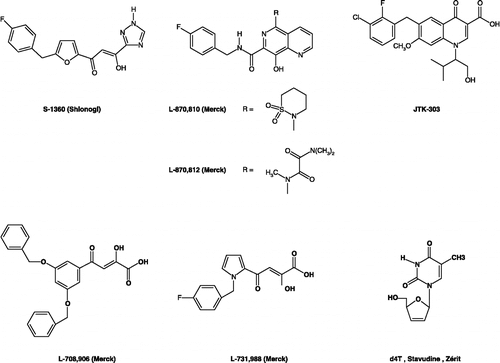
Therefore, HIV-1 integrase, one of the three constitutive viral enzymes required for replication, has recently emerged as an attractive target for chemotherapeutic intervention in the treatment of AIDS since no human counterpart of the enzyme is known Citation10-14. The viral integrase mediates the integration of the proviral DNA into the genome of the host cell. Integration is a multistep process which includes three different biochemical processes: assembly of proviral DNA on integrase, endonucleolytic processing of the proviral DNA, and strand transfer of the proviral DNA into host chromosomal DNA. Several classes of HIV IN inhibitors (INIs) have been developed, including dinucleotides [Citation15], hydroxylated aromatics Citation16-20, and α,γ-diketo acids compounds (DKAs) Citation21-27. While a number of structurally diverse compounds have been reported to be inhibitors of HIV integrase, only four drugs have reached the clinical trials Citation28-29: S-1360 designed by Shionogi-GSK companies, L-870,810 and L-870,812 and 8-hydroxynaphthyridines designed by Merck Research laboratories, and JTK-303 designed by Japan Tobacco and developed by Gilead (). In fact, the interest in studying DKAs started with reports of the selective inhibitory properties of L-708,906 and L-731,988 toward strand transfer and the crystal structure of the catalytic domain of HIV-1 IN with 5-CITEP [Citation21,Citation22]. Also, the α,γ-diketo acids represent the most convincing, biologically validated inhibitors of this viral enzyme [Citation21,Citation25]. The DKAs compete with host DNA for the binding sites on HIV integrase, thus selectively inhibiting the strand transfer process [Citation22]. This inhibition is dependent on the presence of divalent magnesium ion, or manganese ion in vitro assays [Citation25]. The L-731,988 compound showed good potency against integrase, particularly in the strand transfer step with an IC50 of 0.08 μM compared with 6 μM in the 3′-processing step [Citation22].
Figure 1 Chemical structures of representative α,γ-Diketo-Based integrase inhibitors and the nucleoside reverse transcriptase inhibitor (the 2′,3′,didehydro-2′,3′-dideoxythymidine).
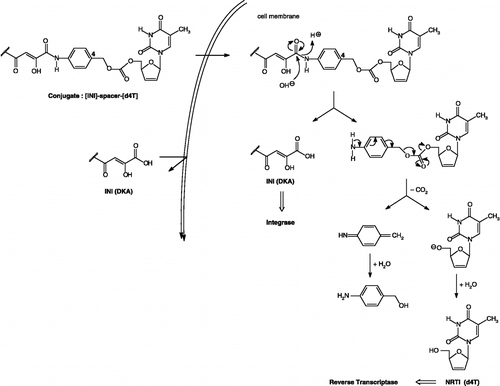
However, the antiviral efficacy of inhibitors depends not only on the enzyme inhibitory activity, but also on their intracellular concentrations closely related to the membrane permeability. Also, the presence of a free carboxyl group on DKAs could lead to insufficient cell membrane permeability. To improve antiviral activity, we have suggested «double-drug» strategy that combined an effective anti-HIV agent such as a nucleoside RT inhibitor (NRTI) used for masking the free carboxylic acid of the HIV IN inhibitor in a single molecule. This strategy has been advocated for three main reasons: (1) the low membrane permeability of IN inhibitors would be improved while the cell membrane has affinity for nucleosides Citation30-31; (2) the conjugation of HIV IN inhibitor with the nucleoside RT inhibitor would facilitate the penetration through the biological membrane mediated by the nucleoside transporter Citation32-33; (3) once the prodrugs avoided the extracellular hydrolysis, the intracellular hydrolysis would regenerate the parent inhibitors, which could act on two separate targets and exhibit synergistic anti-HIV efficacy. Also, there are several attractive features to such an approach since the “double-drug” could act on two separate targets and exhibit synergistic anti-HIV efficacy (). One essential criteria in the design of such prodrugs is the linker that must be stable outside the target cell and regenerate the parent compounds in the cytoplasm to act on their respective targets.
Figure 2 Design and proposed mechanism of conversion of double-drugs to d4T and IN inhibitor.
With this aim based on the “double-drug” strategy, we reported in 2004 the synthesis and the anti-HIV activity assessment of a series of heterodimers which combine in their structure an NRTI analogue [2′,3′-didehydro-2′,3′-dideoxyuridine (d4U) or 2′,3′-dideoxyuridine (d2U) or 2′,3′-didehydro-2′,3′-dideoxythymidine (d4T)] and an INI (belonging to the β-diketo acids class) connected through an amino acid (glycine or β-alanine) as a cleavable linker [Citation34]. The most active compound in this series was the [4-[1-(2-fluorobenzyl)-1H-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic]-GLYCYL-C5′[d4T] heterodimer (IC50 3 μM, HIV-1LAI / CEM-SS cells and IC50 0.73 μM, HIV-1IIIB / MT4 cells). However, they were less potent inhibitors than the parent compounds from which they derived in lymphocytic cell lines (CEM-SS and MT-4).
Taking into account our previous studies focused on prodrugs containing conventional spacers, we decided to prepare new prodrug-type conjugates including a linker which has the ability to cleave spontaneously in physiological environment between the NRTI (the 2′,3′-didehydro-2′,3′-dideoxythymidine, d4T, Stavudine, Zerit® Citation35-39) and the INI, taking into consideration the affinity of nucleosides towards cell membrane [Citation30]. The most prominent example of an electronic cascade spacer is the 1,6-elimination spacer developed by Carl et al.[Citation40]. The p-aminobenzyl alcohol (PAB) moiety was designed in the literature to hydrolytically decompose upon enzymatic deacylation releasing the free drug. After unmasking the aromatic amine, the amine group becomes electron-donating and initiates virtually instantaneously an electronic cascade that leads to the expulsion of the leaving group, which release the free drug after elimination of carbon dioxide () This type of spacer has been largely studied in preceding investigations in cancer chemotherapy for enhanced drug release by Scheeren et al. Citation41-43, Monneret et al. Citation44-46, Senter et al. [Citation47], Dubowchick et al. [Citation48] and Deny et al. [Citation49].
Two types of prodrugs were synthesized bearing either an ortho- or para-hydroxybenzylcarbonate linkage, both spacers expected to spontaneously decompose after enzymatic cleavage. The linking group was conjugated to the DKA through amide bond formation and to the 5′-hydroxyl group of d4T by a carbonate group. This arrangement should cleave by amide hydrolysis followed by degradation of the remaining para- or ortho-aminobenzylcarbonate as proposed mechanism described in .
Materials and methods
Chemistry
Instrumentation
Commercial reagents (acetone /VWR and toluene/Aldrich) were used as received without additional purification. Tetrahydrofuran (THF) was distilled from sodium/benzophenone immediately prior to use. Reagent grade dichloromethane (CH2Cl2) was refluxed and distilled from phosphorus pentoxide. Unless otherwise stated, reactions were run under argon and monitored by thin-layer chromatography (TLC) using precoated silica gel 60 F254 sheets (0.2 mm layer) purchased from Macherey-Nagel, and compounds were detected by UV absorption at 254 nm. Column chromatography was achieved by using Merck silica gel 60 (0.063–0.200 mm). All samples were kept in a drying oven at 30°C over P4O10 for at least 24 h prior to analysis.
Melting points were determined using a Kofler melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin Elmer Spectrum BX FT-IR and only noteworthy absorptions are listed. 1H and 13C-NMR spectra were recorded on a JEOL NMR LA 400 (400 MHz) spectrometer using TMS as an internal standard. Chemical shifts were reported as δ values in parts per million units, downfield from TMS. The splitting pattern abbreviations are as follows: s = singlet, d = doublet, br = broad, m = multiplet. Coupling constants J are given in hertz (Hz). NH and OH signals appeared as broad singlets exchangeable with D2O. Elemental analysis of new compounds was performed at the Institute de Recherche en Chimie Organique Fine (Rouen, France).
1-(3,5-Dibenzyloxyphenyl)ethanone (2a) [Citation34]
1-(3-Benzyloxyphenyl)ethanone (2b)
To a stirred and cooled (0°C) solution of 3-hydroxyacetophenone (10 g, 73.45 mmol) 1b in anhydrous acetone (140 mL) was added, respectively: anhydrous sodium carbonate (15.22 g, 137 mmol) and benzyl bromide (10.48 mL, 88 mmol). The reaction mixture was stirred at room temperature for 24 h before the solvent was removed in vacuo. The residue was dissolved in ethyl acetate (150 mL) and the resulting solution was washed with water (150 mL), dried over magnesium sulfate, filtered and concentrated under reduced pressure. The aryl methyl ketone 2b was isolated as a brown oil (15.9 g) that was taken on to the next step without further purification; Yield: 96%; TLC Rf (n-hexane: acetone = 70: 30) 0.50; IR υmax (KBr)/cm− 1 3065, 3033, 1683 (C = O), 1581, 1437, 1270, 1026, 739; 1H-NMR δ (d6-DMSO) 7.54-7.20 (m, 9H, H-Bn and H-Ph), 5.08 (s, 2H, CH2), 2.57 (s, 3H, CH3).
1-(4-Benzyloxyphenyl)ethanone (2c)
Compound 2c was prepared from 4-hydroxyacetophenone 1c (10 g, 73.45 mmol) by the same procedure as that described for compound 2b. Yield: 80%, white solid (13.3 g); TLC Rf (n-hexane: acetone = 60: 40) 0.71; mp = 92°C; IR υmax (KBr)/cm− 1 2940, 1676 (C = O), 1598, 1248, 1009, 759; 1H-NMR δ (d6-DMSO) 7.94 (d, 2H, J 8.7 Hz, H-Ph), 7.44-7.32 (m, 5H, H-Bn), 7.01 (d, 2H, J 8.7 Hz, H-Ph), 5.13 (s, 2H, CH2), 2,55 (s, 3H, CH3).
Ethyl 4-(3,5-dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoate (3a) [Citation34].
