Abstract
A series of novel benzothiepin-derived compounds are described as potent selective modulators of the human estrogen receptor (SERMs). The objective of the study is to evaluate the antiproliferative effects of the compounds on human MCF-7 breast tumor cells. These heterocyclic compounds contain the traditional triarylethylene arrangement exemplified by tamoxifen, conformationally restrained through the incorporation of the benzothiepin ring system. The compounds demonstrated potency at nanomolar concentrations in antiproliferative assays against an MCF-7 human breast cancer cell line with low cytotoxicity. The compounds exhibited low nanomolar binding affinity for the estrogen receptor (ER) with some specificity for ERβ, and also demonstrate potent antiestrogenic properties in the human uterine Ishikawa cell line. The effect of a number of functional group substitutions on the ER binding properties of the benzothiepin molecular scaffold is explored through a brief computational structure-activity relationship investigation with molecular simulation.
Introduction
The estrogen receptor (comprising of two subtypes ERα and ERβ) is a ligand inducible nuclear receptor which plays a critical physiological role as mediator of the actions of the estrogen hormones[Citation1]. Tamoxifen 1 is a well established antagonist for the estrogen receptor and is the endocrine drug of choice in the treatment of ER positive breast cancer.Citation2-4 While the DNA binding domains of the two ER subtypes are well conserved, the amino acid sequence conservation in the ligand binding domain(LBD) is only 59%. The LBD volume for ERβ is smaller than for ERα and there are important differences in the amino acids of the LBD including replacement of Met421 and Leu384 in ERα with Ile and Met in ERβ[Citation5,Citation6]. Recent studies indicate that ERβ expression may have a potential protective effect on normal cells against ERα induced hyperproliferation[Citation7].
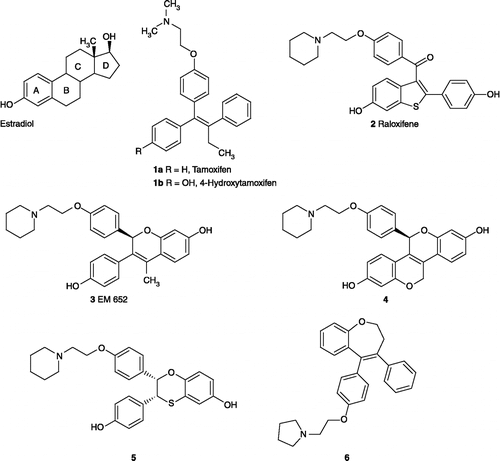
The determination of a number of crystal structures of ER-ligand complexes in recent years (e.g. 4-hydroxytamoxifen 1b and raloxifene 2)Citation8-12 together with the discovery of alternative novel scaffolds for the estrogen receptor modulators through “scaffold hopping” protocols[Citation13] has facilitated the design of several novel agonist and antagonist-type ligands for the ER. Many examples of ER modulator scaffolds based on oxygen-containing core heterocyclic systems have been reported e.g. centchroman[Citation14], benzopyrans such as EM-652 3 which is the active metabolite of EM-800[Citation15], benzopyranones, dibenzo[b,d]pyran-6-ones, bisbenzopyrans[Citation16] (e.g. 4), dihydrobenzoxathins[Citation17], (e.g. 5), bicyclo[3.3.1]nonene[Citation18] and oxachrysenol[Citation19] while mono[Citation20] and bis-benzo[b]oxepines[Citation21] have been designed as agonists of the estrogen receptor. The structures of estradiol together with selected SERMs are illustrated in . We have recently identified a novel estrogen receptor modulator core containing a central benzoxepin scaffold, (e.g. compound 6), [Citation20] which demonstrated some potential as antiproliferative and antagonist ER binding ligands and now report the further development of this novel core scaffold structure as tissue and subtype selective estrogen receptor modulators[Citation22]. The synthesis of benzothiepin derivatives which are substituted at C-4, C-5 and C-8 positions is now described together with an evaluation of their antiproliferative activity and the relative binding affinities for ERα and ERβ.
Figure 1 Structure of estradiol and SERMs.
Materials and methods
Chemistry
IR spectra were recorded as thin films on NaCl plates on a Perkin-Elmer Paragon 100 FT-IR spectrometer. 1H and 13C NMR spectra were obtained on a Bruker Avance DPX 400 instrument at 20°C, 400.13 MHz for 1H spectra, 100.61 MHz for 13C spectra, in either CDCl3 (internal standard tetramethylsilane) or CD3OD. Low resolution mass spectra were run on a Hewlett-Packard 5973 MSD GC–MS system in an electron impact mode, while high resolution accurate mass determinations for all final target compounds were obtained on a Micromass Time of Flight mass spectrometer (TOF) equipped with electrospray ionization (ES) interface operated in the positive ion mode at the High Resolution Mass Spectrometry Laboratory in the Department of Chemistry, Trinity College Dublin. Flash chromatography was carried out using standard silica gel 60 (230–400 mesh) obtained from Merck. All products isolated were homogenous on TLC. Compound 6 was prepared as we previously reported[Citation20].
2,3-Dihydro-5-(4-hydroxyphenyl)-benzo[b]thiepin 9a
Under nitrogen atmosphere at − 78°C, n-butyllithium (10 mL, 2.5 M in hexane) was added dropwise over 15 min to a solution of p-tetrahydropyranyloxy-bromobenzene 7 (25.0 mmol) in dry THF (40 mL) and stirred for 30 min. A solution of the 3,4-dihydro-1-(2HNMR)-benzo[b]thiepin-5-one 8a[Citation23] (25 mmol) in dry THF (40 mL) was added dropwise via a syringe. The reaction mixture was kept at this temperature for another 2 h and stirring continued overnight at room temperature. The mixture was partitioned between water (100 mL) and ethyl acetate (100 mL), the layers separated and the aqueous layer extracted with ethyl acetate (2 × 100 mL) to afford the tertiary alcohol as a yellow oil. The crude product was taken up in MeOH (100 mL) and concentrated HCl (4 mL) and stirred at 60–70°C for about 30 min, then partitioned between ethyl acetate and water (100 mL each), dried over MgSO4 and concentrated in vacuo. Purification of the crude product was achieved by column chromatography (SiO2; ethyl acetate/hexane 1:10, v/v). The product was obtained as a yellow oil (60%) which was used in subsequent reactions without further purification. IR νmax (CHCl3) cm− 1 3364 (w, br; O–H), 1610 (s; C = C); 1H NMR (400 MHz, MeOD-d4) δ 2.21 (q; 2H; 3J = 6.5 Hz; –CH–CH2–), 3.43 (t; 2H; 3J = 6.5 Hz; –S–CH2–), 6.46 (t; 1H; 3J = 7.8 Hz; –CH–CH2–), 6.73/7.03 (AA'BB'; 4H; 3J = 8.8 Hz; Phenyl-H), 7.03 (1H merged; H9), 7.22 (dt; 1H; 3J = 7.4 Hz, 4J = 1.8Hz; H7), 7.30 (dt; 1H; 3J = 7.4 Hz, 4J = 1.4 Hz; H8), 7.64 (dd; 1H; 3J = 7.5 Hz, 4J = 1.5 Hz; H6); 13C NMR (MeOD-d4, 100 MHz) δ 26.0 (CH2), 42.8 (CH2), 114.1 (2 × CH), 126.1(CH), 126.7 (CH), 127.3 (CH), 128.3 (2 × CH), 130.0 (CH), 133.0 (C), 134.0 (CH), 134.3 (C), 143.2 (C), 145.4 (C), 156.2 (C–O).
