Abstract
The flip regression procedure that we used earlier for handling the xanthones system has been applied to phenylaminoquinazoline analogues. It is known that the substituents at the 6- and 7- positions of the polycyclic system have been identified as the most important structural features. The steric as well as the electrostatic interactions proved to be the most important for the inhibitory effect. In this contribution it is shown that the orientation of nodes in their occupied π orbitals, and also the energies of these orbitals explains a further large portion of the variance in their inhibitory activity.
Introduction:
It is accepted that there is a critical need for new targets, in addition to DNA, for anticancer drug development [Citation1]. Epidermal growth factor receptor (EGFR) that has been identified as a kind of protein tyrosine kinase (PTK) and has been demonstrated to be related to many human cancers such as breast and liver cancers [Citation2–4], leading many to believe that EGFR is an attractive target for anti-tumor drug discovery [Citation5].
In the past few years, quantitative structure-activity relationships (QSAR) for different groups of compounds, which have been evaluated as EGFR inhibitors, have been reported [Citation6–9].
It has been demonstrated that in most cases the orientations of nodes in π -like orbitals of aromatic molecules are a critically important feature in understanding their activity. This was first found in phenylalkylamine hallucinogens [Citation10], carbonic anhydrase, trypsin, thrombin and bacterial collagenase inhibitors [Citation11], tryptamine hallucinogens [Citation12] as well as polychlorodibenzofurans [Citation13]. The present contribution extends this to some quinazoline analogues.
A QSAR and 3D QSAR of 134 structurally diverse inhibitors of the EGFR was recently reported, using comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA), in which the steric and electrostatic interactions proved to be the most important for the inhibitory effect [Citation14].
In this contribution it is hoped to improve the correlation by including the nodal orientations. The calculation of nodal orientation is done with the program NODANGLE [Citation15], which has been described previously. NODANGLE calculates the angle between the nodes in π-like orbitals and a reference point on the aromatic ring. NODANGLE works by for each ring analytically least-squares fitting the coefficients of the pz orbitals on the ring atoms to those of the degenerate HOMO and LUMO of benzene. The 10 highest occupied and 10 lowest unoccupied orbitals of the compound in question are searched, and an error term is calculated for each - a scaled sum of squares of the difference between the coefficients of the pz orbitals of the compound and those of benzene with the same nodal orientation as the ring in question. For an exact match to benzene this error term is zero. For an exact match for an orbital of the wrong symmetry it is unity. NODANGLE prints out angles and orbital energies for those orbitals for which the error term is less than 0.5. These are π-like orbitals, and provided the error term is small have nodal orientations that closely match benzene of the appropriate nodal orientation. This calculation is done for each of the three rings of the quinazoline analogues.
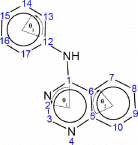
For the quinazoline, calculating the angles in the three rings can be accomplished in one MOPAC calculations by entering the atom as numbered in . The three rings are 6-membered rings numbered 1–6, 5–10 for ring 1 and 2, respectively. Ring 3 is also a 6-membered ring numbered 12–17. The angles calculated by NODANGLE are then Θ1, Θ2 and Θ3 in that figure, measured at atoms 1, 5 and 12 respectively.
Figure 1. Numbering of quinazolines skeleton used in the HyperChem and NODANGLE calculations and angles used in the interpretations. Angles shown are for compound 1. The angle subscript indicates the ring number.
summarizes the quinazoline-inhibitory activity of 63 analogues [Citation14] expressed as log IC50, where IC50 is the effective concentration of the compound required to inhibit by 50% the phosphorylation of a 14-residue fragment of phosphorylase Cγ − 1 (prepared from A431 human epidermoid carcinoma cells through immunoaffinity chromatography) by EGFR.
Table I. Structures and activities (log 1/IC50) of 4-(3-bromo-aniline)- 6,7-dimethoxyquinazoline analogues.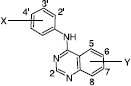
A problem arises from the symmetry of the parent molecule; to deal with this problem we use the program FLIPSTEP, a component of the MARTHA [Citation16] statistical package, which has been described previously [Citation16,17]. The phenyl ring in our system has symmetry, and the two o- positions, and also the two m- positions are related. We refer to these as flippable, and FLIPSTEP calculates regressions for all possible combinations of each flippable descriptor (a property of one of the two o- or m- substituents) exchanged with its flippable partner (the corresponding property of the other of the two), and selects that combination with the regression with the best Fisher F-ratio, after eliminating descriptors that are either collinear with other descriptors or are of poor statistical significance. In the present case only two flippable variables are present, and these are the angles the nodes in the HOPO and LUPO in the phenyl ring make with the 1-position of the phenyl ring. Flipping in this case consists of changing the signs of these angles.