Ethyl 4-(3-benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoate (3b)
To a stirred and cooled (0°C) solution of aryl methyl ketone 2b (11.3 g, 50 mmol) in anhydrous toluene (80 mL) under an argon atmosphere was added respectively: sodium hydride (60% dispersion in mineral oil, 2.5 g, 62 mmol), diethyloxalate (7 mL, 51 mmol) and a drop of ethanol. The reaction mixture was heated under reflux for 2 h, then cooled at room temperature and concentrated to dryness in vacuo. The residue was dissolved in ethyl acetate (500 mL) and the resulting solution was washed respectively with 1N HCl/H2O (200 mL), water (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude residue was crystallized from diethyl ether to give the ketoenol ester 3b (5.0 g) as a yellow powder; Yield: 31%; mp = 82°C; TLC Rf (n-hexane: acetone = 60: 40) 0.70; IR υmax (KBr)/cm− 1 1749 (C = O), 1577, 1271, 1204, 1072, 1025, 771, 734, 696, 677; 1H-NMR δ (d6-DMSO) 7.66-7.33 (m, 9H, H-Bn and H-Ph), 7.10 (s, 1H, CH = C(OH)), 5.19 (s, 2H, CH2-Bn), 4.30 (q, 2H, J 7.1 Hz, CH2-CH3), 1.30 (t, 3H, J 7.1 Hz, CH2-CH3).
Ethyl 4-(4-benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoate (3c)
Compound 3c was prepared from precursor 2c (5.9 g, 26.1 mmol) by the same procedure as that described for compound 3b. Yield: 90%, yellow solid (7.7 g); TLC Rf (dichloromethane: methanol = 97: 3) 0.86; mp = 188°C; IR υmax (KBr)/cm− 1 1743 (C = O), 1593, 1250, 1174, 1021, 988, 852, 778, 699; 1H-NMR δ (d6-DMSO) 8.08 (d, 2H, J 9.0 Hz, H-Ph), 7.47-7.34 (m, 5H, H-Bn), 7.18 (d, 2H, J 9.0 Hz, H-Ph), 7.09 (s, 1H, CH = C(OH)), 5.23 (s, 2H, CH2-Bn), 4.30 (q, 2H, J 7.1 Hz, CH2-CH3), 1.30 (t, 3H, J 7.1 Hz, CH2-CH3).
4-(3,5-Dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic acid (4a) [Citation34]
4-(3-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic acid (4b)
To a solution of ketoenol ester 3b (5 g, 15.3 mmol) in 1:1 tetrahydrofuran/methanol (100 mL) was added a solution of 1N NaOH/H2O (25 mL) and the mixture was stirred at room temperature for 2 h. Then, the reaction mixture was washed with diethyl ether (100 mL), acidified to pH = 2 with 1N HCl/H2O and extracted several times with ethyl acetate (3 × 100 mL). The combined organic phases were washed successively with 1N HCl/H2O (100 mL) and water (100 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude residue was crystallized from diethyl ether to give the ketoenol acid 4b (3.5 g) as a yellow crystals; Yield: 77%, mp = 180°C; TLC Rf (dichloromethane: methanol = 97: 3) 0.56; IR υmax (KBr)/cm− 1 3039-2635 (OH), 1708 (C = O), 1637, 1582, 1439, 1260, 1018, 772; 1H-NMR δ (CDCl3) 7.66-7.32 (m, 9H, H-Bn and H-Ph), 7.09 (s, 1H, CH = C(OH)), 5.19 (s, 2H, CH2); 13C-NMR δ (CDCl3) 190.7 (C-4), 170.5 (C-1), 163.6 (C-2), 159.1 (C-3:phenyl), 137.1 (C-1:phenyl), 136.5 (C-1:Bn), 130.8 (C-5:phenyl), 128.9 (C-3 and C-5:Bn), 128.4 (C-4:Bn), 128.2 (C-2 and C-6:Bn), 121.4 (C-6:phenyl), 121.0 (C-4:phenyl), 113.6 (C-2:phenyl), 98.5 (CH = C(OH)), 69.9 (CH2).
4-(4-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic acid (4c)
Compound 4c was prepared from precursor 3c (3.84 g, 11.8 mmol) by the same procedure as that described for compound 4b. Yield: 94%, yellow powder (3.3 g); TLC Rf (n-hexane: ethyl acetate = 60: 40) 0.40; mp = 202°C; IR υmax (KBr)/cm− 1 3034-2635 (OH), 1712 (C = O), 1595, 1404, 1243, 1174, 1003, 833; 1H-NMR δ (d6-DMSO) 8.06 (d, 2H, J 8.5 Hz, H-Ph), 7.47-7.34 (m, 5H, H-Bn), 7.16 (d, 2H, J 8.5 Hz, H-Ph), 7.05 (s, 1H, CH = C(OH)), 5.22 (s, 2H, CH2); 13C-NMR δ (d6-DMSO) 190.0 (C-4), 169.9 (C-1), 163.3, 163.1, 136.3, 130.4 (C-2 and C-6:phenyl), 128.5, 128.1, 127.8, 127.4, 115.2 (C-3 and C-5:phenyl), 97.6 (CH = C(OH)), 69.6 (CH2).
1-[1-(4-Fluorobenzyl)-1H-pyrrol-2-yl]ethanone (6a) [Citation34]
1-[1-(2-Fluorobenzyl)-1H-pyrrol-2-yl]ethanone (6b)
To a stirred and cooled (0°C) solution of 2-acetylpyrrole (13.08 g, 120 mmol) in dimethylformamide (120 mL) under an argon atmosphere wad added respectively: sodium hydride (60% dispersion in oil, 5.75 g, 144 mmol), 2-flurobenzyl bromide (17.24 mL, 144 mmol). The reaction mixture was stirred at room temperature overnight, poured into a saturated solution of NaHCO3/H2O (200 mL) and extracted with ethyl acetate (3 × 150 mL). The combined organic phases were washed with water (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The title compound 6b, obtained as a clear yellow oil (26.34 g), was taken on to the next step without purification; Yield: 84%; TLC Rf (hexane: ethyl acetate = 80: 20) 0.55; IR υmax (KBr)/cm− 1 1652 (C = O), 1491, 1407, 1328, 1229, 1086, 945, 754; 1H-NMR δ (d6-DMSO) 7.30-7.24 (m, 2H, H-5:pyrrole and H-4:Bn), 7.20-7.16 (m, 2H, H-3:pyrrole and H-5:Bn), 7.06 (t, 1H, J 7.5 Hz, H-3:Bn), 6.61 (t, 1H, J 7.5 Hz, H-6:Bn), 6.22 (t, 1H, J 3.3 Hz, H-4:pyrrole), 5.60 (s, 2H, CH2-Bn), 2.32 (s, 3H, CH3); 13C-NMR δ (d6-DMSO) 187.6 (C = O), 159.4 (d, 1JC-F 244.5 Hz, C-2:Bn), 131.5 (C-2:pyrrole), 129.7 (C-5:pyrrole), 129.0 (d, 3JC-F 8.2 Hz, C-6:Bn), 127.8 (d, 3JC-F 4.1 Hz, C-4:Bn), 126.1 (d, 2JC-F 14.9, C-1:Bn), 124.5 (d, 4JC-F 3.3 Hz, C-5:Bn), 120.7 (C-3:pyrrole), 115.0 (d, 2JC-F 21.5 Hz, C-3:Bn), 108.5 (C-4:pyrrole), 45.7 (d, 3JC-F 4.9 Hz, CH2-Bn), 27.0 (CH3).
Ethyl 4-[1-(4-fluorobenzyl)-1H-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoate (7a) [Citation34]
Ethyl 4-[1-(2-fluorobenzyl)-1H-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoate (7b)
To a stirred and cooled (0°C) solution of compound 6b (13.02 g, 60 mmol) in anhydrous dimethoxyethane (120 mL) under an argon atmosphere was added respectively: sodium hydride (60% dispersion in oil, 2.88 g, 72 mmol), diethyl oxalate (9.78 mL, 72 mmol) and a drop of ethanol. The reaction mixture was heated at reflux for 18 h, stirred overnight at room temperature, poured into a saturated into a solution of 1N NaHCO3/H2O (200 mL) and extracted with ethyl acetate (3 × 150 mL). The combined organic phases were washed with water (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was crystallized with diethyl ether to give the title compound 7b as yellow crystals (5.20 g); Yield: 28%; TLC Rf (hexane: ethyl acetate = 80: 20) 0.39; mp = 98°C; IR υmax (KBr)/cm− 1 1734 (C = O), 1613, 1381, 1330, 1264, 1223, 1086, 1065, 754; 1H-NMR δ (d6-DMSO) 7.48 (m, 2H, H-5:pyrrole and H-4:Bn), 7.30-7.18 (m, 2H, H-3:pyrrole and H-5:Bn), 7.08 (t, 1H, J 7.6 Hz, H-3:Bn), 6.87 (s, 1H, CH = CHOH), 6.64 (t, 1H, J 7.1 Hz, H-6:Bn), 6.35 (t, 1H, J 3.4 Hz, H-4:pyrrole), 5.69 (s, 2H, CH2-Bn), 4.25 (q, 2H, J 7.1 Hz, CH2CH3), 2.32 (t, 3H, J 7.1 Hz, CH3); 13C-NMR δ (d6-DMSO) 183.7 (C-4), 161.8 (C-1), 160.3 (C-2), 159.4 (d, 1JC-F 243.7 Hz, C-2:Bn), 134.8 (C-2:pyrrole), 129.2 (d, 3JC-F 8.2 Hz, C-6:Bn), 128.6 (C-5:pyrrole), 127.6 (d, 3JC-F 4.2 Hz, C-4:Bn), 125.7 (d, 2JC-F 14.1, C-1:Bn), 124.6 (d, 4JC-F 3.3 Hz, C-5:Bn), 122.4 (C-3:pyrrole), 115.1 (d, 2JC-F 21.5 Hz, C-3:Bn), 110.2 (C-4:pyrrole), 101.1 (C-3), 61.9 (CH2CH3), 45.7 (d, 3JC-F 4.9 Hz, CH2-Bn), 13.8 (CH3).
4-[1-(4-Fluorobenzyl)-1H-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic acid (8a) [Citation34]
4-[1-(2-Fluorobenzyl)-1H-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic acid (8b)
To a solution of compound 7b (5.20 g, 17 mmol) in 1:1 tetrahydrofuran / methanol (120 mL) was added a solution of 1N NaOH / H2O (80 mL) and the mixture was stirred at room temperature for 4 h. Then, the reaction mixture was washed with diethyl ether (150 mL), acidified to pH = 2 with 1N HCl / H2O and extracted several times with ethyl acetate (3 × 150 mL). The combined organic phases were washed with 1N HCl / H2O (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was crystallized with diethyl ether to give the title compound 8b as yellow crystals (2.70 g); Yield: 96%; TLC Rf (dihloromethane: methanol = 97: 3) 0.56; mp = 162°C; IR υmax (KBr)/cm− 1 2360, 1712 (C = O), 1623 (C = O), 1410, 1323, 1262, 1067, 755; 1H-NMR δ (d6-DMSO) 7.45-7.41 (m, 2H, H-5:pyrrole and H-4:Bn), 7.31-7.26 (m, 2H, H-3:pyrrole and H-5:Bn), 7.08 (t, 1H, J 7.6 Hz, H-3:Bn), 6.82 (s, 1H, CH = CHOH), 6.64 (t, 1H, J 7.8 Hz, H-6:Bn), 6.33 (dd, 1H, J 4.4 Hz J 2.4 Hz, H-4:pyrrole), 5.68 (s, 2H, CH2-Bn); 13C-NMR δ (d6-DMSO) 183.9 (C-4), 163.3 (C-1), 161.4 (C-2), 159.4 (d, 1JC-F 244.5 Hz, C-2:Bn), 134.5 (C-2:pyrrole), 129.2 (d, 3JC-F 8.3 Hz, C-6:Bn), 128.7 (C-5:pyrrole), 127.7 (d, 3JC-F 4.1 Hz, C-4:Bn), 125.7 (d, 2JC-F 14.0, C-1:Bn), 124.6 (d, 4JC-F 3.3 Hz, C-5:Bn), 122.0 (C-3:pyrrole), 115.1 (d, 2JC-F 20.7 Hz, C-3:Bn), 110.1 (C-4:pyrrole), 100.8 (C-3), 46.4 (d, 3JC-F 4.9 Hz, CH2-Bn).