2,3-Dihydro-5-(4-hydroxyphenyl)-8-methoxybenzo[b]thiepin 9b
The procedure was carried out as above using 3,4-dihydro-8-methoxy-1-(2H)-benzo[b]thiepin-5-one 8b and p-tetrahydropyranyloxy-bromobenzene. Purification was achieved by column chromatography (SiO2; ethyl acetate / petroleum ether 40–60 1:10, v/v). The product was obtained as a yellow oil (60%) which was used in subsequent reactions without further purification. IR νmax (film) cm− 1 3390 (w, br; O–H), 1609 (s; C = C). 1H NMR (400 MHz, MeOD-d4) δ 2.22 (q; 2H; 3J = 6.9 Hz; –CH–CH2–), 3.42 (t; 2H; 3J = 6.5 Hz; –S–CH2–), 3.82 (s; 3H; OCH3), 6.38 (t; 1H; 3J = 7.8 Hz; –CH–CH2–), 6.72/7.02 (AA'BB'; 4H; 3J = 8.8 Hz; Phenyl-H), 6.88 (dd; 1H; 3J = 8.5 Hz, 4J = 2.5 Hz; H7), 6.94 (d; 1H; 3J = 8.5 Hz; H6), 7.21 (d; 1H; 4J = 2.5 Hz; H9) 13C NMR (MeOD-d4, 100 MHz) δ 26.1 (CH2), 42.7 (CH2), 54.0 (CH3), 113.2 (CH), 114.0 (2 × CH), 118.5 (CH), 125.2 (CH), 128.3 (2 × CH), 131.0 (CH), 133.3 (C), 135.5 (C), 137.3 (C), 143.0 (C), 156.2 (C–O), 158.0 (C–O).
2,3-Dihydro-8-fluoro-5-(4-hydroxyphenyl)-benzo[b]thiepin 9c
Preparation as above from 8-fluoro-3,4-dihydro-2H-1-benzothiepin-5-one,8c (0.925 g, 4.72 mmol) The product was purified by chromatography (silica, 5% diethyl ether/hexane) to give the product as a white solid (0.564 g, 44%) which was used in subsequent reactions without further purification. ν (KBr): 3326 (OH), 1266 (CM− 1); 1H (400 MHz, CDCl3, Me4Si): δ 7.40 (1 H, dd, J = , 9.0, 2.5), 7.10–6.97 (4 H, m, aromatic CH), 6.76 (2 H, d, J 8.5, aromatic CH), 6.47 (1 H, t, J = 7.5, CH), 3.48 (2 H, t, J = 7.0, OCH2), 2.25 (2 H, t, J = 7.0, CH2), ppm; 13C NMR(101 MHz, CDCl3, Me4Si): δ155.1 (C), 142.7 (C), 134.6 (C), 132.2 (C), 132.1 (CH), 129.3 (CH), 127.5 (CH), 121.4 (CH), 121.2 (CH), 115.3 (CH), 115.1 (CH), 44.1 (CH2), 26.9 (CH2) ppm; 19F (376 MHz, CDCl3, Me4Si): − 114.7 ppm.
2,3-Dihydro-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 11a
A mixture of 2,3-dihydro-5-(4-hydroxyphenyl)-benzo[b]thiepin 9a (3.52 mmol) and potassium carbonate (17.60 mmol) in acetone (40 mL) were heated for 1 h. Then, 1-(2-chloroethyl)-pyrrolidine hydrochloride (7.03 mmol) was added in portions and the mixture refluxed for 24 h. Finally the slightly brown solution was cooled to room temperature and filtered. The solid remainders were washed thoroughly with acetone and the solvent of the combined filtrates removed in vacuo. The crude product was purified by column chromatography (SiO2; methanol/dichloromethane 1:25, v/v).to afford the product as a yellow oil (66%) which was used in subsequent reactions without further purification. 1H NMR (400 MHz, MeOD-d4) δ 1.84–1.87 (m; 4H; –(CH2)2–CH2–N–), 2.23 (q; 2H; 3J = 7.0 Hz; –CH–CH2–), 2.70 (m; 4H; CH2–N–CH2–), 2.95 (t; 2H; 3J = 5.8 Hz; –N–CH2–), 3.44 (t; 2H; 3J = 6.5 Hz; –S–CH2–), 4.14 (t; 2H; 3J = 5.5 Hz; –O–CH2–), 6.50 (t; 1H; 3J = 7.8 Hz; –CH–CH2–), 6.89/7.13 (AA'BB'; 4H; 3J = 8.8 Hz; Phenyl-H), 7.01 (dd; 1H; 3J = 7.5 Hz, 4J = 1.5 Hz; H9), 7.24 (dt; 1H; 3J = 7.5 Hz, 4J = 1.5 Hz; H7), 7.31 (dt; 1H; 3J = 7.5 Hz, 4J = 1.5 Hz; H8), 7.65 (d; 1H; 3J = 7.5 Hz; H6). 13C NMR (MeOD-d4, 100 MHz) δ 22.3 (2 × CH2), 26.0 (CH2), 42.7 (CH2), 53.7 (2 × CH2–N), 54.0 (CH3), 54.2 (CH2–N), 65.7 (CH2–O), 113.4 (2 × CH), 126.8 (CH), 126.8 (CH), 127.3 (CH), 128.3 (2 × CH), 130.0 (CH), 134.0 (CH), 134.4 (C), 142.9 (C), 145.2 (C), 157.8 (C–O)(M+)C22H25NOS (351.51)
2,3-Dihydro-8-methoxy-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 11b
Preparation as above from 2,3-dihydro-5-(4-hydroxyphenyl)-8-methoxy-benzo[b]thiepin 9b. The crude product was purified by column chromatography (SiO2; methanol/dichloromethane 1:25, v/v) to afford the product as a yellow oil (74%) which was used in subsequent reactions without further purification. 1H NMR (400 MHz, MeOD-d4) δ 1.84–1.88 (m; 4H; –(CH2)2–CH2–N–), 2.25 (q; 2H; 3J = 7.2 Hz; –CH–CH2–), 2.72 (m; 4H; CH2–N–CH2–), 2.97 (t; 2H; 3J = 5.5 Hz; –N–CH2–), 3.44 (t; 2H; 3J = 7.0 Hz; –S–CH2–), 3.85 (s; 3H; OCH3), 4.14 (t; 2H; 3J = 5.8 Hz; –O–CH2–), 6.42 (t; 1H; 3J = 7.8 Hz; –CH–CH2–), 6.88 (1H merged; H7), 6.88/7.13 (AA'BB'; 4H; 3J = 8.8 Hz; Phenyl-H), 6.93 (d; 1H; 3J = 8.5 Hz, H6), 7.22 (d; 1H; 4J = 2.5 Hz, H9) 13C NMR (MeOD-d4, 100 MHz) δ 22.3 (2 × CH2), 26.2 (CH2), 42.6 (CH2), 53.7 (2 × CH2–N), 54.0 (CH3), 54.1 (CH2–N), 65.6 (CH2–O), 113.2 (CH), 113.3 (2 × CH), 118.5 (CH), 126.0 (CH), 128.3 (2 × CH), 130.9 (CH), 134.7 (C), 135.6 (C), 137.1 (C), 142.7 (C), 157.7 (C–O), 158.1 (C–O).
