Abstract
Histidinol dehydrogenase (HDH, EC EC1.1.1.23) catalyses the final step in the biosynthesis of histidine and constitutes an attractive novel target for the development of new agents against the pathogenous, bacteria Brucella suis. A small library of new HDH inhibitors based on the L-histidinylphenylsulfonyl hydrazide scaffold has been synthesized and their inhibitory activity investigated. The obtained results demonstrate that modification of the group between the histidinyl moiety and the phenyl ring constitutes an important structural factor for the design of effective HDH inhibitors.
Introduction
Brucella sp. is the causative agent of brucellosis, the most important anthropozoonotic disease worldwide Citation1-3. This extremely infectious pathogen is traditionally considered as a biological warfare agent classified in the category B [Citation4]. It is responsible for a highly disabling and incapacitating disease that, without treatment, is lethal in 5 to 10% of the cases. There is currently no vaccine available for humans, and even if antibiotic treatment is actually efficient, occurrence of resistant strains is easy. However, human brucellosis remains a threat because curing of the disease is long and persistent forms may appear [Citation5].
Brucella sp. is a facultative intracellular pathogen that multiplies inside the macrophages of its host [Citation6], The “virulome” of this bacteria has been defined as the whole set of genes required for its virulence, i.e. involved in the invasion of the host by the pathogen and in the adaptation to the environment provided by the host [Citation7]. Among them, the gene hisD (BR0252) encoding the enzyme histidinol dehydrogenase, was recently identified Citation7-8.
Histidinol dehydrogenase (HDH, EC1.1.1.23) is a dimeric metalloenzyme containing one Zn2 + ion in each subunit, which catalyzes the last two steps in the biosynthesis of L-histidine: sequential NAD-dependent oxidations of L-histidinol lo L-histidine, via L-histidinal. This enzyme is present only in bacteria and in plants. To date, histidinol dehydrogenases have been cloned and characterized from only two species of bacteria, Salmonella typhimurium and Escherichia coli Citation9-11.
The histidine biosynthesis pathway being absent in mammalian cells, this metalloenzyme represents a selective and promising target for the development of new antibacterial agents avoiding secondary effects of potential inhibitors on the host. Therefore, the potential attractiveness of HDH as a target for antibacterial agents encourages the development of HDH inhibitors.
Recently our group succeeded in cloning and overexpression of the B. suis HDH, and purification of the protein. We designed and developed a series of effective inhibitors which exhibited excellent inhibition profiles with inhibition constants (IC50) in the nanomolar range [Citation12]. We also observed that bulky groups R were advantageous for obtaining increased inhibitory activities [Citation12].
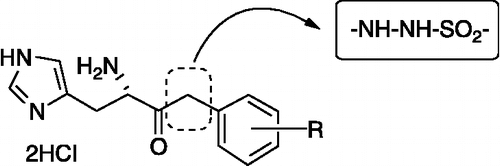
Starting from these previously reported compounds as lead molecules, we report here the effect of replacing the methylene group by a sulfonyl hydrazide moiety. Several reasons led us to incorporate such moiety in our HDH targeting compounds: (i) the easy preparation and subsequent derivatization of such products leading to a chemical diversity necessary when such new targets are considered; (ii) the presence of such moiety in some compounds of biological interest recently described in literature Citation13-14 ().
Figure 1 Chemical structure of new Brucella suis HDH inhibitors.
Materials and methods
Chemistry
All reagents and solvents were of commercial quality and used without further purification. All reactions were carried out under an inert nitrogen atmosphere. TLC analyses were performed on silica gel 60 F254 plates (Merck Art.1.05554). Spots were visualized under 254 nm UV illumination, or by ninhydrin solution spraying. Melting points were determined on a Büchi Melting Point 510 and are uncorrected. 1H and 13C NMR spectra were recorded on Bruker DRX-400 spectrometer using DMSO-d6 as solvent and tetramethylsilane as internal standard. For 1H NMR spectra, chemical shifts are expressed in δ (ppm) downfield from tetramethylsilane, and coupling constants (J) are expressed in Hertz. Electron Ionization mass spectra were recorded in positive or negative mode on a Water MicroMass ZQ.