4-(3,5-Dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-pab-oh (9A)
To a stirred and cooled (0°C) solution of 4a (404 mg, 1 mmol) in anhydrous tetrahydrofuran (8 mL) under an argon atmosphere were added 4-aminobenzyl alcohol (148 mg, 1.2 mmol, 1.2 equiv.) and 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ, 260 mg, 1.1 mmol, 1.05 equiv.), and the mixture was stirred at room temperature for 20 h. After removal of the solvent in vacuo, the residue was dissolved in ethyl acetate (50 mL), washed with 10% citric acid / H2O (40 mL), and water (40 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was crystallized with diethyl ether to give the title compound 9a as yellow crystals (361 mg); Yield: 71%; TLC Rf (chloroform: methanol = 95: 5) 0.47; mp = 154°C; IR υmax (KBr)/cm− 1 3357 (OH and NH), 1695 (C = O), 1584, 1532, 1164, 1013, 823, 698; 1H-NMR δ (d6-DMSO) 10.64 (s, 1H, NH), 7.78 (d, 2H, J 7.6 Hz, H-PAB), 7.46-7.00 (15H, m, H-Bn and H-Ph and H-PAB), 6.77 (s, 1H, CH = C(OH)), 5.20 (s, 4H, 2xCH2-Bn), 5.17 (s, 1H, PAB-OH), 4.61 (s, 1H, CH = C(OH)), 4.47 (s, 2H, CH2-PAB).
4-(3-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-pab-oh (9B)
Compound 9b was prepared from precursor 4b (600 mg, 2.01 mmol) by the same procedure as that described for compound 9a; Yield: 60%, yellow crystals (486 mg); mp = 150°C; TLC Rf (chloroform: methanol = 95: 5) 0.39; IR υmax (KBr)/cm− 1 3355 (OH and NH), 1696 (C = O), 1575, 1531, 1258, 1189, 1023, 819, 774; 1H-NMR δ (d6-DMSO) 10.68 (s, 1H, NH), 7.83 (d, J 8.3 Hz, 2H, H-PAB), 7.72-7.38 (m, 9H, H-Bn and H-Ph), 7.35 (d, J 8.3 Hz, 2H, H-PAB), 7.27 (s, 1H, CH = C(OH)), 5.26 (s, 2H, CH2-Bn), 5.23 (s, 1H, PAB-OH), 4.67 (s, 1H, CH = C(OH)), 4.51 (s, 2H, CH2-PAB).
4-(4-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-pab-oh (9C)
Compound 9c was prepared from precursor 4c (587 mg, 2.03 mmol) by the same procedure as that described for compound 9a; Yield: 69%, yellow crystals (565 mg); mp = 162°C; TLC Rf (chloroform: methanol = 95: 5) 0.41; IR υmax (KBr)/cm− 1 3362 (OH + NH),1697 (C = O), 1605, 1588, 1527, 1508, 1255, 1181, 1014, 822, 773; 1H-NMR δ (d6-DMSO) 10.58 (s, 1H, NH), 8.08 (d, J 8.6 Hz, 2H, H-Ph), 7.77 (d, J 8.3 Hz, 2H, H-PAB), 7.48-7.34 (m, 5H, H-Bn), 7.29 (d, J 8.3 Hz, 2H, H-PAB), 7.19 (d, J 8.6 Hz, 2H, H-Ph), 7.16 (s, 1H, CH = C(OH)), 5.23 (s, 2H, CH2-Bn), 5.16 (s, 1H, PAB-OH), 4.56 (s, 1H, CH = C(OH)), 4.45 (s, 2H, CH2-PAB); 13C-NMR δ (d6-DMSO) 185.5 (C-4), 178.4, 163.1, 159.9, 150.5, 138.9, 136.3, 136.0, 130.0, 129.5, 128.9, 128.5, 128.1, 127.8, 126.8, 126.5, 121.4, 120.4, 120.1, 115.3 (C-3 and C-5:phenyl), 93.7 (CH = C(OH)), 69.7 (CH2-Bn), 62.6 (CH2-PAB).
4-(3,5-Dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-oab-oh (10A)
Compound 10a was prepared from precursor 4a (400 mg, 0.99 mmol) by the same procedure as that described for compound 9a, except that the reaction mixture was added with 2-aminobenzyl alcohol (1.2 equiv.) instead of 4-aminobenzyl alcohol; Yield: 71%, yellow powder (358 mg); mp = 170°C; TLC Rf (ethyl acetate: n-hexane = 1: 1) 0.41; IR υmax (KBr)/cm− 1 3345 (OH), 3319 (NH), 1679 (C = O), 1589, 1524, 1455, 1167, 1056, 1020, 697; 1H-NMR δ (d6-DMSO) 10.77 (1H, s, NH), 8.03 (1H, d, J 8.0 Hz, H-OAB), 7.48-7.16 (16H, m, H-Bn and H-Ph and H-OAB), 7.01 (1H, s, CH = C(OH)), 5.21 (4H, s, 2xCH2-Bn), 5.17 (1H, s, PAB-OH), 4.62 (2H, s, CH2-PAB).
4-(3-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-oab-oh (10B)
Compound 10b was prepared from precursor 4b (642 mg, 2.15 mmol) by the same procedure as that described for compound 9a, except that the reaction mixture was added with 2-aminobenzyl alcohol (1.2 equiv.) instead of 4-aminobenzyl alcohol; Yield: 78%, yellow powder (633 mg); mp = 144°C; TLC Rf (chloroform: methanol = 97: 3) 0.64; IR υmax (KBr)/cm− 1 3413 (OH and NH), 3115 (NH), 1674 (C = O), 1587, 1531, 1388, 1264, 1026, 759, 694, 572; 1H-NMR δ (d6-DMSO) 10.31 (s, 1H, NH), 8.20 (d, 2H, J 8.3 Hz, H-OAB), 7.56-7.06 (m, 12H, H-Ph and H-Bn and H-OAB and CH = C(OH)), 5.06 (2H, s, CH2-Bn), 4.74 (2H, s, CH2-OAB).
4-(4-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-oab-oh (10C)
Compound 10c was prepared from precursor 4c (600 mg, 2.01 mmol) by the same procedure as that described for compound 9a, except that the reaction mixture was added with 2-aminobenzyl alcohol (1.2 equiv.) instead of 4- aminobenzyl alcohol; Yield: 78%, yellow powder (632 mg); mp = 180°C; TLC Rf (ethyl acetate: n-hexane = 1: 1) 0.50; IR υmax (KBr)/cm− 1 3437 (OH), 3259 (NH), 1670 (C = O), 1600, 1583, 1506, 1455, 1258, 1230, 1171, 1007, 824, 777, 756, 716; 1H-NMR δ (d6-DMSO) 10.76 (s, 1H, NH), 8.08 (d, 2H, J 9.0 Hz, H-Ph), 7.20 (s, 1H, CH = C(OH)), 7.48-7.31 (m, 7H, H-Bn and H-OAB), 7.19 (d, 2H, J 9.0 Hz, H-Ph), 7.15 (d, 1H, J 6.8 Hz, H-OAB), 5.83 (s, 1H, OAB-OH), 5.23 (s, 2H, CH2-Bn), 4.62 (s, 2H, CH2-OAB), 4.58 (s, 1H, CH = C(OH)); 13C-NMR δ (d6-DMSO) 185.5 (C-4), 177.6, 163.2, 159.3, 136.3, 135.9, 132.3, 130.2 (C-2 and C-6:Ph), 128.5 (C-II and C-VI:Bn), 128.2, 128.1, 127.9 (C-III and C-V:Bn), 127.8, 124.8 (C-2:OAB), 115.4 (C-3 and C-5:Ph), 93.5 (CH = C(OH)), 69.6 (CH2-Bn), 61.9 (CH2-OAB).
4-(3,5-Dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-pabc-pnp (11A)
To a stirred and cooled (0°C) solution of 9a (403 mg, 0.79 mmol) in anhydrous tetrahydrofuran (10 mL) under an argon atmosphere was added respectively: N-N-diisopropylethylamine (DIEA, 1.2 equiv., 0.16 mL, 0.95 mmol), bis-p-nitrophenyl carbonate (2 equiv., 481 mg, 1.58 mmol) and molecular sieves (3 g). The reaction mixture was stirred at room temperature for 2 days and washed respectively with 10% citric acid / H2O (50 mL), water (50 mL), brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (n-hexane: dichloromethane: = 40: 60) yielding the title compound 11a as yellow crystals (144 mg); Yield: 27%; TLC Rf (dichloromethane: methanol = 92: 8) 0.87; mp = 212°C; IR υmax (KBr)/cm− 1 3363 (NH), 1760 (C = O), 1594, 1348, 1266, 1216, 1163, 1045; 1H-NMR δ (CDCl3) 9.06 (s, 1H, NH), 8.28 (d, 2H, J 9.0 Hz, H-PNP), 7.75 (d, 2H, J 8.3 Hz, H-PABC), 7.51-7.37 (m, 18H, H-Bn and H-Ph and H-PABC and H-PNP), 7.23 (s, 1H, CH = C(OH)), 6.85 (s, 2H, CH2O(C = O)O), 5.10 (s, 4H, 2xCH2-Bn); 13C-NMR δ (CDCl3) 185.5 (C-4), 179.5 (C-2), 159.1 (C-1), 155.5 (C-3:Ph), 154.8 (OC( = O)O), 152.4, 137.4, 135.5, 131.0, 129.9, 128.7, 128.3, 127.6, 126.2, 125.6, 125.3 (C-PNP), 121.8, 121.7, 120.1 (C-PABC), 115.6, 107.8, 106.7, 100.6, 94.2 (CH = C(OH)), 70.5 (CH2OC( = O)O), 70.3 (CH2-Bn).