4-Bromo-2,3-Dihydro-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 12a
To a solution of 2,3-dihydro-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 11a (2.11 mmol) in dry dichloromethane (30 mL) at − 10°C was added in small portions pyridiniumbromide perbromide (2.11 mmol) at such a rate, that the reagent was readily dissolved before the next addition. The reaction mixture was then stirred at room temperature for 8 h. A solution of sodium hydrogen carbonate (10%, 50 mL) was added and the aqueous layer extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with water and brine (50 mL each), dried over MgSO4 and the solvent removed under reduced pressure. The crude yellow liquid (containing one equivalent of pyridine) was purified by column chromatography (SiO2; methanol/dichloromethane 1:25, v/v) to afford the product as a yellow oil (82%), which was used in subsequent reactions without further purification. IR νmax (film) cm− 1 2787 (s), 1606 (s; C = C); 1H NMR (400 MHz, MeOD-d4) δ 1.84–1.88 (m; 4H; –(CH2)2–CH2–N–), 2.72 (m; 4H; CH2–N–CH2–), 2.80 (t; 2H; 3J = 6.5 Hz; –CH2–CH2–S–), 2.97 (t; 2H; 3J = 5.5 Hz; –N–CH2–), 3.59 (t; 2H; 3J = 6.5 Hz; –S–CH2–), 4.15 (t; 2H; 3J = 5.5 Hz; –O–CH2–), 6.92/7.13 (AA'BB'; 4H; 3J = 8.8 Hz; Phenyl-H), 6.90–6.92 (m; 1H; H9), 7.20–7.27 (m; 2H; H7, H8), 7.61–7.64 (m; 1H; H6); 13C NMR (MeOD-d4, 100 MHz) δ 22.3 (2 × CH2), 37.6 (CH2), 40.5 (CH2), 53.7 (2 × CH2–N), 54.1 (CH2–N), 65.7 (CH2–O), 113.1 (2 × CH), 122.4 (C–Br), 127.3 (CH), 127.3 (CH), 130.0 (CH), 130.3 (2 × CH), 132.8 (C), 134.1 (C), 134.2 (CH), 141.2 (C), 146.0 (C), 157.7 (C–O).
4-Bromo-2,3-dihydro-8-methoxy-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 12b
Preparation from 2,3-dihydro-8-methoxy-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 11b as described above. The crude material was purified by column chromatography (SiO2; methanol/dichloromethane 1:25, v/v) to afford the product as a yellow oil (74%) which was used in subsequent reactions without further purification. IR νmax (film) cm− 1 2784 (s), 1606 (s; C = C); 1H NMR (400 MHz, MeOD-d4) δ 1.85–1.88 (m; 4H; –(CH2)2–CH2–N–), 2.73 (m; 4H; CH2–N–CH2–), 2.82 (t; 2H; 3J = 6.5 Hz; –CH2–CH2–S–), 2.97 (t; 2H; 3J = 5.5 Hz; –N–CH2–), 3.58 (t; 2H; 3J = 6.5 Hz; –S–CH2–), 3.80 (s; 3H; OCH3), 4.16 (t; 2H; 3J = 5.5 Hz; –O–CH2–), 6.80–6.82 (m; 2H; H6, H7), 6.91/7.12 (AA'BB'; 4H; 3J = 9.0 Hz; Phenyl-H), 7.18–7.19 (m; 1H; H9). 13C NMR (MeOD-d4, 100 MHz) δ 22.3 (2 × CH2), 37.7 (CH2), 40.2 (CH2), 53.7 (2 × CH2–N), 54.0 (CH3), 54.1 (CH2–N), 65.6 (CH2–O), 113.0 (2 × CH), 113.4 (CH), 118.7 (CH), 121.2 (C–Br), 130.3 (2 × CH), 131.1 (CH), 134.1 (C), 134.3 (C), 137.9 (C), 140.9 (C), 157.6 (C–O), 158.4 (C–O).
4-Bromo-8-fluoro-5-[4-(2-pyrrolidinylethoxy)-phenyl-2,3-dihydrobenzo[b]thiepin 10
Preparation from 9c (0.360 g, 1.32 mmol) as described above for the synthesis of compounds 12a–b. The solvent was removed under reduced pressure to give the bromide as a yellow oil which required no purification (91%). νmax (film): 3355 (OH), 1258 NMR, 731 NMR; 1H (400 MHz, CDCl3, Me4Si): δ 7.08 (2 H, d, J = 8.5), 6.84–6.63 (5 H, m, aromatic CH), 4.58 (2 H, t, J = 6.0, OCH2CH2), 3.00 (2 H, t, J = 6.0, OCH2CH2) ppm; 13C NMR(101 MHz, CDCl3, Me4Si): δ155.1 (C), 142.7 (C), 134.6 (C), 132.2 (C), 132.1 (CH), 129.3 (CH), 127.5 (CH), 121.4 (CH), 121.2 (CH), 115.3 (CH), 44.1 (CH2), 26.9 (CH2) ppm; 19F (376 MHz, CDCl3, Me4Si): − 113.9 ppm.
2,3-Dihydro-4-(-4hydroxyphenyl)-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 13a
Pd(PPh3)4 (0.035 mmol) was added to a solution of 4-bromo-2,3-dihydro-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 12a. (1.16 mmol), 4-hydroxyphenylboronic acid (1.74 mmol), and 2 M Na2CO3 (5.80 mmol) in THF (20 mL) and heated to reflux for 5–6 h. After cooling, the mixture was partitioned between water and ethyl acetate (40 mL each), filtered and extracted with ethyl acetate (3 × 40 mL). The combined organic layers were washed with water and brine (40 mL each), dried over MgSO4 and the solvent removed in vacuo.The orange product was purified by column chromatography (SiO2; methanol/dichloromethane 1:15, v/v) to afford the product which was recrystallised from MeOH (and few drops of DCM) at 4°C as white solid; (72%), M.p. 178–180°C (dec.); IR νmax (KBr) cm− 1 (3491w, br; –OH), 1608(s; C = C)1508,1239. 1H NMR (400 MHz, DMSO-d6) δ1.67–1.70 (m; 4H; –(CH2)2–CH2–N–), 2.54–2.57 (m; 6H; –CH2–CH2–S–, –CH2–N–CH2–), 2.81 (t; 2H; 3J = 5.6 Hz; –N–CH2–), 3.35 (t; 2H; 3J = 6.0 Hz; –S–CH2–), 3.99 (t; 2H; 3J = 5.5 Hz; –O–CH2–), 6.59 (d; 2H; 3J = 8.5 Hz; Phenyl-H), 6.68–6.72 (m; 4H; Phenyl-H), 7.00 (d; 2H; 3J = 8.5 Hz; Phenyl-H), 6.85 (dd; 1H; 3J = 8.5 Hz, 4J = 1.5 Hz; H9), 7.23 (dt; 1H; 3J = 7.5 Hz, 4J = 1.8 Hz; H7), 7.30 (dt; 1H; 3J = 7.5 Hz, 4J = 1.3 Hz; H8), 7.61 (dd; 1H; 3J = 7.5Hz, 4J = 1.5 Hz; H6), 9.39 (s; 1H; –OH). 13C NMR (DMSO-d6, 100 MHz) δ 23.0 (2 × CH2), 34.3 (CH2), 42.1 (CH2), 54.0 (2 × CH2–N), 54.9 (CH2–N), 66.0 (CH2–O), 113.7 (2 × CH), 114.9 (2 × CH), 127.5 (CH), 128.3 (CH), 130.4 (2 × CH), 130.9 (CH), 131.8 (2 × CH), 132.0 (C), 133.2 (C), 134.4 (CH), 134.5 (C), 136.9 (C), 140.4 (C), 148.0 (C), 156.1 (C–O), 156.7 (C–O). C28H29NO2S. 0.3 CH2Cl2 Calculated: C: 72.46H: 6.36, N: 2.99; Found: C: 72.15, H: 6.40, N: 2.86. HRMS: Found: 444.1999 (M++1); C28H30NO2S requires 444.1997.