Calculations
The molecules were setup with HyperChem [Citation18] and optimized at the AM1 level with MOPAC 6 [Citation19]. An AM1 optimization was considered adequate for these compounds, as AM1 was developed and parameterized for common organic structures, and also because the calculated angles (but not the orbital energies) are extremely insensitive to the level of theory. A NODANGLE calculation was run on the MOPAC output file to identify the relevant orbital and obtain the angles and corresponding orbital energies. The angles and orbital energies were correlated with the activities taken from the literature [Citation14] with the program FLIPSTEP.
The classical descriptors LDI, MEANQ and solvation energy were calculated with Mopac 93, while the diagonal components of the polarizability tensor were obtained from Mopac 6. MEANQ is the mean absolute Mulliken charge calculated over all atoms in the compound, while LDI (local dipole index) is the mean of the absolute difference in Mulliken charge between bonded pairs of atoms, calculated over all bonded pairs of atoms in the molecule. Both are measures of charge polarization in the molecule.
Results
In this study the HOP (highest occupied π orbital) is not identical to HOMO and also LUP (lowest unoccupied π orbital) is not identical to LUMO. In general we restrict our attention to π – like orbitals, and in particular, to those four orbitals that most resemble the degenerate HOMO and LUMO of benzene. SHOP and SLUP refer to the energies of the second highest occupied and second lowest unoccupied π-orbital respectively, and they are not necessarily the same for all rings, hence HOP1, HOP2 and HOP3 refer to orbital energies for the relevant orbitals for the three rings of the quinazoline analogues. and summarizes the orbital energies and angles and other variables of the compounds in Table 1.
Table IIA. Orbital energies (eV) and angles (°) of the compounds in Table I.
Table IIB. Table IIA continued.
NODANGLE does not print out values of angles or energies for non π – like orbitals for which the error term for the angle exceeds 0.5, or for which coefficients of s, px or py atomic orbitals are large compared with those of pz. summarizes the calculated angles and their error terms and the classical descriptors. The different descriptors used in this study are summarized in .
Table III. Calculated angles and their error terms.
Table IV. Calculated classical descriptors.
Table V. Descriptors used in this study
The best model as given in [Citation7] for quinazoline analogues was:
where n is the number of compounds used in the fit, R2 is the squared correlation coefficient, S is the standard deviation, and Q2 is the square of the multiple correlation coefficients based on the leave-one-out residuals. The indicator variable I = 1 is for 6,7-OMe derivatives. B1Y,7 is the steric effect (y- substituent at 7-position). B1X,3 is the same but for x- substituent at the 3-position of the 4-phenylamino moiety. σ− Y are electron donating groups as y-substituent. ClogP is the hydrophobicity.
The nodal angles are converted to sin 2θH, cos 2θH, sin 4θL and cos 4θL and then θH and θL are removed from the variables set. As a result, we will have a set of variables consists of: HOP1, SHOP1, LUP1, SLUP1, HOP2, SHOP2, LUP2, SLUP2, HOP3, SHOP3, LUP3, SLUP3, C2θ1H, S2θ1H, C2θ2H, S2θ2H, C2θ3H, S2θ 3H, C4θ1L, S4θ 1L, C4θ2L, S4θ2L, C4θ3L, S4θ3L. These variables are defined in .
NODANGLE did not print out a value for either the energy of SHOP1 for compound 48 or the energy of SHOP3 for compounds 15 and 16 because the error term was larger than 0.5. Hence, these variables were excluded from the regression analysis.
A FLIPSTEP calculation, that performs a backward-stepwise variable selection, was carried out using the default setting of VIFMAX value of 35. VIFMAX is the criterion for excluding variables that are collinear with other variables in the regression. The variance inflation factor (VIF) is defined for each independent variable i as 1/(1-Ri2), where Ri2 is the R2 for independent variable i regressed on all of the other independent variables. VIFMAX is the value of VIF above which a variable will be removed from the regression early in the procedure. In general a value of VIF above 10 is cause for concern. By default VIFMAX is set to 35. With this value, the maximum VIF in the final equation is usually much less than 35.