4-(3-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-pabc-pnp (11B)
Compound 11b was prepared from precursor 9b (584 mg, 1.45 mmol) by the same procedure as that described for compound 11a; Yield: 17%, yellow crystals (140 mg); mp = 108°C; TLC Rf (dichloromethane: methanol = 92: 8) 0.86; IR υmax (KBr)/cm− 1 3342 (OH and NH), 1758 (C = O), 1682 (C = O), 1530, 1277, 1217, 1111, 1015, 678; 1H-NMR δ (CDCl3) 9.06 (s, 1H, NH), 8.28 (d, 1H, J 9.1 Hz, H-PNP), 7.76 (d, 1H, J 8.1 Hz, H-PABC), 7.63 (m, 2H, H-Ph), 7.50-7.29 (m, 11H, H-Bn and H-Ph and H-PABC and H-PNP), 7.23 (s, 1H, CH = C-OH), 5.29 (s, 2H, CH2OC( = O)O), 5.15 (s, 2H, CH2-Bn); 13C-NMR δ (CDCl3) 185.4 (C-4), 179.7 (C-2), 159.2 (C-1), 155.5 (C-3:Ph), 154.8 (OC( = O)O), 152.4, 145.9, 145.4, 137.4, 136.3, 134.8, 131.0, 130.0, 129.9, 128.7, 128.2, 127.5, 125.5 (C-PNP), 125.3 (C-PNP), 121.8, 121.6, 120.9, 120.5 (C-Ph), 120.1 (C-PABC), 113.3 (C-2:Ph), 94.0 (CH = C(OH)), 70.5 (CH2OC( = O)O), 70.3 (CH2-Bn).
4-(4-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic-pabc-pnp (11C)
Compound 11c was prepared from precursor 9c (592 mg, 1.47 mmol) by the same procedure as that described for compound 11a; Yield: 20%, yellow crystals (167 mg); mp = 120°C; TLC Rf (dichloromethane: methanol = 92: 8) 0.88; IR υmax (KBr)/cm− 1 3365 (OH + NH), 1755 (C = O), 1696 (C = O), 1613, 1527, 1266, 1231, 776; 1H-NMR δ (d6-DMSO) 10.70 (s, 1H, NH), 8.31 (d, 2H, J 9.1 Hz, H-PNP), 8.07 (d, J 8.8 Hz, 2H, H-Ph), 7.88 (d, 2H, J 8.5 Hz, H-PABC), 7.57 (d, 2H, J 9.1 Hz, H-PNP), 7.47 (d, 2H, J 8.5 Hz, H-PABC), 7.42-7.34 (m, 5H, H-Bn), 7.19 (d, 2H, J 8.8 Hz, H-Ph), 7.15 (s, 1H, CH = C(OH)), 5.25 (s, 2H, CH2OC( = O)O), 5.24 (s, 2H, CH2-Bn); 13C-NMR δ (d6-DMSO) 185.4 (C-4), 182.2 (C-2), 160.1 (C-1), 155.2 (C-3:Ph), 151.9 (OC( = O)O), 145.1, 138.2, 136.3, 129.9 (C-Ph), 129.2 (C-PABC), 128.4, 128.0, 127.8, 126.1, 125.3 (C-PNP), 122.6 (C-PNP), 120.4 (C-PABC), 115.7, 115.2, 114.8, 94.0 (CH = C(OH)), 70.1 (CH2OC( = O)O), 69.6 (CH2-Bn).
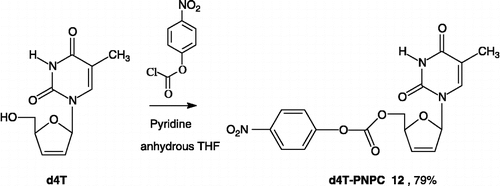
5′-PNPC-2′,3′-didehydro-2′,3′-dieoxythymidine (12)
To a stirred and cooled (0°C) solution of d4T (500 mg, 2.23 mmol) in anhydrous tetrahydrofuran (30 mL) under an argon atmosphere was added respectively: pyridine (0.27 mL, 3.34 mmol) and a solution of 4-nitrophenyl chloroformate (675 mg, 3.34 mmol) in anhydrous tetrahydrofuran (8 mL). The reaction mixture was stirred at room temperature and after 2 h TLC (silica, chloroform: methanol = 97: 3) indicated completion. The mixture was diluted with ethyl acetate (100 mL) and washed respectively with water (100 mL), 5% NaHCO3 / H2O (200 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was crystallized with diethyl ether to give the title compound 12 as white crystals (685 mg); Yield: 79%; TLC Rf (chloroform: methanol = 97: 3) 0.57; mp = 190°C; IR υmax (KBr)/cm− 1 3039 (NH), 1770 (C = O), 1694 (C = O), 1524, 1466, 1351, 1270, 1216, 1084, 859; 1H-NMR δ (CDCl3) 8.77 (s, 1H, NH), 8.30 (d, 2H, J 8.5 Hz, H-III:PNP and H-V:PNP), 7.39 (s, 1H, H-6), 7.35 (d, 2H, J 8.5 Hz, H-II:PNP and H-VI:PNP), 7.10 (s, 1H, H-1′), 6.38 (d, 1H, J 6.1 Hz, H-2′), 5.98 (d, 1H, J 6.1 Hz, H-3′), 5.13 (s, 1H, H-4′), 4.54 (s, 2H, H-5′a and H-5′b), 1.87 (s, 3H, CH3); 13C-NMR δ (CDCl3) 163.6 (C-4), 155.0 (C-I:PNP), 152.3 (OC = O), 150.1 (C-2), 145.7 (C-IV:PNP), 135.9 (C-6), 132.7 (C-2′), 128.0 (C-3′), 125.5 (C-II and C-V:PNP), 121.7 (C-II and C-VI:PNP), 111.3 (C-5), 89.6 (C-1′), 83.5 (C-4′), 68.8 (C-5′), 12.4 (CH3).
Heterodimer [4-(3,5-Dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic]-pabc-c5′[d4t] (13A)
To a stirred and cooled (0°C) solution of 10a (228 mg, 0.45 mmol) in anhydrous dichloromethane (10 mL) under an argon atmosphere was added respectively: 4-dimethylaminopyridine (1.3 equiv., 71 mg, 0.58 mmol), 5′-PNPC-d4T (12, 1 equiv.) and a catalytic amount of N,N-diisopropylethylamine. The reaction mixture was stirred at room temperature and after 3 days TLC (silica, ethyl acetate: n-hexane = 80: 20) indicated completion. The mixture was diluted with ethyl acetate: water (1/1, 200 mL). The organic layer was separated, washed with a solution of 10% citric acid/H2O (200 mL), then water (200 mL), dried over MgSO4 and concentrated to dryness under reduced pressure. The residue was crystallized with diethyl ether to give the title compound 13a as yellow crystals (163 mg); Yield: 48%, mp = 156°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.45; IR υmax (KBr)/cm− 1 3310 (OH and NH), 1753 (C = O), 1691 (C = O), 1583, 1264, 1159, 1080, 1058, 775, 736, 697; 1H-NMR δ (CDCl3) 9.80 (s, 1H, NH-d4T), 8.28 (s, 1H, NH-PAB), 7.81 (d, 1H, J 8.6 Hz, H-PAB), 7.46-7.34 (m, 16H, H-Bn and H-Ph and H-PAB and CH = C(OH)), 7.02 (s, 1H, H-6:d4T), 6.84 (s, 1H, H-1′:d4T), 6.28 (d, 1H, J 5.8 Hz, H-2′:d4T), 5.86 (d, 1H, J 5.8 Hz, H-2′:d4T), 5.18 (s, 4H, CH2-Bn), 5.01 (s, 2H, CH2OC( = O)O), 5.00 (s, 1H, H-4′:d4T), 4.31 (s, 2H, H-5′a and H-5′b:d4T), 1.55 (s, 3H, CH3); Anal. Calcd. For C42H37N3O11: C, 66.40; H, 4.91; N, 5.53. Found: C, 66.34; H, 4.87; N, 5.46%.
Heterodimer [4-(3-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic]-pabc-c5′[d4t] (13B)
Compound 13b was prepared from precursor 10b (200 mg, 0.50 mmol) by the same procedure as that described for compound 13a; Yield: 36%, yellow crystals (117 mg); mp = 130°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.51; IR υmax (KBr)/cm− 1 3309 (OH and NH), 1749 (C = O), 1694 (C = O), 1525, 1264, 777, 697; 1H-NMR δ (CDCl3) 11.32 (s, 1H, NH-d4T), 10.73 (s, 1H, NH-PAB), 7.63-7.11 (m, 15H, H-Bn and H-Ph and H-PAB and CH = C(OH) and H-6:d4T), 6.90 (s, 1H, H-1′:d4T), 6.19 (d, 1H, J 5.8 Hz, H-2′:d4T), 5.74 (d, 1H, J 5.8 Hz, H-2′:d4T), 5.03 (s, 4H, CH2-Bn and CH2OC( = O)O), 4.93 (br s, 1H, H-4′:d4T), 4.39 (dd, 1H, J 2.4 Hz, J 12.3 Hz, H-5′a:d4T), 4.28 (dd, 1H, J 2.4 Hz, J 12.3 Hz, H-5′b:d4T), 1.66 (s, 3H, CH3); Anal. Calcd. For C35H31N3O10: C, 64.31; H, 4.78; N, 6.43. Found: C, 64.29; H, 4.74; N, 6.39%.
Heterodimer [4-(4-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic]-pabc-c5′[d4t] (13C)
Compound 13c was prepared from precursor 10c (251 mg, 0.62 mmol) by the same procedure as that described for compound 13a; Yield: 45%, yellow crystals (183 mg); mp = 162°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.32; IR υmax (KBr)/cm− 1 3327 (OH and NH), 1749 (C = O), 1695 (C = O), 1591, 1499, 1337, 1286, 1169, 1112, 852, 754, 630; 1H-NMR δ (d6-DMSO) 11.42 (s, 1H, NH-d4T), 10.77 (s, 1H, NH-PAB), 8.19-6.98 (m, 15H, H-Bn and H-Ph and H-PAB and CH = C(OH) and H-6:d4T), 6.89 (s, 1H, H-1′:d4T), 6.47 (d, 1H, J 5.8 Hz, H-2′:d4T), 6.04 (d, 1H, J 5.8 Hz, H-3′:d4T), 5.30 (s, 2H, CH2-Bn), 5.17 (s, 2H, CH2OC( = O)O), 5.06 (br s, 1H, H-4′:d4T), 4.44 (dd, 1H, J 2.7 Hz, J 12.2 Hz, H-5′a:d4T), 4.34 (dd, 1H, J 2.7 Hz, J 12.2 Hz, H-5′b:d4T), 1.62 (s, 3H, CH3); 13C-NMR δ (d6-DMSO) 185.3 (C-4), 177.9, 163.7 (C-4:d4T), 163.6, 162.9, 159.9, 154.1, 150.6 (C-2:d4T), 137.8, 136.2 (C-6:d4T), 135.8, 133.1 (C-2′:d4T), 131.0, 130.8, 129.9, 129.1, 128.4, 127.9, 127.6, 126.9 (C-3′:d4T), 126.0, 120.4, 120.1, 115.6, 115.2, 114.7, 109.4 (C-5:d4T), 93.6 (C-3), 88.7 (C-1′:d4T), 83.4 (C-4′:d4T), 69.5 (CH2OC( = O)O), 68.9 (CH2-Bn), 67.7 (C-5′:d4T), 11.7 (CH3); Anal. Calcd. For C35H31N3O10: C, 64.31; H, 4.78; N, 6.43. Found: C, 64.25; H, 4.72; N, 6.35%.