2,3-Dihydro-4-hydroxyphenyl-8-methoxy-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo [b] thiepin 13b
Preparation as above from 4-bromo-2,3-dihydro-8-methoxy-5-[4-(2-pyrrolidinylethoxy)-phenyl]benzo[b]thiepin 12b. The dark red crude product was purified by column chromatography (SiO2; methanol / dichloromethane 1:15, v/v) to afford the product as a beige solid, which was dissolved in DCM, treated with 6M HCl and set aside at 4°C to achieve crystallisation of product as the hydrochloride salt. The product was obtained as a white solid; (58%), M.p. 246–249°C (dec.); IR νmax (KBr) cm− 1 3743(w, br; –OH)1608, (s; C = C)1508,1239. 1H NMR (400 MHz, DMSO-d6) δ 1.92 (br; 4H; –(CH2)2–CH2–N–), 2.55 (t; 2H; 3J = 6.5 Hz;–CH2–CH2–S–), 3.09 (br; 4H; –CH2–N–CH2–), 3.50 (m; 4H; –N–CH2–, –S–CH2–), 3.79 (s; 3H; –O–CH3), 4.23 (t; 2H; 3J = 4.8 Hz; –O–CH2–), 6.59 (d; 2H; 3J = 8.5 Hz; Phenyl-H), 6.74–6.76 (m; 5H; Phenyl-H), 6.89 (dd; 1H; 3J = 8.5 Hz, 4J = 2.5 Hz; H7), 6.99 (d; 2H; 3J = 8.0 Hz; Phenyl-H), 7.17 (d; 1H; 4J = 2.5 Hz; H9), 9.40 (s; 1H; –OH) 13C NMR (DMSO-d6, 100 MHz) δ 23.0 (2 × CH2), 34.4 (CH2), 41.9 (CH2), 54.0 (2 × CH2–N), 54.9 (CH2–N), 55.2 (CH3), 66.1 (CH2–O), 113.6 (2 × CH), 114.4 (CH), 114.9 (2 × CH), 118.7 (CH), 130.4 (2 × CH), 131.8 (2 × CH), 131.9 (CH), 132.2 (C), 134.4 (C), 134.6 (C), 136.7 (C), 139.4 (C), 140.0 (C), 156.0 (C–O), 156.6 (C–O), 157.7 (C–O). C29H31NO3S.0.3 CH2Cl2; Calculated:C: 65.70, H: 6.13, N: 2.63; Found: C: 65.76; H: 6.20, N: 2.58. HRMS: Found: 474.2107, (M++1), C29H32NO3S requires 474.2103.
1-{2-[4-(4-Bromo-8-fluoro-2,3-dihydro-1-benzo[b]thiepin-5-yl)-phenoxy]-ethyl} -pyrrolidine 12c
4-(4-Bromo-8-fluoro-2,3-dihydro-1-benzothiepin-5-yl)-phenol 10 (0.46 g, 1.32 mmol) was treated with potassium carbonate (0.91 g, 6.60 mmol) and 1-(2-chloroethyl)pyrroldine hydrochloride (0.34 g, 1.98 mmol) in acetone (20 ml) as above The crude product was purified by chromatography (2.5% methanol/dichloromethane) to give the product as a yellow oil (0.236 g, 40%). IR νmax (film)cm− 1: 2960, 1606 (C = C), 1508, 1472, 1246, 732; 1H (400 MHz, CDCl3, Me4Si): δ 7.35 (1 H, dd, J = 8.0, 1.5, C6H4), 7.14 (2 H, d, J = 8.5, C6H4), 6.86–6.91 (4 H, m, C6H4), 4.11 (2 H, t, avg. J = 6.0, OCH2CH2N–), 3.62, (2 H, t, J = 6.5, OCH2CH2), 2.91 (2 H, t, J = 6.0, OCH2CH2N–), 2.82 (2 H, J = 6.5, OCH2CH2), 2.62 (4 H, m, CH2NCH2), 1.81, (4 H, m, CH2CH2); 13C (101 MHz, CDCl3, Me4Si): δ 162.2 (C), 159.7 (C), 158.2 (C), 140.7 (C), 134.1 (C), 132.4(CH), 130.9 (CH), 123.0 (C), 121.5 (CH), 121.3 (CH), 115.5(CH), 115.3 (CH), 113.9 (CH), 66.8 (CH2), 54.9 (CH2), 41.6 (CH2), 38.4 (CH2), 23.4 (CH2) ppm; 19F (376 MHz, CDCl3, Me4Si): − 113.7 ppm; HRMS Found 448.0760; C22H24BrFNOS requires 448.0746.
4-{8-Fluoro-5-[4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-2,3-dihydro-1-benzo[b]thiepin-4-yl}-phenol 13c
Pd(PPh3)4 (48 mg, 31 mmol) was added to a solution of 1-{2-[4-(4-bromo-8-fluoro-2,3-dihydro-1-benzo[b]thiepin-5-yl)-phenoxy]-ethyl}-pyrrolidine 12c (138 mg, 310 μmol) in dry THF under nitrogen and stirred for 15 min. 4-Hydroxyphenylboronic acid (56 mg, 0.4 mmol) and 2 M sodium carbonate (0.77 ml, 1.55 mmol) was added and the solution refluxed for 6 h. The solution was cooled and acidified with 2 M hydrochloric acid. The aqueous layer was extracted with dichloromethane and the combined organic layers were washed with water (30 mL), brine (30 mL), dried over sodium sulfate and the solvent removed under reduced pressure. The product was purified by column chromatography (silica gel, 2.5% methanol/dichloromethane) to give the product as a brown solid (23 mg, 16%). νmax (KBr)cm− 1 3437 (OH), 1608 (C = C), 1508, 1247 (C = C); 1H (400 MHz, d-DMSO, Me4Si): δ 6.96 (2 H, d, J = 8.5, C6H3), 6.86–6.67 (7 H, m, aromatic CH), 6.56 (2 H, d, J = , 9.0,), 4.60 (2 H, t, avg. J = 6.2, OCH2CH2N–), 4.20 (2 H, t, J = avg. 5.0, OCH2CH2), 3.22 (6 H, m, 4 × CH2NCH2, 2 × CH2NCH2), 2.68 (4 H, m, CH2–CH2); 13C (101 MHz, CDCl3, Me4Si): δ 155.9 (C), 144.3 (C), 140.5 (C), 136.3(C), 135.7 (C), 135.6 (C), 134.1 (C), 132.2 (C), 131.6 (CH), 130.2 (CH), 129.9 (CH), 120.2, 119.9, 114.8, 144.6 (C–F), 114.4 (CH), 113.2 (CH), 65.6 (CH2), 53.8 (CH2), 53.7(CH2) 41.9 (CH2), 34.0 (CH2), 22.7 (CH2); 19F (376 MHz, CDCl3, Me4Si): − 117.17; Found 462.1914 C28H29NO2F S requires 462.1903.