This set of quinazolines separates into two parts (symmetry wise): the singly-substituted quinazoline ring system, and the singly substituted phenyl ring. The quinazoline ring system has no vertical mirror planes or axes. Hence, ring 1 cannot be flipped into ring 2. Thus for this part of the molecule flip regression is not applied. The singly substituted phenyl ring has C2v symmetry, so flip regression is applicable to this. Thus only ring 3 should be flipped. Applying FLIPSTEP on this model resulted in removing S4θ2L, C4θ2L, LUP2 and SLUP1 due to colinearity. C4θ1L, HOP1, LUP1, C2θ1H and S2θ1H were deleted because they are statistically insignificant. FLIPSTEP stepwise regression gives:
Here, F is the Fisher variance ratio. The numbers in parentheses are Student's t values; a value greater than approximately 2 is indicative of significance at the 0.05 level. shows the progress of FLIPSTEP for this model.
Table VI. progress Of Flipstep For Quantum Descriptors.
Multi-linear regression analysis was carried out on the flipped variables selected by FLIPSTEP regression using the program MULTLR from the Martha package [Citation16]. Multilinear regression analysis applied to all variables in without removing the variables with high significance leads to precisely the values given by FLIPSTEP. Removal of the two variables of poor significance (C2θ3H and S2θ3H) leads to Equation (3). For example, C2θ3H and its flippable partner S2θ3H are of very poor significance, and their removal is justified. These variables are not independent, but are orthogonal to their partner terms. If only one of them were removed because of insignificance it would become impossible to carry out the prediction, because there is nothing to be flip into. If FLIPSTEP is to be used predicatively, then each variable and its flippable partner should be retained if one of them is significant. Removing one or other of the sine or cosine terms would force the angle to be either 0 or 90 degrees, which is not justifiable. The angle may well be 0 or 90 degrees as far as it can be determined, within the confidence limits set, but saying that the optimum angle is 0 is not the same thing as saying the angle does not matter. Deleting S2θ3H and retaining C2θ3H would have the effect of forcing the angle θ3H to be 0 degrees, rather than its maximum likelihood value of approximately 4°. In the present case both variables are deleted, because of their very poor significance, and the functions of θ3L, which are both very highly significant are retained. In regression analysis it is usual to delete variables that are not significant, usually taking the 0.05 significance level as the criterion. In the special case of flip regression we consider the flippable pairs as a whole, deleting them only if both are nonsignificant, as in the case of 2θ3H they are.
Multilinear regression analysis shows that SLUP3 and S4θ3L are the most critical variables, by the magnitude of the standardized regression coefficient and Student's t criteria respectively. Both the HOP and LUP orbital energies on ring 3 are very significant. The orientation of the node in the HOP orbital present on ring 3 is not very significant while the orientation of the node in the LUP orbital present on ring 3 is very highly significant, and is in fact the most significant term in the regression. The orientation of the nodes in the occupied and virtual orbitals present on rings 1 and 2, except for θ2H, are not very significant. Regression of Log 1/IC50 with the previous variables but deleting the nonsignificant θ3H terms gives:
P is the significance level based on a randomization test, using the Martha routine FLIPRAND. This procedure involves repeatedly running randomization of dependent variable followed by flip regression 1000 times, preserving the independent variables. The correlation coefficient for each randomization is saved, and the significance, calculated after Fisher normalization. In contrast to Equation (2) the coefficients given here are the standardized regression coefficients. Their magnitudes are a measure of the relative importance of the contributions of the terms in the model to Log 1/IC50. In applying Equation 3 it must be remembered that not the raw descriptor values should be used, but rather the values standardized to zero mean and unit variance. It should be noted that in contrast to Equation (1) no compound has here been deleted. Deleting the two insignificant terms, leads to very little deterioration of the model; it gives a correlation coefficient of 0.9431 that is very close to that obtained when retaining these variables in Equation (2), 0.9450, which justifies our choice for removing these variables.
Equation (2) and Table III show that R2 obtained from FLIPSTEP is the same as that obtained from multilinear regression analysis. The quality of the correlations is quite good and in Equation (3) we have about 6 points per variable, which is well within acceptable limits [Citation20–22]. This entire regression was done with only the orbital energies and nodal angles, and could probably be substantially improved by including classical variables.