Heterodimer [4-(3,5-dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic]-OABC- C5′[d4T] (14a)
Compound 14a was prepared from precursor 11a (226 mg, 0.44 mmol) by the same procedure as that described for compound 13a; Yield: 49%, yellow crystals (165 mg); mp = 138°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.40; IR υmax (KBr)/cm− 1 3365 (OH), 3262 (NH), 1733 (C = O), 1697(C = O), 1585, 1526, 1453, 1280, 1163, 1056, 936, 912; 1H-NMR δ (d6-DMSO) 9.80 (s, 1H, NH-d4T), 8.28 (s, 1H, NH-OAB), 8.06 (d, 1H, J 8.0 Hz, H-OAB), 7.46-7.34 (m, 16H, H-Bn and H-Ph and H-OAB), 7.25 (s, 1H, CH = C(OH), 7.06 (s, 1H, H-6:d4T), 6.84 (s, 1H, H-1′:d4T), 6.28 (d, 1H, J 6.0 Hz, H-2′:d4T), 5.86 (d, 1H, J 6.0 Hz, H-3′:d4T), 5.21 (m, 2H, CH2OC( = O)O), 5.10 (s, 4H, CH2-Bn), 5.00 (s, 1H, H-4′:d4T), 4.50 (br s, 1H, H-5′a:d4T), 4.3 (br s, 1H, H-5′b:d4T), 1.78 (s, 3H, CH3); Anal. Calcd. For C42H37N3O11: C, 66.40; H, 4.91; N, 5.53. Found: C, 66.32; H, 4.84; N, 5.47%.
Heterodimer [4-(3-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic]-oabc-c5′[d4t] (14B)
Compound 14b was prepared from precursor 11b (178 mg, 0.44 mmol) by the same procedure as that described for compound 13a; Yield: 41%, yellow crystals (118 mg); mp = 147°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.48; IR υmax (KBr)/cm− 1 3369 (OH), 3065 (NH), 1748 (C = O), 1694 (C = O), 1582, 1526, 1454, 1264, 1080, 1025, 757, 697; 1H-NMR δ (d6-DMSO) 11.39 (s, 1H, NH-d4T), 10.45 (s, 1H, NH-OAB), 7.67-7.28 (m, 14H, H-Bn and H-Ph and H-OAB and CH = C(OH), 7.24 (s, 1H, H-6:d4T), 6.87 (s, 1H, H-1′:d4T), 6.44 (d, 1H, J 5.6 Hz, H-2′:d4T), 6.04 (d, 1H, J 5.6 Hz, H-3′:d4T), 5.27 (s, 2H, CH2-Bn), 5.22 (s, 2H, CH2OC( = O)O), 5.04 (s, 1H, H-4′:d4T),), 4.43 (d, 1H, J 12.2 Hz, H-5′a:d4T), 4.33 (d, 1H, J 12.2 Hz, H-5′b:d4T), 1.60 (s, 3H, CH3); Anal. Calcd. For C35H31N3O10: C, 64.31; H, 4.78; N, 6.43. Found: C, 64.27; H, 4.70; N, 6.37%.
Heterodimer [4-(4-Benzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic]-oabc-c5′[d4t] (14C)
Compound 14c was prepared from precursor 11c (88 mg, 0.22 mmol) by the same procedure as that described for compound 13a; Yield: 42%, yellow crystals (60 mg); mp = 17°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.39; IR υmax (KBr)/cm1 3373 (OH), 3035 (NH), 1754 (C = O), 1694 (C = O), 1454, 1252, 1174, 1068, 775, 759; 1H-NMR δ (d6-DMSO) 9.78 (s, 1H, NH-d4T), 8.38 (s, 1H, NH-OAB), 8.02 (d, 2H, J 9.0 Hz, H-Ph), 7.46-7.18 (m, 11H, H-Bn and H-OAB and CH = C(OH) and H-6:d4T), 7.07 (d, 2H, J 9.0 Hz, H-Ph), 7.03 (s, 1H, H-1′:d4T), 6.28 (d, 1H, J 5.6 Hz, H-2′:d4T), 5.87 (d, 1H, J 5.6 Hz, H-3′:d4T), 5.30 (s, 2H, CH2OC( = O)O), 5.16 (s, 2H, CH2-Bn), 5.04 (br s, 1H, H-4′:d4T),), 4.50 (dd, 1H, J 2.7 Hz J 12.2 Hz, H-5′a:d4T), 4.38 (dd, 1H, J 2.7 Hz J 12.2 Hz, H-5′b:d4T), 1.78 (s, 3H, CH3); Anal. Calcd. For C35H31N3O10: C, 64.31; H, 4.78; N, 6.43. Found: C, 64.25; H, 4.71; N, 6.38%.
4-[1-(4-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic-pab-oh (15A)
Compound 15a was prepared from precursor 8a (300 mg, 1.04 mmol) by the same procedure as that described for compound 10a; Yield: 24%, yellow crystals (100 mg); mp = 170°C; TLC Rf (chloroform: methanol = 95: 5) 0.32; IR υmax (KBr)/cm-1 3405 (OH and NH), 1695 (C = O), 1639 (C = O), 1591, 1525, 1414, 1326, 1242, 1222, 1086, 1065, 824; 1H-NMR δ (d6-DMSO) 10.35 (s, 1H, NH), 7.71 (d, 2H, J 8.4 Hz, H-PAB), 7.55 (s, 1H, H-5:pyrrole), 7.45 (s, 1H, H-3:pyrrole), 7.27 (d, 2H, J 8.4 Hz, H-PAB), 7.15-7.06 (m, 4H, H-Bn), 6.90 (s, 1H, CH = C(OH)), 5.62 (s, 2H, CH2-Bn), 4.44 (s, 2H, CH2-PAB); 13C-NMR δ (d6-DMSO) 184.7 (C-4), 167.1 (C-1), 160.6 (d, 1JC-F 202.2 Hz, C-4:Bn), 160.4 (C-2), 139.6, 137.2, 135.5, 133.5, 130.2, 129.2, 128.5, 127.7, 126.7, 126.6, 125.6, 122.9, 121.3, 120.9, 116.0 (d, 2JC-F 20.7 Hz, C-3:Bn), 111.2 (C-4:pyrrole), 98.6 (CH = C(OH)), 63.4 (CH2-Bn), 47.4 (CH2-PAB).
4-[1-(2-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic-pab-oh (15B)
Compound 15b was prepared from precursor 8b (300 mg, 1.04 mmol) by the same procedure as that described for compound 10a; Yield: 24%, yellow crystals (100 mg); mp = 140°C; TLC Rf (chloroform: methanol = 95: 5) 0.33; IR υmax (KBr)/cm− 1 3390 (OH and NH); 1690 (C = O), 1628 (C = O), 1592,1526, 1412, 1327, 1230, 1086, 1368, 1009, 754,677; 1H-NMR δ (d6-DMSO) 10.48 (s, 1H, NH), 7.71 (d, J 8.5 Hz, 2H, H-PAB), 7.47-7.04 (m, 8H, H-5 and H-3:pyrrole and H-Bn and H-PAB), 6.91 (s, 1H, CH = CHOH), 6.64 (t, 1H, J 7.8 Hz, H-6:Bn), 6.37 (dd, 1H, J 3.6 Hz J 2.9 Hz, H-4:pyrrole), 5.72 (s, 2H, CH2-Bn), 4.45 (s, 2H, CH2-PAB).
4-[1-(4-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic-oab-oh (16A)
Compound 16a was prepared from precursor 8a (500 mg, 1.73 mmol) by the same procedure as that described for compound 10a, except that the reaction mixture was added with 2-aminobenzyl alcohol (1.2 equiv.) instead of 4- aminobenzyl alcohol; Yield: 34%, yellow powder (230 mg); mp = 190°C; TLC Rf (chloroform: methanol = 95: 5) 0.69; IR υmax (KBr)/cm− 1 3390 (OH + NH), 1690 (C = O), 1628 (C = O), 1592, 1526, 1412, 1327, 1230, 1086, 1368, 1009, 754, 677; 1H-NMR δ (d6-DMSO) 10.57 (s, 1H, NH), 7.99 (d, 2H, J 7.4 Hz, H-PAB), 7.57 (s, 1H, H-5:pyrrole), 7.46 (s, 1H, H-3:pyrrole), 7.99 (d, 2H, J 7.4 Hz, H-PAB), 7.16-7.14 (m, 4H, H-Bn), 6.95 (s, 1H, CH = C(OH)), 6.34 (s, 1H, H-4:pyrrole); 5.62 (s, 2H, CH2-Bn), 4.58 (s, 2H, CH2-PAB); 13C-NMR δ (d6-DMSO) 183.2 (C-4), 165.5 (C-1), 160.9 (d, 1JC-F 244.5 Hz, C-2:Bn), 159.1 (C-2), 136.0, 134.7, 134.5, 132.3, 128.9, 128.8, 128.0, 127.7, 124.8, 122.2 (C-3:pyrrole), 115.1 (d, 2JC-F 21.5 Hz, C-3:Bn), 110.4 (C-4:pyrrole), 97.7 (C-3), 61.9 (CH2-Bn), 51.4 (CH2-PAB).
4-[1-(2-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic-oab-oh (16B)
Compound 16b was prepared from precursor 8b (579 mg, 2.00 mmol) by the same procedure as that described for compound 10a, except that the reaction mixture was added with 2-aminobenzyl alcohol (1.2 equiv.) instead of 4- aminobenzyl alcohol; Yield: 40%, yellow powder (316 mg); mp = 172°C; TLC Rf (chloroform: methanol = 95: 5) 0.52; IR υmax (KBr)/cm− 1 3298 (OH and NH),1678 (C = O),1633, 1591, 1537, 1454, 1325, 1244, 1230, 1084, 1064, 1010, 754; 1H-NMR δ (d6-DMSO) 10.53 (s, 1H, NH), 7.99-7.03 (m, 10H, H-5:pyrrole and H-3:pyrrole and H-Bn and H-OAB), 6.94 (s, 1H, CH = CHOH), 6.36 (s, 1H, H-4:pyrrole), 5.72 (s, 2H, CH2-Bn), 4.57 (s, 2H, CH2-OAB); 13C-NMR δ (d6-DMSO) 183.6 (C-4), 165.7 (C-1), 159.2 (d, 1JC-F 217.9 Hz, C-2:Bn), 158.2 (C-2), 157.8, 135.9, 135.5, 134.5, 132.3, 129.3; 129.2, 128.8, 128.0, 127.8, 127.6, 126.5, 125.6,124.7, 124.6, 124.5, 124.4, 122.0, 121.5, 115.0 (d, JCIII-F 20.6 Hz, CIII-OFP), 114.8, 113.8, 111.7, 110.2 (C3-pyrrole), 109.1 (C-4:pyrrole), 97.3 (C-3), 61.8 (CH2-OAB), 46.4 (CH2-Bn).