2,3-Dihydro-8-hydroxy-4-hydroxyphenyl-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b] thiepin 14
A solution of 2,3-dihydro-8-methoxy-4-hydroxyphenyl-5-[4-(2-pyrrolidinylethoxy)-phenyl]-benzo[b]thiepin 13b (52.8 μmol) in dry DCM (5 mL) at 0°C, was added dropwise to BBr3 (0.26 mmol, 1 M solution in DCM) diluted with dry DCM (3 mL). After 10 h of stirring at 0°C the mixture was quenched with 10% (w/w) NaHCO3 (20 mL) and the dark precipitate dissolved in a mixture of EtOAc/MeOH 10:1 (v/v) (20 mL) with vigorous stirring. The aqueous phase was separated and extracted with this EtOAc/MeOH-mixture. The combined organic fractions were dried with MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (SiO2; methanol/dichloromethane 1:7, v/v) to separate the product as a solid; (62%). 1H NMR (400 MHz, DMSO-d6) δ 1.73 (s, br; 4H; –(CH2)2–CH2–N–), 2.53 (t; 2H; 3J = 6.0 Hz; –CH2–CH2–S–), 2.71 (s, br; 4H; –CH2–N–CH2–), 2.95 (s, br; 2H; –CH2–N–), 3.31 (t; 2H; 3J = 6.0 Hz; –S–CH2–), 4.03 (t; 2H; 3J = 5.1 Hz; –O–CH2–), 6.57 (d; 2H; 3J = 8.5 Hz; Phenyl–H), 6.67–6.73 (m; 5H; Phenyl–H), 6.64 (d; 1H; 3J = 8.5 Hz, H6), 6.97 (d; 2H; 3J = 8.5 Hz; Phenyl–H), 7.03 (d; 1H; 4J = 2.5 Hz; H9), 9.34 (s; 1H; –OH), 9.70 (s; 1H; –OH) 13C NMR (DMSO-d6, 100 MHz) δ 22.9 (2 × CH2), 34.5 (CH2), 41.6 (CH2), 53.9 (2 × CH2–N), 54.8 (CH2–N), 65.5 (CH2–O), 113.5 (2 × CH), 114.9 (2 × CH), 115.5 (CH), 120.4 (CH), 130.4 (2 × CH), 131.8 (2 × CH), 131.9 (CH), 132.4 (C), 134.1 (C), 135.0 (C), 136.9 (C), 138.2 (C), 138.9 (Aromatic C), 155.9 (C–O), 156.1 (C–O), 156.4 (C–O). HRMS Found 460.1959 (M++1); C28H30NO3S requires 460.1946.
Biochemical evaluation of activity:
Antiproliferation studies
All assays were performed in triplicate for the determination of mean values reported. Compounds were assayed as the free bases isolated from reaction. The human breast tumor cell line MCF-7 was cultured in Eagles minimum essential medium in a 95%O2/5% CO2 atmosphere with 10% fetal calf serum. The medium was supplemented with 1% non-essential amino acids. Cells were trypsinised and seeded at a density of 1.5 × 104 into a 96-well plate and incubated at 37°C, 95%O2/5% CO2 atmosphere for 24 h. After this time they were treated with 2 μL volumes of test compound which had been pre-prepared as stock solutions in ethanol to furnish the final concentration range of study, 1 nM–100 μM, and re-incubated for a further 72 h. Control wells contained the equivalent volume of the vehicle ethanol (1% v/v). The culture medium was then removed and the cells washed with 100 μL phosphate buffered saline (PBS) and 50 μL MTT added, to reach a final concentration of 1 mg/mL MTT added. Cells were incubated for 2 h in darkness at 37°C. At this point solubilization was begun through the addition of 200 μL DMSO and the cells maintained at room temperature in darkness for 20 min to ensure thorough colour diffusion before reading the absorbance. The absorbance value of control cells (vehicle teated) was set to 100% cell viability and from this graphs of absorbance versus cell density per well were prepared to assess cell viability and from these, graphs of percentage cell viability versus concentration of subject compound added were drawn.
Cytotoxicity studies
Human MCF-7 breast cancer cells were plated at a density of 1.5 × 104 per well in a 96-well plate, then incubated at 37°C, 95%O2/5% CO2 atmosphere for 24 h. Cells were treated with the compound of choice at varying concentrations (1 nM–100 μM), then incubated for a further 72 h. Following incubation 50 μL aliquots of medium were removed to a fresh 96-well plate. Cytotoxicity was determined using and LDH assay kit obtained from Promega, following the manufacturer's instructions for use. A 50 μL per well LDH substrate mixture was added and the plate left in darkness at room temperature for equilibration. Stop solution (50 μL) was added to all wells before reading the absorbance at 490 nm. A control of 100% lysis was determined for a set of untreated cells which were lysed through the addition of 20 μL lysis solution to the media 45 min prior to harvesting. Data was presented following calculation, as percentage cell lysis versus concentration of subject compound.
Estrogen receptor binding assay
ERα and ERβ fluorescence polarization based competitor assay kits were obtained from Panvera at Invitrogen Life Technologies. The recombinant ER(insect expressed, full length, untagged human ER obtained from recombinant baculovirus-infected insect cells) and the fluorescent estrogen ligand were removed from the − 80 C freezer and thawed on ice for 1 h prior to use. The fluorescent estrogen ligand(2 nM) was added to the ER (30 nM for ERα and 20 nM for ERβ) and screening buffer (100 mM potassium phosphate (pH 7.4), 100 μg/mL BGG, 0.02%NaN3 was added to make up to a final volume that was dependant on the number of tubes used (number of tubes (e.g. 50) × volume of complex in each tube (50 μL) = total volume (e.g.2500 μL). Test compound (1 μL, concentration range 1 nM) to 100μM) was added to 49 μL screening buffer in each borosilicate tube (6 mm diameter). To this 50 μL of the fluorescent estrogen/ER complex was added to make up a final volume of 100μL and final concentration range for the test compound of 0.01 nM to 1 μM. A vehicle control contained 1% (v/v) of ethanol and a negative control contained 50 μL screening buffer and 50 μL fluorescent estrogen/ER complex. The negative control was used to determine the polarisation value when no competitor was present (theoretical maximum polarization). The tubes were incubated in the dark at room temperature for 2 hours and were mixed by shaking on a plate shaker. The polarization values were read on a Beacon single-tube fluorescent polarization instrument with 485 nm excitation and 530 nm emission interference filters. For ERα and ERβ, graphs of anisotropy (mA) versus competitor concentration were obtained for determination of IC50 values.
Estrogenic activity: alkaline phosphatase assay
Following the procedure of Littlefield et al[Citation24], human Ishikawa cells were maintained in Eagle's Minimum Essential Medium (MEM containing 10% vol/vol fetal bovine serum (FBS) and supplemented with 100 U/mL penicillin and 10 μg/mL streptomycin, 2 mM glutamine and 1 mM sodium pyruvate. Twenty four hours before the start of the experiment, near confluent cells were changed to an estrogen-free medium (EFBM), A 1:1 mixture of phenol-free Ham's F-12 and Dulbecco's Modified Eagles Medium, together with the supplements listed above, and 5% calf serum, stripped of endogenous estrogens with dextran coated charcoal. On the day of the experiment, cells were harvested with 0.25% trypsin and plated in 96-well flat bottomed microtitre plates in EFBM at a density of 2.5 × 104 cells/well. Test compounds were dissolved in ethanol at 10− 3 M, diluted with EFBM (final concentration of ethanol 0.1%) and filter sterilised. After addition of the test compounds, (plated in 50 μL, added estradiol in 50 μL, and blank medium to give a final volume 150 μL) the cells were incubated at 37°C in a humidified atmosphere containing 95%O2/5% CO2 for 72 h. All experimental values were obtained in triplicate. The microtitre plates were then inverted and the growth medium removed. The plates were then rinsed by gentle immersion and swirling in 2 L of PBS (0.15 M NaCl, 10 mM sodium phosphate, pH 7.4). The plates were removed from the container, the residual saline in the plate was not removed, and the wash was repeated. The buffered saline was then shaken out, and the plate blotted on paper towel. The covers were replaced and the plates were placed at − 80 C for at least 15 min. then thawed at room temperature for 5–10 min. The plates were then placed on ice and 50μL ice cold solution containing 50 mM p-nitrophenyl phosphate, 0.24 mM MgCl2 and 1 M diethanolamine (pH 9.8) was added. The plates were warmed to room temperature (time zero), and the yellow colour from the production of p-nitrophenol was allowed to develop. The plates were monitored at 405 nm until maximum stimulation of the cells showed an absorbance of approximately 1.2.