Preparation of Nα-(tert-butoxycarbonyl)-Nτ-methoxytrityl-L-histidine hydrazide (2)
To a solution of methyl Nα -(tert-butoxycarbonyl- Nτ-methoxytritryl-L-histidinate 1 (1eq.) (prepared as previously described [Citation12]) in methanol was added 10 eq. of hydrazine monohydrate and the solution was stirred at room temperature. The reaction was monitored by TLC until complete consumption of the starting material. The crude product was then co-evaporated with toluene to give yellowish oil which was used in the next step without further purification. 1H NMR (DMSO- d6, 400 MHz) 7.37 (s, 6H), 7.22 (s, 1H), 7.00 (dd, J = 30.4, 6.9 Hz, 8H), 6.74–6.6 (s, 1H),4.13 (s, 1H), 3.75 (s, 3H), 3.37 (dd, J = 13.3, 6.5 Hz, IH), 2.71 (td, J = 22.7, 14.1, 14.12 Hz, 2H), 1.31 (s, 9H). 13C NMR (DMSO-d6, 101MHz) 170.74, 158.58, 154.95, 142.67, 137.45, 136.95, 134.05, 130.60, 129.08, 128.00, 127.78, 118.92, 113.28, 77.68, 73.84, 54.93, 52.88, 30.88, 28.09; MS ESI+ m/z 542.26 (M + H)+ 564.24. ESI− m/z 541.19 (M − H)− , 576.20 (M + Cl)− .
Synthesis of substituted N-L-histidinyl-phenylsulfonylhydrazide. General procedure
To 1eq. of Nα-(tert-butoxycarbonyl)-1-methoxytrityl-L-histidine hydrazide 2 in distilled pyridine was added 1eq. of the corresponding substituted phenylsulfonylchloride.
The reaction was stirred for two hours. Upon completion, the solvent was evaporated under reduced pressure. The crude product was purified on silica gel (CH2Cl2/ Methanol: 95/5) to afford the expected compound as a white powder. The different compounds were deprotected under acidic conditions (HCl solution in dioxane 4M) to remove both the methoxytrityl and the tert-butoxycarbonyl groups.
N-L-histidinyl-4-methylphenylsulfonyl hydrazide (3a)
mp 141–143°C; 1H NMR (DMSO-d6, 400MHz) 14.67 (s, 1H), 10.70 (s, 1H), 10.11 (d, J = 1.5 Hz, 1H), 9.10 (d, J = 0.97 Hz, 1H), 8.66 (s, 2H), 7.62 (d, J = 8.2 Hz, 2H), 7.44 (s, 1H), 7.37 (d, J = 8.1 Hz, 2H), 4.21 (s, 1H), 3.13 (dd, J = 6.3, 3.01 Hz, 2H), 2.38 (s, 3H). 13C NMR (DMSO-d6, 101MHz) 166.68, 143.36, 135.96, 133.83, 129.41, 127.41, 126.16, 117.58, 49.53, 26.09, 21.03; MS ESI+ m/z 324 (M + H)+, 364.22 (M + K)+. ESI− m/z 322.15 (M − H)− , 358.14 (M + Cl)− .
N-L-histidinyl-4-tert-butylphenylsulfonyl hydrazide (3b)
1H NMR (DMSO-d6, 400MHz) 14.69 (s, 1H), 10.64 (d, lH, J = 0.6Hz), 10.10 (d, lH, J = 0.6Hz), 9.10 (d, lH, J = 1.3Hz), 8.70 (s, 2H), 7.67 (d, 2H, J = 8.7Hz), 7.60 (d, 2H, J = 8.8Hz), 7.45 (d, 1H, J = 1.2Hz), 4.20 (t, 1H, J = 6.8Hz), 3.15 (d, 2H, J = 6.9Hz), 1.30 (s, 9H). I3C NMR (DMSO-d6, 101MHz) 166.92, 156.03, 136.12, 133.91, 127.21, 126.23, 125.84, 117.61, 49.66, 34.83, 30.75, 26.11; MS ESI+ m/z 366.18 (M + H)+. ESI+ m/z 364.16 (M − H)− , 400.09 (M + Cl)− , 765.18 (2M + Cl)− .
N-L-histidinyl-4-methoxyphenylsulfonyl hydrazide (3c)
mp 187–189°C; 1H NMR (DMSO-d6, 400MHz) 14.73 (s, 1H), 10.69 (d, 1H, J = 1.6Hz), 10.00 (d, 1H, J = 2.1Hz), 9.11 (d, 1H, J = 1.2Hz), 8.68 (s, 2H), 7.64 (d, 2H, J = 8.9Hz), 7.48 (d, 1H, J = 0.5Hz), 7.08 (d, 2H, J = 9.0Hz), 4.22 (s, IH), 3.84 (s, 3H), 3.14 (d, 2H, J = 7.0Hz). l3C NMR (DMSO-d6, 101MHz) 166.63, 162.66, 133.79, 130.22, 129.66, 126.24, 117.57, 114.17, 55.68, 49.51, 26.14; MS ESI+ m/z 340.24 (M + H)+. ESI− m/z 338.20 (M − H)− , 374.19 (M + Cl)− , 713.23 (2M + Cl)− .