Heterodimer [4-[1-(4-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic]-pabc-c5′[d4t] (17A)
Compound 17a was prepared from precursor 15a (198 mg, 0.50 mmol) by the same procedure as that described for compound 13a; Yield: 33%, yellow crystals (107 mg); mp = 142°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.38; IR υmax (KBr)/cm− 1 3310 (OH), 3075 (NH), 1750 (C = O), 1693 (C = O), 1525, 1266, 1241, 1085, 1068, 750, 675; 1H-NMR δ (d6-DMSO) 11.29 (s, 1H, NH-d4T), 10.41 (s, 1H, NH-PAB), 7.78 (d, 2H, J 8.3 Hz, H-PAB), 7.52 (s, 1H, H-5:pyrrole), 7.43 (s, 1H, H-3:pyrrole), 7.35 (d, 2H, J 8. 3 Hz, H-PAB), 7.26 s, 1H, H-6:d4T), 7.16-7.05 (m, 4H, H-PFP), 6.91 (s, 1H, CH = C(OH)), 6.81 (s, 1H, H-1′:d4T), 6.39 (d, 1H, J 5.4 Hz, H-2′:d4T), 6.34 (s, 1H, H-4:pyrrole), 5.96 (d, 1H, J 5.4 Hz, H-3′:d4T), 5.63 (s, 2H, CH2-PFP), 5.50 (s, 1H, CH = C(OH)), 5.10 (s, 2H, CH2OC( = O)O), 4.99 (s, 1H, H-4′:d4T), 4.37 (d, 1H, J 11.7 Hz, H-5′a:d4T), 4.27 (d, 1H, J 11.7 Hz, H-5′b:d4T), 1.56 (s, 3H, CH3); 13C-NMR δ (d6-DMSO) 183.8 (C-4), 183.3 (C-1), 164.9 (d, 1JC-F 237.8 Hz, C-IV:PFP), 159.8, 154.2, 150.7 (C-2), 138.0, 135.9 (C-6:d4T), 134.3 (C-5:pyrrole), 133.2 (C-2′:d4T), 131.1, 127.0 (C-3′:d4T), 129.1 (C-2 and C-6:PAB), 128.7(d, 3JC-F 8.3 Hz, C-II and C-VI:PFP), 127.0 (C-3pyrrole), 120.6 (C-3 and C-5:PAB), 115.2(d, 2JC-F 21.5 Hz, C-III and C-V PFP), 110.3 (C-4:pyrrole), 97.8 (CH = C(OH)), 88.9 (C-1′:d4T), 83.5 (C-4′:d4T), 69.0 (CH2OC( = O)O), 67.8 (C-5′:d4T), 51.3 (CH2-PFP), 11.8 (CH3); Anal. Calcd. For C33H29FN4O9: C, 61.49; H, 4.53; N, 8.69. Found: C, 61.42; H, 4.49; N, 8.64%.
Heterodimer [4-[1-(2-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic]-pabc-c5′[d4t] (17B)
Compound 17b was prepared from precursor 15b (130 mg, 0.33 mmol) by the same procedure as that described for compound 13a; Yield: 53%, yellow crystals (113 mg); mp = 140°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.43; IR υmax (KBr)/cm− 1 3298 (OH), 3070 (NH),1749 (C = O),1693 (C = O), 1525, 1414, 1266, 1085, 1068, 755, 675; 1H-NMR δ (d6-DMSO) 11.31 (s, 1H, NH-d4T), 10.44 (s, 1H, NH-PAB), 7.77 (d, 2H, J 8.5 Hz, H-PAB), 7.45 (s, 1H, H-5:pyrrole), 7.35 (d, 2H, J 8.5 Hz, H-PAB), 7.33-7.10 (m, 8H, H-OFP and H-3:pyrrole and H-PAB and H-6:d4T), 6.91 (s, 1H, CH = C(OH)), 6.81 (s, 1H, H-1′:d4T), 6.37 (br s, 2H, H-2′:d4T and H-4:pyrrole), 5.97 (br s, 3H, H-3′:d4T and CH2-OFP), 5.09 (s, 2H, CH2OC( = O)O), 4.99 (s, 1H, H-4′:d4T), 4.37 (d, 1H, J 12.2 Hz, H-5′a:d4T), 4.27 (d, 1H, J 12.2 Hz, H-5′b:d4T), 1.54 (s, 3H, CH3); 13C-NMR δ (d6-DMSO) 183.3 (C-4), 166.1, 163.7, 154.2, 150.7, 135.9 (C-6:d4T), 133.2, 131.1 (C-2′:d4T), 129.1 (C-2 and C-6:PAB), 127.7 (C-3′:d4T), 127.0 (C-3:pyrrole), 124.6, 122.0, 120.6 (C-3 and C-5:PAB), 120.2, 115.2, 115.0, 110.3 (C-4:pyrrole), 109.5, 97.8 (CH = C(OH)), 88.9 (C-1′:d4T), 83.5 (C-4′:d4T), 69.0 (CH2OC( = O)O), 67.8 (C-5′:d4T), 30.6 (CH2-OFP), 11.8 (CH3); Anal. Calcd. For C33H29FN4O9: C, 61.49; H, 4.53; N, 8.69. Found: C, 61.44; H, 4.47; N, 8.62%.
Heterodimer [4-[1-(4-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic]-oabc-c5′[d4t] (18A)
Compound 18a was prepared from precursor 16a (195 mg, 0.49 mmol) by the same procedure as that described for compound 13a; Yield: 22%, yellow crystals (70 mg); mp = 145°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.54; IR υmax (KBr)/cm− 1 3384(OH), 3044 (NH), 1753 (C = O), 1693 (C = O), 1510, 1267, 1241, 1084, 1068, 756, 682; 1H-NMR δ (d6-DMSO) 11.32 (s, 1H, NH-d4T), 10.19 (s, 1H, NH-OAB), 7.55-7.07 (m, 11H, H-PFP and H-5:pyrrole and H-3:pyrrole and H-POB and H-6:d4T), 6.90 (s, 1H, CH = C(OH)), 6.80 (s, 1H, H-1′:d4T), 6.34 (br s, 2H, H-4:pyrrole and H-2′:d4T), 5.94 (s, 1H, H-3′:d4T), 5.62 (s, 2H, CH2-PFP), 5.18 (m, 2H, CH2OC( = O)O), 4.06 (s, 1H, H-4′:d4T), 4.35 (d, 1H, J 11.9 Hz, H-5′a:d4T), 4.30 (d, 1H, J 11.9 Hz, H-5′b:d4T), 1.52 (s, 3H, CH3); 13C-NMR δ (d6-DMSO) 183.3 (C-4), 166.4, 163.7, 160.1, 154.2, 150.7, 135.9 (C-5:pyrrole), 134.9, 134.4, 129.9, 129.6, 129.4, 129.0, 128.96, 128.9, 128.8, 128.7, 128.0, 127.0 (C-3:pyrrole), 126.4, 125.9, 122.2, 115.4, 115.1, 110.4 (C-4:pyrrole), 109.6, 97.9 (C-6:d4T), 88.9 (C-1′:d4T), 83.5 (C-4′:d4T), 68.0 (CH2OC( = O)O), 66.0 (C-5′:d4T), 51.4 (CH2-PFP), 11.7 (CH3); Anal. Calcd. For C33H29FN4O9: C, 61.49; H, 4.53; N, 8.69. Found: C, 61.43; H, 4.49; N, 8.62%.
Heterodimer [4-[1-(2-Fluorobenzyl)-1h-pyrrol-2-yl]-2-hydroxy-4-oxobut-2-enoic]-oabc-c5′[d4t] (18B)
Compound 18b was prepared from precursor 16b (196 mg, 0.50 mmol) by the same procedure as that described for compound 13a; Yield: 19%, yellow crystals (61 mg); mp = 135°C; TLC Rf (ethyl acetate: n-hexane = 80: 20) 0.46; IR υmax (KBr)/cm− 1 3492 (OH), 3292 (NH), 1747 (C = O), 1695 (C = O), 1590, 1526, 1457, 1411, 1263, 1244, 1086, 756; 1H-NMR δ (d6-DMSO) 11.31 (s, 1H, NH-d4T), 10.17 (s, 1H, NH-OAB), 7.49-7.10 (m, 11H, H-OFP and H-5:pyrrole and H-3:pyrrole and H-OAB and H-6:d4T), 6.90 (s, 1H, CH = C(OH)), 6.80 (s, 1H, H-1′:d4T), 6.37 (s, 1H, H-4:pyrrole), 6.33 (d, 1H, J 5.8 Hz, H-2′:d4T), 5.93 (d, 1H, J 5.8 Hz, H-3′:d4T), 5.72 (s, 2H, CH2-OFP), 5.15 (m, 2H, CH2OC( = O)O), 4.96 (s, 1H, H-4′:d4T), 4.34 (d, 1H, J 10.7 Hz, H-5′a:d4T), 4.25 (d, 1H, J 10.7 Hz, H-5′b:d4T), 1.52 (s, 3H, CH3); Anal. Calcd. For C33H29FN4O9: C, 61.49; H, 4.53; N, 8.69. Found: C, 61.40; H, 4.47; N, 8.62%.
Antiviral Test Procedures
The cultures of CEM-SS and MT4 cells were maintained at 37°C in a 5% CO2 atmosphere in RPMI-1640 medium supplemented with 10% complement-depleted foetal bovine serum (FBS). The antiviral HIV-1 activity of a given compound in CEM-SS cells was measured by quantification of the Reverse Transcriptase activity (RT) associated with particles released from HIV-1LAI-infected cells in the culture medium. CEM-SS cells were infected with 100 TCDI50 (the virus stock was titrated under the same experimental conditions); after 30 min adsorption, free virus particles were washed out and cells were re-suspended in RPMI-1640 plus 10% calf foetal serum at a final concentration of 105 cells mL− 1 in the presence of different concentrations of test compounds. After 5 days, virus production was measured by RT assay as already described [Citation50]. The 50% inhibitory concentration (IC50) was derived from the computer generated median effect plot of the dose-effect data [program: therapeutic and safety indexes, 51]. The cytotoxicity of the drugs was evaluated in parallel by incubating uninfected cells in the presence of different concentrations of antiviral products. The cell viability was determined by a measure of mitochondrial dehydrogenase activity, enzymes reducing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) into formazan (whose quantity was measured by the absorbance at 540 nm) [Citation52]. The 50% cytotoxic concentration (CC50) is the concentration of drug which reduces cell viability by 50% and was calculated with the program used in the determination of the IC50. The assays using different cells and virus isolates were done according to previously published protocols [Citation50,Citation53]; virus production was quantified by the RT activity associated to virus particles released from the cells in the culture medium. Conditions in which the inhibitory properties were measured on HIV-1 Reverse Transcriptase in vitro has also been described [Citation50]. The CEM-SS cells were obtained from P. Nara through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (Bethesda, Md., USA).