Computational procedure
Ligand preparation:
Compound 13c was drawn using ACD/Chemsketch v10 [Citation25] and converted to a SMILES string. A single conformer was generated with a final MMFF optimisation step for refinement of the compound, using Omega v2.1 [Citation26].
Receptor preparation:
PDB entries 3ERT and 1QKN were downloaded from the Protein Data Bank (PDB). A single bridging water molecule held between Glu353 (Glu305) and Arg394 (Arg346) was retained in both isoforms. Addition and optimisation of hydrogen positions was carried out using MOE 2005.06 [Citation27] ensuring all other atom positions remained fixed.
Docking:
FRED v2.11 [Citation28] was utilized in this study to dock the conformer in both estrogen receptor isoforms. All default values were applied with rigid-body optimisation of each ligand pose with Chemgauss2. The docked complexes for both isoforms were imported to Sybyl v6.91 [Citation29] and a treated to a flexible ligand and active site docking run. The active site residues assessed previously using MOE 2005.06 were allowed to be flexible during the docking calculation. All other values were kept as default except the number of iterations, which was increased to 10,000. Finally the docked complex was optimised under the MMFF force field using Szybki v1.1 [Citation30], with optimisation of free rotor torsions and of polar hydrogen positions close to the ligand.
Results and discussion
Chemistry
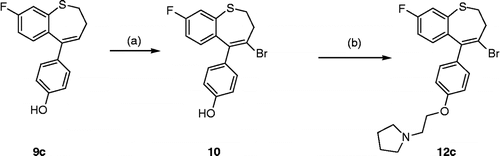
The synthesis of the benzothiepin type compounds is illustrated in Scheme . The phenylsulfanylbutyric acids 7a–c (obtained by alkylation of the corresponding thiols with ethyl 4-bromobutyrate) were cyclised with polyphosphoric acid to afford the benzothiepin-5-ones 8a–c respectively. Treatment of 8a–c sequentially with n-butyllithium and the THP protected 4-bromophenol followed by acid workup afforded the phenolic products 9a–c in good yield. Alkylation of the phenols 9a and 9b with 1-(2-chloroethyl)pyrrolidine, followed by bromination of the alkenes 11a and 11b with pyridinium hydrobromide perbromide afforded the vinyl bromide products 12a and 12b respectively. Subsequent Suzuki arylation of these bromides 12a–b with 4-hydroxyphenyl boronic acid resulted in isolation of the required benzothiepin products 13a and 13b in good yield (58–72%), (). The fluoro substituted product 12c required initial vinyl bromination of the phenol 9c and subsequent alkylation of the resulting bromide 10 to afford the required product 12c, (Scheme ). The diphenolic compound 14 was obtained in 62% yield by demethylation of 13b with boron tribromide in dichloromethane. This reaction required careful tlc monitoring as some C7- brominated product was also formed in the reaction. Alternative demethylation reagents such as pyridine hydrochloride and boron trifluoride dimethylsulfide were less successful in this reaction.
Scheme 1 Synthesis of Benzothiepins 13a–c and 14. Scheme reagents: (a) R = H, F; PPA, 110°C, 4h; R = OMe, Eaton's reagent, 80°C, 2h; (b) i nBuLi, 4-Br-C6H5-OTHP, THF; ii HCI, MeOH.(c) K2CO3, 1-(2-chloroethyl)pyrrolidine hydrochloride, acetone, 24h, 85°C;(d) PyHBr3, CH2 Cl2, 20°, 18th; (e) Pd(PPh3)3, 4-HOC6H5B(OH)2, Na2CO3(aq), THF; (f) BBr3, CH2Cl2.
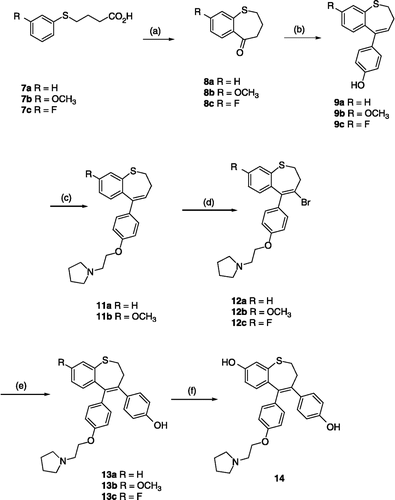
Table I. Antiproliferative effects and estrogen receptor binding affinities for benzothiepin type SERMs.
Scheme 2 Synthesis of Benzothiepin 12c. Scheme reagents: (a) PyHBr3, CH2Cl2, 20°, 18h; (b) K2CO3, 1-(2-chloroethyl)pyrrolidine hydrochloride, acetone, 24h, 85°C.
Biochemistry
The compounds were evaluated in a series of estrogen dependent in vitro assays which measured their affinity for the estrogen receptors and also their ability to act as functional antagonists or agonists of estrogen.
Antiproliferative activity in MCF-7 breast cancer cells
Compounds 13a–c, 14 containing the benzothiepin-derived molecular scaffold were initially assessed for their antiproliferative action using the ER expressing (ER dependent) human MCF-7 breast cancer cell line and the results are displayed in . These compounds are considerably more potent than tamoxifen, (IC50 = 4.120 μM) and the initial benzoxepin compound 6 (IC50 = 1.584 μM). The 8-fluoro substituted compound 13c proved to be the most active compound in the series with IC50 = 18.5 nM. The fluoro substituent at C-8 would prevent metabolic hydroxylation occurring at this position. The 8-unsubstituted compound 13a also displayed potent activity with IC50 = 69 nM. Compounds13b (methoxy substituted) and 14 (containing phenolic substituents on both Rings B and C) were moderately active (IC50 = 209 and 306 nM respectively). The replacement of the OH group on compound 14 (cLog P = 5.99) with lipophilic fluorine as in 13c (cLog P = 6.86) results in a decrease in the IC50 value from 306 nM to 18.5 nM indicating the positive effect of the fluorine on the antiproliferative activity. These results compare very favourably with the antiproliferative activity reported for tamoxifen [Citation31], fluorotamoxifen[Citation32,Citation33], benzocycloheptenes and related dibenzothiepins. Citation34-36.