Including classical variables
Some classical descriptors were added to the previous set of variables. These variables are LDI, MEANQ, polarizabilities and solvation energies. These variables are shown in Table IV. Flip regression was applied on the new set of quantum and classical variables. Adding the classical variables would result in a set of 28 variables defined in Table V. The variables that entered the FLIPSTEP calculations are: HOP1, LUP1, SLUP1, HOP2, SHOP2, LUP2, SLUP2, HOP3, LUP3, SLUP3, C2θ1H, S2θ1H, C4θ1L, S4θ1L, C2θ2H, S2θ2H, C4θ2L, S4θ2L, C2θ3H, S2θ3H, C4θ3L, S4θ3L, MEANQ, LDI, Pxx, Pyy, Pzz and SE. These variables are then flipped as described above. Applying FLIPSTEP stepwise regression resulted in deleting S4θ2L, C4θ2L, LUP2, SLUP1, MEANQ and HOP2 due to colinearity. S2θ1H, Pyy, SE, HOP1, S4θ1L, SLUP2, LDI, LUP1, C2θ2H and HOP3 were deleted because they are statistically insignificant. Flip regression with quantum and classical descriptors gives:
shows the progress of FLIPSTEP for this model.
Adding the classical variables to the set of variables improves R2 value, but only marginally. The best descriptor is still SLUP3. LUP3 is highly significant but HOP3 is not significant now. The orientation of the node in the LUP orbital present on ring 1 is very significant while the orientation of the node in the HOP orbital present on rings 2 and 3 is not very significant..Pxx and Pzz which are the polarizability tensor component in the directions of highest and lowest polarizability (and highest and lowest inertia) are very highly significant. We have one fewer variables than in Equation (2).
Finally, following the same argument when performing multilinear regression for the quantum descriptors, a multi-linear regression analysis was carried out on Log 1/IC50 with the variables in , without removing the variables with poor significance (S2θ3H and C4θ3L), because their flippable partners are very significant.
Table VII. progress of FLIPSTEP for quantum and classical descriptors.
Multiple regression analysis shows that SLUP3 is the most critical variable. LUP3 has very high significance while SHOP2 has a low significance, as measured by the standardized coefficients in Equation (5). The orientations of the nodes in the LUP orbital in rings 1 and 3 have a very high significance. Also, Pzz and Pxx have high significance. Regression of Log 1/IC50 with the previous variables gives:
As with Equation (3) the coefficients are the standardized values.
Equations (4) and (5) show that R2 obtained from FLIPSTEP the same as that obtained from multilinear regression analysis. In this equation, unlike Equation (3), no terms have been deleted due to non-significance. Equation (5) gives an R2 of 0.9017, compared with 0.8895 for Equation (3), at the expense of increasing the number of descriptors by 1.
shows the flip status and flip significance from FLIPSTEP calculations carried out on quantum variables alone and that carried on quantum and classical variables together. A value of the significance greater than 0.05 is indicative that flipping the corresponding compound makes little difference to the quality of the regression. A flip status of 1 indicates that the compound has not flipped in the final regression, and − 1 that it has. This has only relative significance, and flipping all of the compounds in a flip regression has no effect.
Table VIII. Flip status and flip significance for Equations (2) and (4).
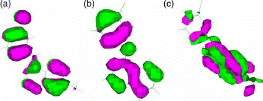
shows HOMO orbitals for three sample compounds of the 63 compounds. HOMO orbitals for the 63 compounds are similar to the molecular orbitals shown in either (a), (b) or (c). As shows, the pz orbitals of ring 3 looks more similar to benzene π-orbitals than those for rings 1 and 2. This enhances the high coefficient for ring 3 LUP orbitals.
Figure 2. HOMO orbitals for quinazolines.
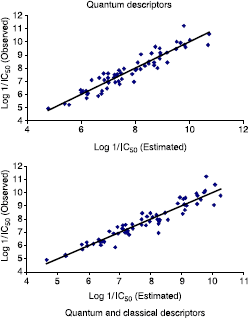
summarizes the observed activity as well as the estimated activity according to the multilinear regression carried out on quantum variables alone and that carried on quantum and classical variables together, while shows the plot of the logarithm of the observed activity against the logarithm of the estimated activities in both cases.
Table IX. Observed Log 1/IC50 versus estimated.