Results and discussion
Chemistry
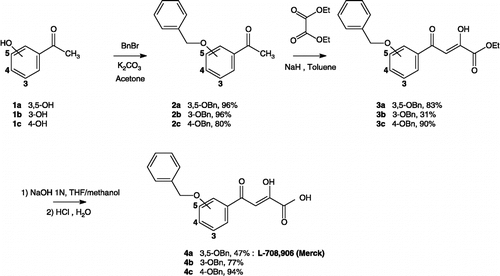
The target prodrugs of the general formula [INI]-PABC-[d4T] (13a-c, 17a-b) and [INI]-OABC-[d4T] (14a-c, 18a-b) described in the present paper were prepared by attaching the nucleoside inhibitor moiety to the spacer end of the IN inhibitor-spacer moieties. The synthetic method for these target compounds involved the formation of the key precursors: the NRTI (d4T) and the INIs (DKAs). The synthesis of the d4T was achieved according to a method previously reported in the literature [Citation54]. For the INIs, we have developed the synthesis of the α,γ-diketo acids 4a-c and 8a-b respectively as outlined in Scheme and . The synthesis of the 4-(3,5-dibenzyloxyphenyl)-2-hydroxy-4-oxobut-2-enoic acid (4a) was described in a preceding paper [Citation34]. Since the DKAs 4b and 4c have not been previously reported, we developed the synthesis of these compounds using a synthetic approach outlined in a preliminary account by oxalylation of the corresponding aryl methyl ketones in the presence of base followed by alkaline hydrolysis (Scheme 1) [Citation34]. Thus, the DKAs 4b and 4c were obtained in three steps starting respectively from commercially available 3-hydroxyacetophenone (1b) and 4-hydroxyacetophenone (1c). In the first step, the acetophenone was subjected to a benzylation reaction using benzyl bromide in acetone in the presence of anhydrous sodium carbonate. The resulting aryl methyl ketones 2b-c were then converted to the corresponding ketoenol esters 3b-c using diethyl oxalate in the presence of NaH (in toluene) respectively in 31% and 90% yields. In the last step, hydrolysis of the ester intermediates 3b-c provided the corresponding ketoenol acids 4b-c, isolated respectively in 77% and 94% yields.
Scheme 1 Preparation of ketoenol acids 4a–c.
Scheme 2 Preparation of ketoenol acids 8a–b.
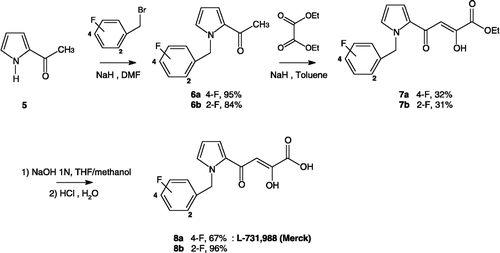
For comparative studies, the DKAs 8a-b were prepared by a similar synthetic approach as indicated in Scheme 2. Firstly, 2-acetylpyrrole (5) was alkylated with the appropriate benzyl bromide in alkaline medium (sodium hydride), obtaining the N-alkylated derivatives 6a–b that underwent condensation with diethyl oxalate in the presence of sodium hydride. Then, the ketoenol esters 7a–b that formed were easily hydrolyzed in 1N sodium hydroxide to afford the required α,γ-diketo acids 8a–b respectively in 20% and 22% overall yields.
The second part was devoted to the insertion of a p- or o-aminobenzyl alcohol (PAB-OH and OAB-OH) spacer between the α,γ-diketoacids 4a–c and 8a–b and the NRTI. Thus, the selective coupling reactions of 4a–c with PAB-OH or OAB-OH were carried out in anhydrous THF under an argon atmosphere in the presence of EDDQ (2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline) inducing the formation of peptide linkage and the expected products 9a–c and 10a–c were isolated (60 to 78%) (Scheme ) [Citation55]. Following this, the linker of compounds 9a–c was activated toward coupling with d4T as the corresponding p-nitrophenyl carbonate derivatives 11a–c. The benzylic alcohols 9a–c were then converted subsequently into the corresponding activated analogues 11a–c by reacting them with bis-(4-nitrophenyl) carbonate in anhydrous THF in the presence of N,N-diisopropylamine and were isolated in low yields (17 to 27%) after purification by silica-gel chromatography [Citation56]. In a final step, reaction of 5′-hydroxyl of d4T with these activated carbonate derivatives 11a–c in the presence of dimethylaminopyridine (DMAP) led unfortunately to a mixture consisting mainly of the unreacted products 11a–c.
Scheme 3 Synthesis of [INI]-PABC-[d4T] (13a–c) and [INI]-OABC-[d4T] (14a–c) conjugates.
![Scheme 3 Synthesis of [INI]-PABC-[d4T] (13a–c) and [INI]-OABC-[d4T] (14a–c) conjugates.](/cms/asset/7c9b70bf-1da5-4b1d-8915-7ef32fed5738/ienz_a_242422_f0005_b.gif)
These data prompted us to devise a different strategy of linking the drugs, as depicted in Scheme . In an alternative approach, the activated carbonate d4T 12 is linked directly to the DKAs functionalized with respectively the p- and o-aminobenzyl alcohol spacers 9a–c and 10a–c. Thus, when d4T was treated with 1.5 equiv. of 4-nitrophenyl chloroformate in the presence of pyridine, the desired activated d4T-carbonate 12 was formed in a reasonable yield (79%) (Scheme ) [Citation57]. The final coupling step was performed by addition of activated d4T-carbonate 12 to a solution of the intermediates 9a–c and 10a–c in anhydrous dichloromethane in the presence of DMAP to yield the expected target prodrugs 13a–c and 14a–c (36 to 49%) (Scheme 3).
Scheme 4 Preparation of the 5′-PNPC-d4T 12.
Finally, for comparative studies, we focused on modifications on the INI in the target heterodimers with the introduction of the L-731,988 compound based on our previous biological results [Citation34]. The synthesis of the prodrugs 17a–b and 18a–b was carried out in a similar way starting from the corresponding DKAs 8a–b as shown in Scheme . Reaction of DKAs 8a–b with commercially available PAB-OH and OAB-OH in the presence of EEDQ yielded the intermediates 15a–b (24%) and 16a–b (34 and 40%). Treatment of these intermediates with activated d4T-carbonate 12 gave the corresponding heterodimers 17a–b and 18a–b (19 to 53%).
Scheme 5 Synthesis of [INI]-PABC-[d4T] (17a–b) and [INI]-OABC-[d4T] (18a–b) conjugates.
![Scheme 5 Synthesis of [INI]-PABC-[d4T] (17a–b) and [INI]-OABC-[d4T] (18a–b) conjugates.](/cms/asset/084412ef-1f50-40f2-8510-5b423d98d1a7/ienz_a_242422_f0007_b.gif)
All structures were confirmed by analytical and spectroscopic data (see chemical procedures in experimental section).
Biological
The synthesized target bis-substrate molecules [INI]-PABC-[d4T] (13a–c, 17a–b) and [INI]-OABC-[d4T] (14a–c, 18a–b) were evaluated in vitro for their ability to inhibit replication of HIV-1 in two human T-4 lymphoblastoid cell lines (CEM-SS and MT-4) and HIV-2 in PBMC cells (). The clinically used nucleoside analogues AZT and d4T were included as reference materials. For the purpose of reference, data for parental DKAs (4a–c, 8a–b) and the d4T analogue precursor (12, d4T-PNPC) were also displayed. We also included corresponding data for the [INI]-PAB-OH (9a–c, 15a–b) and [INI]-OAB-OH (10a–c, 16a–b) precursors.
Table I. Antiviral and cytotoxicity evaluation of the DKAs, the modified DKAs, the d4T-PNPC, and the conjugates [INI]-spacer-[d4T] against selected HIV strains.
Central to the idea of constructing bis-substrate inhibitors connected via an amide and a carbonate functionalities was to determine to what extent the individual NRTI and INI components in each molecule retain their capacity to inhibit HIV-1 replication once the spacers were introduced. As the data displayed in , all active compounds proved to be more potent in the CEM model system also in order to rationalize the analysis, we concentrated on these results. Relative to d4T (IC50 0.273 μM) and parental DKAs (4a: IC50 0.58 μM and 8a: IC50 0.44 μM), the corresponding heterodimers 13a, 17a and 18a were also active at submicromolar concentrations (IC50 0.41 to 0.97 μM); the heterodimer 14a only exhibited a weak activity (IC50 1.47 μM) close to the toxicity (CC50 3.4 μM). Relative to the parental DKA 4b (IC50 1.49 μM), the heterodimer 13b had a similar activity (IC50 1.60 μM) whereas 14b displayed a poor or no specific activity in the CEM-SS cells (IC50 4.67 μM, CC50 7.83 μM). The results for the heterodimers 13c (IC50 0.61 μM) and 14c (IC50 1.85 μM) were remarkable since the parental DKA 4c (IC50 > 100 μM) exhibited no significant anti-HIV activity, suggesting a contribution of d4T. Finally, relative to d4T (IC50 0.273 μM) and parental DKA 8b (IC50 18.5 μM), the heterodimer 17b was active at submicromolar concentrations (IC50 0.82 μM) but 18b exhibited no measurable anti-HIV activity (IC50 > 10 μM). Overall, all prodrugs showed a much higher cytotoxicity in CEM-SS cells in comparison with the corresponding free parent drugs.
Concerning the [INI]-spacer compounds, evaluation of the [INI]-PAB-OH (9a–c, 15a–b) and [INI]-OAB-OH (10a–c, 16a–b) precursors revealed that their antiviral activity dramatically decreased in comparison with the corresponding parent compounds or even was lost; this effect was accompanied, for some of the precursors, by an increase in cytotoxicity. The lack of antiviral activity for several precursors indicates that the parental DKA was not released neither extracellularly (the molecules are stable outside the cell) nor intracellularly, possibly in the latter case as a result of non internalization into the cell.