In order to determine whether the antiproliferative effects elicited by the benzothiepin compounds was mediated through the ER, the most potent examples(compounds 13c and 14) were tested in hormone independent MDA-MB 231 breast cancer cells and displayed IC50 values of 120 nM and 1.47μM respectively. The value obtained for compound 13c in this assay suggests that that compound may function through alternative antiproliferative mechanisms, independent of the ER. The cytotoxicity of the compounds was also determined in the standard LDH assay as previously reported as we wished to confirm that the antiproliferative effects of the compounds were due to cytostasis rather than cellular necrosis. The compounds all demonstrated low cytotoxicity profiles suggesting that their action is cytostatic rather than cytotoxic, (). Typical values for cytotoxic induced antiproliferative effects for compound 13a at 10 μM is 8%, whereas value for tamoxifen is determined to be 24% at testing concentration of 10 μM. The cytotoxicity values obtained were less than that for tamoxifen for all compounds except for compound 13c (31% at 10 μM) which is one of the most potent antiproliferative compounds in the series evaluated. The corresponding values in the MDA-MB 231 assay for compounds 13c and 14 were 17% and 11% respectively again indicating a relatively low level of cytotoxicity for these benzothiepins.
Estrogen receptor binding studies
Estrogen receptor binding studies were carried out with ERα and ERβ using a fluorescence polarization procedure[Citation37]. The displacement of fluorescein labeled estradiol (fluoromone) in a competitive binding assay from the human recombinant full length receptor proteins ERα and ERβ expressed from baculovirus – infected insect cells by the synthesized ligands was observed as a decrease in polarization values. Compounds 13a–c and 14 containing the benzothiepin-derived molecular scaffolds were effective ERα ligands with IC50 values in the range 8–10 nM (), indicating that the benzothiepin sulfur is equally as effective as the benzoxepin oxygen in facilitating binding of the ligands to the ERα as also reported for benzoxathiins[Citation17]. The ERα and ERβ binding values for compounds 13a,13b and 14 were also confirmed in a competitive radiometric binding assay. The introduction of the more lipophilic fluorine at C-8 in compound 13c (cLog P = 6.86) resulted in considerably more potent activity in the MTT assay and ERα and ERβ binding assays than compound 14 (cLog P = 6.99). The benzothiepin-derived compounds were all potent ligands for ERβ with IC50 values in the range 0.7–6.5 nM. The twelve-fold selectivity of compound 13c for ERβ was attributed to the presence of the F substituent at position 8 in Ring B of the rigid structure (as discussed in the molecular modeling and docking section below) and compare very favourably with the ER binding data reported for fluorotamoxifen (IC50 5 μM)[Citation32]. The corresponding diphenolic compound 14 showed much lower selectivity with β/α ratio of 1.5 respectively which is closer to the β/α ratio value of 1.67 obtained for 4-hydroxytamoxifen in this experiment.
Estrogenic stimulation
The estrogen stimulation and antagonistic properties of the most active compounds 13c and 14 were determined in the estrogen bioassay which is based on the stimulation of alkaline phosphatase (AlkP) in the Ishikawa human endometrial adenocarcinoma cell line [Citation24].Compounds are tested as estrogen antagonists by their effect on the inhibition of estradiol stimulation in the Ishikawa cells in a dose-dependent manner. Compounds 13c and 14 were much more potent than tamoxifen as estrogen antagonists with IC50 values of 7.4 nM and 6.4 nM respectively and correlates with the potent ERα and ERβ binding observed for these compounds. The estrogenic stimulatory properties of these compounds could be determined in the Ishikawa cells by measuring the stimulation of alkaline phosphatase(AlkP) in these cells in the absence of estradiol. Compounds 13c and 14 displayed minimal stimulatory values of 0.2% and 3.3% respectively. The results from this AlkP assay for estrogen antagonism and stimulation provide useful information in selection of the structural features for optimum antiestrogenic activity without associated adverse estrogenic effect on tissues such as the uterus.
Molecular modelling
To rationalize the observed biological activity of the most active compound 13c, a flexible computational investigation was undertaken. Currently there are no crystal structures of human ERα/β with the same antagonist co-crystallized and thus to compare docking of the same ligand in different isoforms remains difficult. To overcome this problem receptor flexibility must be accounted for in the docking process. Utilising Flexidock (Sybyl6.91), both the ligand and active site residues were allowed to move flexibly during docking. Finally the resulting pose was optimised using Szybki v1.1. The resulting pose was selected and atomic interactions were analysed by Ligand Protein Contacts (LPC) software [Citation38]. The residues depicted are those that are known to be key in the binding process for both agonists/antagonists: Asp351, Glu353 and Arg394 (anchor the ligand in the active site), His524 (additionally important in ligand binding process). illustrates the interactions made by each ligand with both receptor isoforms.
Table II. Summary of key Ligand-Protein contactsa.
As illustrated in , compound 13c docks in a typical antiestrogenic manner when compared with 4-hydroxytamoxifen or raloxifene. What is immediately apparent is the inversion or flipping of the rings A and C placing the fluoro and hydroxy substituents at opposite ends. Mewshaw et al [Citation39] have observed this type of ‘ring-flipping’ previously due to the pseudosymmetry of the 2-phenylnaphthalene scaffold. Consequently, selectivity for ERβ was also observed with the series due to this A & C ‘ring-flipping’. In the case of the benzothiepin scaffold, the ligand docks in ERα forming a hydrogen bond with His524 whereas in ERβ none is observed. Instead, the hydroxy group of ring C forms a strong hydrogen bonding interaction with Ala350 and Glu353. This orientation within ERα reduces bonding with Asp351 compared with in ERβ as illustrated in .
Figure 2 Docked complex of compound 13c in active site of ERα and ERβ superposed.
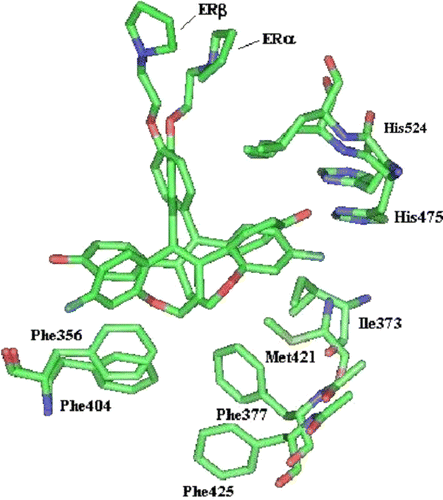
The substitution of Met421 (ERα) with Ile373 (ERβ) causes steric interaction with the ligand, supplementary to that imposed by Phe356 and Phe377, preventing the ligand from docking in the same orientation as observed with ERα. It has been suggested previously by a number of groups [Citation40,Citation41] that enhanced ERβ selectivity is achieved through a ligand that can differentiate between ERα Met421 and ERβ Ile373. Compound 13c exhibits these preferences and thus was observed to be ∼10-fold selective for ERβ.
Conclusion
In summary, this study demonstrates that benzothiepin derived molecular scaffolds can provide useful estrogen receptor modulators. The compounds exhibited potent activity in an antiproliferative assay against the MCF-7 breast cancer cell line and specificity in binding to the ERβ subtype. The potent β-specific binding of compound 13c is rationalized in a docking study which demonstrated the effect of the fluorine substituent in differentiation of ligand binding to the ER receptor subtypes. These results should facilitate the rational design of further ERβ subtype specific ligands.
Acknowledgements
We are very grateful to Professor Richard Hochberg at Yale University Medical School, for kindly facilitating the alkaline phosphatase experiments and for the generous gift of the Ishikawa cells. This work was supported through funding from the Trinity College IITAC research initiative (HEA PRTLI), with additional support for computational facilities from the Wellcome Trust.