N-L-histidinyl-biphenyl-4-ylsulfonyl hydrazide (3d)
mp 220°C (decomposition); 1H NMR (DMSO-d6. 400MHz) 14.73 (s, 1H), 10.78 (s, 1H), 10.25 (s, 1H), 9.11 (d, 1H, J = 1.0Hz), 8.71 (m, 2H), 7.81 (m, 6H), 7.48 (m, 4H), 4.24 (t, 1H, J = 6.8Hz), 3.17 (d, 2H, J = 6.8Hz). l3C NMR (DMSO-d6, 101 MHz) 166.82, 144.38, 138.34, 137.64, 133.86, 129.07, 128.51, 128.03, 127.11, 127.03, 126.29, 117.59, 49.57, 26.16; MS ESI+ m/z 386.13 (M + H)+, 771.24 (2M + H)+. ESI− m/z 384.10 (M − H)− , 419.96 (M + Cl)− , 805.20 (2M + Cl)− .
N-L-histidinyl-pentafluorophenylsulfonyl hydrazide (3e)
mp 215°C (decomposition); 1H NMR (DMSO-d6, 400MHz) 14.55 (s, 1H), 11.36 (s, 1H), 9.07 (d, J = 1.2 Hz, 1H), 8.66 (s, 1H), 7.48 (s, 1H), 4.26 (t, J = 6.5 Hz, 1H), 3.23 (dd, J = 6.1, 3.3 Hz, 1H). 13C NMR (DMSO-d6, 101MHz) 167.52,133.95, 126.01, 117.88, 49.88, 25.99; MS ESI+m/z 400.09 (M + H)+. ESI− m/z 398.11 (M − H)− , 434.17 (M + Cl)− , 833.17 (2M + CL)− .
N-L-histidinyl-4-cyanophenylsulfonyl hydrazide (3f)
mp 205°C (decomposition); 1H NMR (DMSO-d6, 400MHz) 14.70 (s, 1H), 10.96 (s, 1H), 10.59 (s, 1H), 9.09 (d, 1H, J = 1.3Hz), 8.71 (s, 2H), 8.07 (d, 2H, J = 8.6Hz), 7.91 (d, 2H, J = 8.6Hz), 7.48 (d, 1H, J = 0.9Hz), 4.24 (t, 1H, J = 6.8Hz), 3.15 (d, 2H, J = 6.8Hz). l3C NMR (DMSO-d6, 101MHz) 166.91, 143.19, 133.84, 133.13, 128.20, 126.16, 117.74, 117.65, 115.27, 49.54, 26.06; MS ESI+m/z 335.20 (M + H)+. ESI− m/z 333.20 (M − H)− , 369.13 (M + Cl)− , 667.29 (2M − H)− , 703.18 (2M + Cl)− .
N-L-histidinyl-4-trifluoromethylphenylsulfonyl hydrazide (3g)
mp 195°C (decomposition); 1H NMR (DMSO-d6, 400MHz) 14.71 (s, 1H), 10.90 (s, 1H), 10.53 (s, 1H), 9.10 (d, 1H, J = 1.3Hz), 8.72 (s, 2H), 7.97 (s, 4H), 7.48 (d, 1H, J = 1.0Hz), 4.23 (t, 1H, J = 6.8Hz), 3.17 (d, 2H, J = 6.7Hz). l3C NMR (DMSO-d6, 101MHz) 166.97, 143.12 (d, JC–F = 1.32 Hz), 134.17 (s,1C), 133.85, 132.51 (dd, JC–F = 64.18, 31.94 Hz), 128.40, 126.217, 117.68, 49.60, 26.06; MS ESI+m/z 378.16 (M + H)+. ESI− m/z 376.24 (M − H)− , 789.10 (2M + Cl)− .