Figure 3. Observed Log 1/IC50 versus estimated for regression analysis carried out for quantum descriptors and that carried out for quantum and classical descriptors.
Comparison with other QSAR studies of quinazoline analoges
The results that we obtained in this study are better than the results obtained in [Citation7], in which number of compounds are 51 as reported in Equation (1) while we include all the compounds, also the R2 value (0.9011) that we obtained is much higher than they obtained (0.852). In [Citation14], the steric as well as the electrostatic interactions proved to be the most important for the inhibitory effect and this is represented qualitatively while we represent these results quantitatively.
Conclusions
The nodal orientation terms have a powerful explanatory value in that they account for much more of the variance in activity than is possible using the classical descriptors alone. Were it not for the large number of descriptors already in the equation in comparison to number of molecules, a combination of the classical descriptors and the nodal orientation terms gives even better explanatory of activity of the quinazolines analogues.
This study has used two relatively new techniques. The first is flip regression, for handling the symmetry of the phenylaminoquinazoline system. The second is the use of the orbital nodal angle descriptors. Their descriptors are interesting ones but their application is limited: they can be used for the same structural family only. (within one core) This is based on the concept that the stability of stacked aromatic systems is highly orientation dependent, as we have found in previous studies [Citation10–13], and is also dependent on the energies of those orbitals in the two aromatic systems that resemble the degenerate HOMO and LUMO of benzene. It is envisaged that the benzene rings of the quinazolines are interacting with aromatic systems on the receptor and that alignment occurs between the π-orbital nodes on the pair. Precisely which rings are involved becomes apparent from the identity of the descriptors that remain in the equations. SLUP3 was identified to be an important descriptor. Adding classical variables improves the correlation but only marginally. The only classical variables found to be significant were the polarizability components. High polarizability in the highest inertia direction was found to be favorable to high activity, while high polarizability in the lowest inertia direction was detrimental.
References
- G Powis. (1991). Trends Pharmacol Sci 12:188.
- P Traxler, and P Furet. (1999). Pharmacol Ther 82:195.
- WG Cance, and ET Liu. (1995). Breast Cancer Res Treat 35:105.
- EB Yang, DF Wang, LY Cheng, and P Mack. (1997). Cancer J 10:319.
- EB Yang, YJ Guo, K Zhang, YZ Chen, and P Mack. (2001). Biochim Biophys Acta 1550:144.
- C Szantai-kis, I Kovesdi, D Eros, P Banhegyi, A Ulrich, G Keri, and L Ofri. (2006). Curr Med Chem 13 (3):277.
- A Kurup, R Gorg, and C Hansch. (2001). Chem Rev 101:2573.
- A Vema, SK Panigrahi, G Rambabu, B Gopalakrishnan, JA Sarma, and GR Desiraju. (2003). Bioorg Med Chem 11:4643.
- G Chen, X Luo, W Zhu, C Luo, H Liu, CM Puah, K Chen, and H Jiang. (2004). Bioorg Med Chem 12:2409.
- BW Clare. (1998). J Med Chem 41:3845–3856.
- CT Supuran, and BW Clare. (2004). J Enz Inhib Med Chem 19:237–248.
- BW Clare. (2004). THEOCHEM 712:143–148.
- BW Clare. (2006). THEOCHEM 763:207–215.
- P Mariano, T Ivanka, and P Iliza. (2006). Int J Quant Chem 106 (6):1432.
- BW Clare. 2005. nodangle.zip. http://www.chem.biomedchem.uwa.edu.au/staff/homepages/BrianClare.
- BW Clare. 2005. martha.zip. http://www.chem.biomedchem.uwa.edu.au/staff/homepages/BrianClare.
- BW Clare, and CT Supuran. (2005). Bioorg Med Chem 13:2197–2211.
- Hypercube Hyperchem 1115 NW 4th Street, Gainesville, Florida 32601-4256 U.S.A..
- JJP Stewart. (1990). Q.C.P.E. Bull 10:86.
- MS Tute. In: NJ Harter, and AB Simmord, editors. History and objectives of Quantitative Drug Design in Advances in Drug Research, Vol. 6, London: Academic Press; (1971). p. 1.
- C Hansch, editors. Comprehensive Drug Design. New York: Pergamon Press; (1990).
- JG Topliss, and RJ Costello. (1972). J Med Chem 15:1066–1068.