References
- MacGregor JI, Jordan VC. Basic guide to the mechanisms of antiestrogen action. Pharmacol Rev 1998; 50(2)151–196
- Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. J Med Chem 2003; 46(6)883–908, 1. Receptor interactions
- Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. J Med Chem 2003; 46(7)1081–1111, 2. Clinical considerations and new agents
- Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 2006; 6(3)181–202
- Gustafsson JA. Estrogen receptor beta-a new dimension in estrogen mechanism of action. J Endocrinol 1999; 163(3)379–383
- Pearce ST, Jordan VC. The biological role of estrogen receptors alpha and beta in cancer. Crit Rev Oncol Hematol 2004; 50(1)3–22
- Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ERbeta expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer 2004; 11(3)537–551
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998; 95(7)927–937
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997; 389(6652)753–758
- Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001; 9(2)145–153
- Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell 2005; 18(4)413–424
- Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. Embo J 1999; 18(17)4608–4618
- Lloyd DG, Buenemann CL, Todorov NP, Manallack DT, Dean PM. Scaffold hopping in de novo design. Ligand generation in the absence of receptor information. J Med Chem 2004; 47(3)493–496
- Singh MM. Centchroman, a selective estrogen receptor modulator, as a contraceptive and for the management of hormone-related clinical disorders. Med Res Rev 2001; 21(4)302–347
- Labrie F, Labrie C, Belanger A, Simard J, Gauthier S, Luu-The V, Merand Y, Giguere V, Candas B, Luo S, Martel C, Singh SM, Fournier M, Coquet A, Richard V, Charbonneau R, Charpenet G, Tremblay A, Tremblay G, Cusan L, Veilleux R. EM-652 (SCH57068), a third generation SERM acting as pure antiestrogen in the mammary gland and endometrium. J Steroid Biochem Mol Biol 1999; 69(1-6)51–84
- Jain N, Kanojia RM, Xu J, Jian-Zhong G, Pacia E, Lai MT, Du F, Musto A, Allan G, Hahn D, Lundeen S, Sui Z. Novel chromene-derived selective estrogen receptor modulators useful for alleviating hot flushes and vaginal dryness. J Med Chem 2006; 49(11)3056–3059
- Kim S, Wu JY, Birzin ET, Frisch K, Chan W, Pai LY, Yang YT, Mosley RT, Fitzgerald PM, Sharma N, Dahllund J, Thorsell AG, DiNinno F, Rohrer SP, Schaeffer JM, Hammond ML. Estrogen receptor ligands. II. Discovery of benzoxathiins as potent, selective estrogen receptor alpha modulators. J Med Chem 2004; 47(9)2171–2175
- Sibley R, Hatoum-Mokdad H, Schoenleber R, Musza L, Stirtan W, Marrero D, Carley W, Xiao H, Dumas J. A novel estrogen receptor ligand template. Bioorg Med Chem Lett 2003; 13(11)1919–1922
- Wallace OB, Lauwers KS, Dodge JA, May SA, Calvin JR, Hinklin R, Bryant HU, Shetler PK, Adrian MD, Geiser AG. A selective estrogen receptor modulator for the treatment of hot flushes. J Med Chem 2006; 49(3)843–846
- Lloyd DG, Hughes RB, Zisterer DM, Williams DC, Fattorusso C, Catalanotti B, Campiani G, Meegan MJ. Benzoxepin-derived estrogen receptor modulators: a novel molecular scaffold for the estrogen receptor. J Med Chem 2004; 47(23)5612–5615
- Sarkhel S, Sharon A, Trivedi V, Maulik PR, Singh MM, Venugopalan P, Ray S. Structure-based drug design: Synthesis, crystal structure, biological evaluation and docking studies of mono- and bis-benzo[b]oxepines as non-steroidal estrogens. Bioorg Med Chem 2003; 11(23)5025–5033
- Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation: Estrogen receptor alpha and estrogen receptor beta: Regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res 2000; 2(5)335–344
- Nioche JYD, J., Festal D. Synthesis and structure-activity relationships of new ACAT inhibitors. Eur J Med Chem 1995; 30(5)377–385
- Littlefield BA, Gurpide E, Markiewicz L, McKinley B, Hochberg RB. A simple and sensitive microtiter plate estrogen bioassay based on stimulation of alkaline phosphatase in Ishikawa cells: Estrogenic action of delta 5 adrenal steroids. Endocrinology 1990; 127(6)2757–2762
- ACD/Chemsketch v10: Advanced Chemistry Labs., http://wwwacdlabscom/download/chemskhtml
- OMEGA 2.1: distributed by Openeye Scientific Software
- Molecular Operating Environment (MOE): Developed and distributed by Chemical Computing Group., (http://wwwchemcompcom)
- FRED (version 2.11): Developed and distributed by Openeye Scientific Software., http://wwweyesopencom
- Sybyl6.91: Distributed by Tripos Inc http://wwwtriposcom
- Szybki: Developed and distributed by Openeye Scientific Software., http://wwweyesopencom
- Teo CC, Kon OL, Sim KY, Ng SC. Synthesis of 2-(p-chlorobenzyl)-3-aryl-6-methoxybenzofurans as selective ligands for antiestrogen-binding sites. Effects on cell proliferation and cholesterol synthesis. J Med Chem 1992; 35(8)1330–1339
- Shani J, Gazit A, Livshitz T, Biran S. Synthesis and receptor-binding affinity of fluorotamoxifen, a possible estrogen-receptor imaging agent. J Med Chem 1985; 28(10)1504–1511
- Yang DJ, Tewson T, Tansey W, Kuang LR, Reger G, Cherif A, Wright KC, Moult RG, Tilbury RS, Chu K, Kim EE, Wallace S. Halogenated analogues of tamoxifen: Synthesis, receptor assay, and inhibition of MCF7 cells. J Pharm Sci 1992; 81(7)622–625
- McCague R, Leclercq G, Jordan VC. Nonisomerizable analogues of (Z)- and (E)-4-hydroxytamoxifen. Synthesis and endocrinological properties of substituted diphenylbenzocycloheptenes. J Med Chem 1988; 31(7)1285–1290
- Acton D, Hill G, Tait BS. Tricyclic triarylethylene antiestrogens: Dibenz[b,f]oxepins, dibenzo[b,f]thiepins, dibenzo[a,e]cyclooctenes, and dibenzo[b,f]thiocins. J Med Chem 1983; 26(8)1131–1137
- Catherino WH, Wolf DM, Jordan VC. A naturally occurring estrogen receptor mutation results in increased estrogenicity of a tamoxifen analog. Mol Endocrinol 1995; 9(8)1053–1063
- Lloyd DG, Smith HM, O' Sullivan T, Zisterer DM, Meegan MJ. Synthesis, Structure-Activity Relationships and Antagonistic Effects in Human MCF-7 Breast Cancer Cells of Flexible Estrogen Receptor Modulators. Medicinal Chemistry 2005; 1(4)335–353
- Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999; 15(4)327–332
- Mewshaw RE, Edsall RJ, Jr, Yang C, Manas ES, Xu ZB, Henderson RA, Keith JC, Jr, Harris HA. ERbeta ligands. 3. Exploiting two binding orientations of the 2-phenylnaphthalene scaffold to achieve ERbeta selectivity. J Med Chem 2005; 48(12)3953–3979
- Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, Collini MD, Miller CP, Dinh T, Henderson RA, Keith JC, Jr. Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-beta ligands. J Med Chem 2004; 47(21)5021–5040
- Yang C, Edsall R, Jr, Harris HA, Zhang X, Manas ES, Mewshaw RE. ERbeta ligands. Part 2: Synthesis and structure-activity relationships of a series of 4-hydroxy-biphenyl-carbaldehyde oxime derivatives. Bioorg Med Chem 2004; 12(10)2553–2570