N-L-histidinyl-4-nitrophenylsulfonyl hydrazide (3h)
mp 205°C(decomposition); 1H NMR (DMSO-d6, 400MHz)14.56 (s, 1H), 10.97 (s, 1H), 10.65 (s, 1H), 9.07 (d, 1H, J = 1.1Hz), 8.66 (s, 2H), 8.40 (m, 2H), 8.02 (m, 2H), 7.46 (d, 1H, J = 0.7Hz), 4.22 (t, 1H, J = 6.9Hz), 3.14 (d, 2H, J = 6.9Hz). 13C NMR (DMSO-d6, 101MHz) 166.92, 149.81, 144.64, 133.87, 129.14, 126.16, 124.26, 117.66, 49.55, 26.04; MS ESI+m/z 355.14 (M + H)+, ESI− m/z 353.16 (M − H)− , 389.22 (M + Cl)− , 707.16 (2M − H)− , 734.17(2M + Cl)− .
N-L-histidinyl-4-fluorophenylsulfonyl hydrazide (3i)
mp 222–224°C; 1H NMR (DMSO-d6, 400MHz) 14.70 (s, 1H), 10.81 (s, 1H), 10.27 (s, 1H), 9.10 (d, 1H, J = 1.3Hz), 8.69 (s, 2H), 7.80 (dd, 2H, J = 5.2Hz, 8.9Hz), 7.47 (d, 1H, J = 0.9Hz), 7.42 (t, 2H, J = 8.9Hz), 4.23 (t, 1H, J = 6.8Hz), 3.14 (d, 2H, J = 6.9Hz). 13C NMR (DMSO-d6 101 MHz) 166.76, 164.53 (d, JC–F = 251.1 Hz), 135.19, 133.84, 130.56 (d, JC–F = 9.4 Hz), 126.21, 117.60, 116.19 (d, JC–F = 22.9 Hz), 49.55, 26.10; MS ESI+m/z 328.16 (M + H)+, 655.20 (2M + H)+. ESI− m/z 326.14 (M − H)− , 362.14 (M + Cl)− , 689.14 (2M + Cl)− .
N-L-histidinyl-4-chlorophenylsulfonyl hydrazide (3j)
mp 210°C (decomposition); 1H NMR (DMSO-d6, 400MHz) 14.71 (s, 1H), 10.84 (s, 1H), 10.34 (s, 1H), 9.10 (s, 1H), 8.72 (s, 2H), 7.75 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.48 (s, 1H), 4.23 (t, J = 6.4 Hz, 1H), 3.16 (d, J = 6.5 Hz, 2H). 13C NMR (DMSO-d6, 101MHz) 166.80, 137.91, 137.84, 133.79, 129.36, 129.14, 126.17, 117.62, 49.54, 26.08; MS ESI+m/z 344.14 (M + H)+. ESI− m/z 342.16 (M − H)− , 378.09 (M + Cl)− , 723.09 (2M + Cl)− .
N-L-histidinyl-4-bromophenylsulfonyl hydrazide (3k)
mp 93–94°C; 1H NMR (DMSO-d6, 400MHz) 14.60 (s, 1H), 10.80 (s, 1H), 10.34 (s, 1H), 9.08 (d, J = 0.8 Hz, 1H), 8.65 (s, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.64 Hz, 2H), 7.46 (s, 1H), 4.20 (t, J = 6.6 Hz, 1H), 3.14 (d, J = 6.8 Hz, 2H). 13C NMR (DMSO-d6, 101MHz) 166.80, 138.26, 133.93, 132.07, 129.40, 127.03, 126.22, 117.63, 49.59, 26.06; MS ESI+m/z 390.05 (M + H)+. ESI− m/z 386.02 (M − H)− , 424.1 (M + Cl)− , 810.91 (2M + Cl)− .
N-L-histidinyl-4-iodophenylsulfonyl hydrazide (3l)
mp 139–141°C; 1H NMR (DMSO-d6, 400MHz) 14.65 (s, 1H), 10.79 (s, 1H), 10.30 (s, 1H), 9.09 (d, J = 1.1 Hz, 1H), 8.69 (s, 2H), 7.97 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.5 Hz, 2H), 7.47 (s, 1H), 4.22 (t, J = 6.6 Hz, 1H), 3.15 (d, J = 6.7 Hz, 2H). 13C NMR (DMSO-d6, 101 MHz) 166.82, 138.65, 137.86, 133.85, 129.03, 126.17, 117.65, 101.59, 49.57, 26.08; MS ESI+m/z 436.03 (M + H)+. ESI− m/z 434.05 (M − H)− , 469.99 (M + Cl)− .