The d4T analogue precursor 12 (d4T-PNPC) was also evaluated against HIV-1 in CEM-SS cells and exhibited nearly equal antiviral activity (IC50 0.45 μM) as d4T and low cytotoxicity (therapeutic index > 222). As all antiviral nucleoside drugs require metabolic activation in their target cells to the bio-active triphosphorylated forms by kinase-mediated phosphorylation for the expression of their antiviral activity [Citation58] this result suggested the effective cleavage of the carbonate bond of compound 12. The d4T-PNPC analogue (12) either would penetrate the cell membrane and then the d4T generated could, after phosphorylation, bind the RT target in the infected cell or the parent compound 12 might disintegrate outside the cell resulting in the release of d4T intermediate which may penetrate into the cell to inhibit the RT.
Looking next at the bis-substrate inhibitors, the conjugates [INI]-PABC-[d4T] (13a,c and 17a–b) and [INI]-OABC-[d4T] (18a) retained antiviral activity but this effect was accompanied by an increased in cytotoxicity by comparison to d4T, DKA or even some precursors. At this stage, it is not known if the activity results from an incomplete liberation of both parental molecules. The prodrugs were more cytotoxic; this result led us to presume that this increase in toxicity may be due to the partial release of the [INI]-PAB-OH or [INI]-OAB-OH moieties with an uncleaved spacer. In exhaustive studies, it would have been interesting to compare the cytotoxicity of the spacers PAB-OH and OAB-OH alone. Possible interpretations for these results are that the conjugates would penetrate (according to our hypothesis) / or not the cell membrane and might disintegrate inside / or outside the cell, resulting in the release of [INI]-PAB-OH or [INI]-OAB-OH moieties and d4T, this latter could inhibit the RT target in infected cells. Consequently, the liberation of active d4T would be (apparently) due to the simple spontaneous hydrolysis of the carbonate bond connecting the spacer and d4T and the cytotoxicity of the conjugates might be probably due to the stability of [INI]-PAB-OH or [INI]-OAB-OH precursors in physiological conditions which prevents the disintegration to their parent INIs. These observations would lead us to presume that the amide bond connecting the INI and the spacer did not spontaneously hydrolyzed under physiological conditions and perhaps not released enough active INIs.
From this analysis, we cannot conclude that the nucleoside and the INI subunits of our molecules bound simultaneously with their respective sites so as to produce a synergistic effect as expected in our hypothesis. This latter study is however pertinent in view of this work is being pursed in further investigations with new hybrid-type prodrugs conjugating HIV INIs with d4T by self-cleavable spacers containing an amino acid residue.
In conclusion, based on the prodrug concept, novel bipharmacophore drugs associating in a single molecule a reverse transcriptase inhibitor covalently linked to an integrase inhibitor were developed. The antiviral efficacy of conjugated compounds depends on many factors, such as enzyme inhibition, cell membrane permeability, extracellular stability, intracellular disintegration and the correlation between them is very complex.
Acknowledgements
We thank “Ensemble Contre le Sida” (Sidaction) for financial support.
References
- De Clercq E. J Med Chem 1995; 38: 2491–2517
- De Clercq E. Nat Rev Drug Discovery 2002; 1: 13–25
- Imamichi T. Curr Pharm Des 2004; 10: 4039–4053
- Richman DD. Nature 2001; 410: 995–1001
- Dayam R, Neamati N. Biochemistry of HIV therapeutics. Encyclopedia of molecular cell biology and molecular medicine. Wiley-VCH, Weinheim, Germany 2004; 155–178
- Barbaro G, Scozzafava A, Mastrolorenzo A, Supuran CT. Curr Pharm Des 2005; 11: 1805–1843
- Cohen J. Science 2002; 296: 2320–2324
- Mocroft A, Lundgren JD. Antimicrob Chemother 2004; 54: 10–13
- Shehu-Xhilaga M, Tachedjian G, Crowe SM, Kedzierska K. Curr Med Chem 2005; 12: 1705–1719
- Pommier Y, Neamati N. Adv Virus Res 1999; 52: 427–458
- Asante-Appiah E, Skalka AM. Adv Virus Res 1999; 52: 351–369
- Nair V. Rev Med Virol 2002; 12: 179–193
- Hazuda DJ, Young SD. Adv Antiviral Drug Des 2004; 4: 63–77
- Pommier Y, Johnson AA, Marchand C. Nat Rev Drug Disc 2005; 4: 236–248
- Mazumder A, Uchida H, Neamati N, Sunder S, Jaworska-Maslanka M, Wickstrom E, Zeng F, Jones RA, Mandes RF, Chenault HK, Pommier Y. Mol Pharmacol 1997; 51: 567–575
- Burke TR, Jr, Fesen M, Mazumder A, Yung J, Wang J, Carothers AM, Grunberger D, Driscoll J, Pommier Y, Kohn K. J Med Chem 1995; 38: 4171–4178
- Zhao H, Neamati N, Mazumder A, Sunder S, Pommier Y, Burke TR, Jr. J Med Chem 1997; 40: 1186–1194
- Artico M, Di Santo R, Costi R, Novellino E, Greco G, Massa S, Tramontano E, Marongiu ME, De Montis A, La Colla P. J Med Chem 1998; 41: 3948–3960
- King PJ, Ma G, Miao W, Jia Q, McDougall BR, Reinecke MG, Cornell C, Kuan J, Kim TR, Robinson WE, Jr. J Med Chem 1999; 42: 497–509
- Lin Z, Neamati N, Zhao H, Kiryu Y, Turpin JA, Aberham C, Strebel K, Kohn K, Witvrouw M, Pannecouque C, Debyser Z, De Clercq E, Rice WG, Pommier Y, Burke TR, Jr. J Med Chem 1999; 42: 1401–1414
- Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR. Proc Natl Acad Sci U.S.A. 1999; 96: 13040–13043
- Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Science 2000; 287: 646–650
- Fujishita T, Yoshinaga T. WO 9950245 1999
- Wai JS, Egbertson MS, Payne LS, Fisher TE, Embrey MW, Tran LO, Melamed JY, Langford HM, Guare JP, Jr, Zhuang L, Grey VE, Vacca JP, Holloway MK, Naylor-Olsen AM, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Schleif WA, Gabryelski LJ, Young SD. J Med Chem 2000; 43: 4923–4926
- Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. Proc Natl Acad Sci U.S.A. 2002; 99: 6661–6666
- Pais GCG, Zhang X, Marchand C, Neamati N, Cowansage K, Svarovskaia ES, Pathak VK, Tang Y, Nicklaus M, Pommier Y, Burke TR, Jr. J Med Chem 2002; 45: 3184–3194
- Zhang X, Pais GCG, Svarovskaia ES, Marchand C, Johnson AA, Karki RG, Nicklaus MC, Pathak VK, Pommier Y, Burke TR. Bioorg Med Chem Lett 2003; 13: 1215–1219
- Johnson AA, Marchand C, Pommier Y. Curr Top Med Chem 2004; 4: 671–686
- Neamati N. Expert Opin Ther Patents 2002; 12: 709–724
- Cass CE, Young JD, Baldwin SA. Biochem Cell Biol 1998; 76: 761
- Ralevic V, Burnstock G. Pharmacol Rev 1998; 50: 413
- Chan TCK, Shaffer L, Redmond R, Pennington KL. Biochem Pharmacol 1993; 46: 273
- Domin BA, Mahony WB, Zimmerman TP. Biochem Pharmacol 1993; 46: 725
- Sugeac E, Fossey C, Ladurée D, Schmidt S, Laumond G, Aubertin AM. J Enz Inhib Med Chem 2004; 19(6)497–509
- Mansuri MM, Starrett JE, Ghazzouli I, Hitchcock MJM, Sterzycki RZ, Brankovan V, Lin T, August EM, Prusoff WH, Sommadossi J, Martin JC. J Med Chem 1989; 32: 461–466
- Shiragami H, Uchida Y, Izaw K. Nucleosides & Nucleotides 1996; 15: 47–58
- Lin T, Schinazi RF, Prusoff WH. Biochem Pharmacol 1987; 36: 2713–2718
- Balzarini J, Kang GJ, Dalal M, Herdewijn P, De Clercq E, Broder Johns DG. Mol Pharmacol 1987; 32: 162–167
- Browne MJ, Mayer KH, Chafee SB, Dudley MN, Posner MR, Steinberg SM, Graham KK, Geletko SM, Zinner SH, Denman SL, Dunkle LM, Kaul S, Mc Laren C, Skowron G, Kontlab NM, Kennedy TA, Weitberg AB, Curt GA. J Infect Dis 1993; 167: 21–29
- Carl PL, Chakravarty PK, Katzenellenbogen JA. J Med Chem 1981; 24: 479–480
- De Groot FMH, de Bart ACW, Verheijen JH, Scheeren HW. J Med Chem 1999; 42: 5277–5283
- De Groot FMH, van Berkom LWA, Scheeren HW. J Med Chem 2000; 43: 3093–3102
- De Groot FMH, Loos WJ, Koekkoek R, van Berkom LWA, Busscher GF, Seelen AE, Albrecht C, de Bruijn P, Scheeren HW. J Org Chem 2001; 66: 8815–8830
- Florent J-C, Dong X, Gaudel G, Mitaku S, Monneret C, Gesson J-P, Jacquesy J-C, Mondon M, Renoux B, Andrianomenjanahary S, Michel S, Koch M, Tillequin F, Gerken M, Czech J, Straub R, Bosslet K. J Med Chem 1998; 41: 3572–3581
- Bouvier E, Thirot S, Schmidt F, Monneret C. Biorgan & Med Chem 2004; 12: 969–977
- El Alaoui A, Saha N, Schmidt F, Monneret C, Florent J-C. Biorgan Med Chem 2006; 14: 5012–5019
- Jeffrey SC, Torgov MY, Andreyka JB, Boddington L, Cerveny CG, Denny WA, Gordon KA, Gustin D, Haugen J, Kline T, Nuyen MT, Senter PD. J Med Chem 2005; 48: 1344–1358
- Walker MA, King HD, Dalterio RA, Trail P, Firestone R, Dubowchik GM. Biorgan Med Chem 2004; 14: 4323–4327
- Hay MP, Wilson WR, Denny WA. Biorgan Med Chem 2005; 13: 4043–4055
- Moog C, Wick A, Le Ber P, Kirn A, Aubertin AM. Antiviral Res 1994; 24: 275–288
- Chou J, Chou TC. Computer software for Apple II Series and IBM-PC and instruction manual. Elsevier-Biosoft, Cambridge, 19–28
- Mosmann T. J Immunol Meth 1983; 65: 55–63
- Lefebvre I, Perigaud C, Pompon A, Aubertin AM, Girardet JL, Kirn A, Gosselin G, Imbach JL. J Med Chem 1995; 38: 3941–3950
- Joshi BV, Rao TS, Reese CB. J Chem Soc Perkin Trans 1992; I: 2537–2544
- Belleau B, Malek GA. J Am Chem Soc 1968; 90: 1651–1652
- Dobowchik GM. European Patent 0624377 1997.
- Hay MP, Wilson WR, Denny WA. Tetrahedron 2000; 56(4)645–657
- Meier C. Synlett 1998; 3: 233–242