N-L-histidinyl-2-naphthysulfonyl hydrazide (3m)
mp 220°C (decomposition); 1H NMR (DMSO-d6 400MHz) 14.69 (s, 1H), 10.81 (s, 1H), 10.31 (s, 1H), 9.09 (d, J = 1.2 Hz, 1H), 8.65 (s, 2H), 8.44 (d, J = 1.8 Hz, 1H), 8.20 (d, J = 8.1 Hz, 1H), 8.11 (d, J = 8.8 Hz, 1H), 8.04 (d, J = 8.1 Hz, 1H), 7.80 (dd, J = 8.7, 1.9 Hz, 1H), 7.68 (dddd, J = 20.9, 8.1, 6.9, 1.3 Hz, 2H), 7.44 (d, J = 0.8 Hz, 1H), 4.22 (t, J = 6.5 Hz, 1H), 3.13 (ddd, J = 36.6, 15.6, 6.7 Hz, 2H). 13C NMR (DMSO-d6, 101MHz) 166.86, 136.20, 134.46, 133.85, 131.56, 129.34, 129.02, 128.87, 128.44, 127.73, 127.39, 126.16, 122.92, 117.67, 49.64, 26.06; MS ESI+m/z 360.19 (M + H)+, 719.31 (2M + H)+. ESI− m/z 358.21 (M − H)− , 394.14 (M + Cl)− , 753.28 (2M + Cl)− .
Enzyme assays
The activity and specificity of HDH were measured by monitoring the reduction of NAD+ to NADH directly at 340 nm (εM = 6200 M− 1.cm− 1) as previously described [Citation15]. The enzymatic activity was studied at 30°C in the presence of 0.5 mM histidinol, 5 mM NAD+ and 0.5 mM MnCl2 in 50 mM sodium glycine buffer at pH = 9.2. For kinetic studies, experiments were carried out with 150 mM sodium glycine (pH 9.2) and 2 mM NAD+. The Km for the substrate was determined by varying the concentration of histidinol from 10 to 50 μM. Activity (1 unit) is defined as the amount of HDH producing 1 μmol of NADH per min in the reaction. To perform IC50 determinations of the different inhibitors, the latter were added at various concentrations, ranging from 1 μM to 400 μM, and preincubated for 5 min at 30°C with the enzyme solution prior to the initiation of the reaction. The enzyme concentration in the assay system was 4.5 × 10− 11 M.
Results and discussion
Chemistry
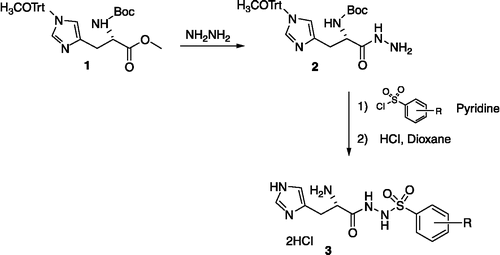
Substituted histidinyl phenylsulfonylhydrazide derivatives 3 were readily prepared according to the synthetic pathway depicted in Scheme . The precursor 1 was synthesized in four steps starting from L-histidine as previously described [Citation12]. Reaction of compound 1 with hydrazine hydrate afforded the corresponding hydrazide 2 which was reacted with various substituted phenylsulfonyl chloride in the presence of pyridine to yield, after acidic treatment the corresponding hydrazides 3.
Scheme 1 The synthetic pathway of 2–3.
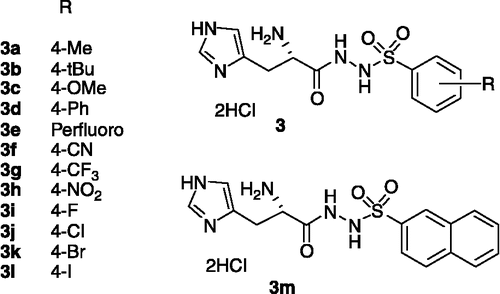
All the synthesized compounds 3 listed in were fully characterized by 1H-NMR, 13C-NMR and mass spectral data.
Figure 2 Structure of inhibitors series 3.
Brucella suis HDH inhibitory activity
All the newly synthesized compounds were assayed for their inhibitory activity against the purified B. suis HDH. Inhibitory data are presented in . The results show a decreased affinity for 3 compared to the previously reported derivatives [Citation12], with IC50 within the range of 25 to more than 400 μM. The most active derivatives in this series were compounds 3d and 3k with an IC50 of 25μM and 70μM respectively. Additionally, some compounds such as 3e, 3i, 3j, 3h were devoid of any inhibitory activity for concentration ≥ 400 μM.
Table I. Inhibition of B. suis histidinol dehydrogenase with compounds 3a to 3m.
The following SAR should be noted from data of : among the compound series, the activity order was 3d>3k>3c>3a>3f, which might reflect the importance of the substituent bulkiness in the position 4 of the phenyl ring.
Compared with the previously reported series [Citation12], compounds 3 are much less effective, indicating that the nature and the length of the linker between the histidinyl moiety and the phenyl ring is an important factor which can be modulated for the design and the discovery of new potential inhibitors.
In conclusion, a small library of substituted L-histidinyl phenylsulfonylhydrazide was synthesized as potential B. suis histidinol dehydrogenase inhibitors. The introduction of a hydrazinosulfonyl scaffold was shown to influence potencies of inhibitors. From these data, compound 3d demonstrated the best inhibitor activity. Nevertheless, this series of compounds remains less active compared to the one previously described. Informations derived from this study indicate that the linker between the histidinyl moiety and the phenyl ring constitute an important structural feature which could be subject to further modifications in order to design more potent inhibitors.
Acknowledgements
This work was supported by a grant from the German Sanitätsamt der Bundeswehr, N° M SAB1 5A002.
References
- Corbel MJ. Brucellosis: An overview. Emerg Infect Dis 1997; 3: 213–221
- Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med 2005; 352: 2325–2336
- Godfroid J, Cloeckaert A, Liautard JP, Köhler S, Fretin D, Walravens K, Garin-Bastuji B, Letesson JJ. From the discovery of the Malta fever's agent to the discovery of a marine mammal reservoir, brucellosis has continuously been a re-emerging zoonosis. Vet Res 2005; 36: 313–326
- Pappas G, Panagopoulou P, Christou L, Akritidis N. Brucella as a biological weapon. Cell Mol Life Sci 2006; 63: 2229–2236
- Pappas G, Panagopoulou P, Christou L, Akritidis N. Future trends in human brucellosis treatment. Expert Opin Investig Drugs. 2006; 15: 1141–1149
- Köhler S, Michaux-Charachon S, Porte F, Ramuz M, Liautard JP. What is the nature of the replicative niche of a stealthy bug named Brucella?. Trends Microbiol 2003; 11: 215–219
- Köhler S, Foulongne V, Ouahrani-Bettache S, Bourg G, Teyssier J, Ramuz M, Liautard JP. The analysis of the intramacrophagic virulome of Brucella suis deciphers the environment encountered by the pathogen inside the macrophage host cell. Proc Natl Acad Sci USA 2002; 99: 15711–15716
- Liautard JP, Jubier-Maurin V, Boigegrain RA, Köhler S. Antimicrobials: Targeting virulence genes necessary for intracellular multiplication. Trends Microbiol 2006; 14: 109–113
- Grubmeyer CT, Insiga S, Bhatia M, Moazami N. Salmonella typhimurium histidinol dehydrogenase: Complete reaction stereochemistry and active site mapping. Biochemistry 1989; 28: 8174–8180
- Teng H, Grubmeyer CT. Mutagenesis of histidinol dehydrogenase reveals roles for conserved histidine residues. Biochemistry 1999; 38: 7363–7371
- Barbosa JA, Sivaraman J, Li Y, Larocque R, Matte A, Schrag JD, Cygler M. Mechanism of action and NAD+ binding mode revealed by the crystal structure of L-histidinol dehydrogenase. Proc Natl Acad Sci USA 2002; 99: 1859–1864
- Abdo MR, Joseph P, Boigegrain RA, Liautard JP, Montero JL, Köhler S, Winum JY. Brucella suis histidinol dehydrogenase: Synthesis and inhibition studies of a series of substituted benzylic ketones derivated from histidine. Bioorg Med Chem 2007; 15: 4427–4433
- Nofal ZM, Fhamy HH, Mohamed HS. Synthesis and antimicrobial activity of new substituted anilinobenzimidazole. Arch Pharm Res 2002; 25: 250–257
- Siemman S, Evanoff DP, Marrone L, Clarke AJ, Viswanatha T, Dmitrienko GI. N-arylsulfonylhydrazones as inhibitors of IMP-1 metallo-beta-lactamase. Antimicrob Agents Chemother 2002; 46: 2450–2457
- Loper JC, Adams E. Purification and properties of histidinol dehydrogenase from Salmonella typhimurium. J Biol Chem 1965; 240: 788–795