Abstract
This present study identifies a number of azolyl-substituted indoles as potent inhibitors of aromatase. In the sub-series of 3-(azolylmethyl)-1H-indoles, four imidazole derivatives and their triazole analogues were tested. Imidazole derivatives 11 and 14 in which the benzyl moiety was substituted by 2-chloro and 4-cyano groups, respectively, were the most active, with IC50 values ranging between 0.054 and 0.050 μM. In the other sub-series, eight 3-(α-azolylbenzyl)-1H-indoles were prepared and tested. Compound 30, the N-ethyl imidazole derivative, proved to be an aromatase inhibitor, showing an IC50 value of 0.052 μM. All target compounds were further evaluated against 17α-hydroxylase/C17,20-lyase to determine their selectivity profile.
Introduction
Estrogens not only play a key role in normal expression of secondary sexual characteristics, and establishment and maintenance of pregnancy but are also involved in the natural history of breast cancer. A high proportion of breast cancer tumors are dependent upon estrogens for growth and respond to therapeutic measures designed to deplete circulating estrogens. Aromatase, a cytochrome P450 enzyme (P450 arom), catalyzes the biosynthesis of these estrogens from androgens. Activity of aromatase results in aromatization of the A ring of androgens with the concomitant loss of the C18 angular methyl group. So, the inhibition of the enzyme becomes a logical aim in breast cancer hormonotherapy [Citation1,Citation2].
The structurally diverse group of aromatase inhibitors are classically categorized into two major group: steroidal and non-steroidal derivatives; they appear to be different on their ability to interact with the enzyme [Citation3,Citation4]. A great number of steroidal compounds structurally related to the natural substrates (androstenedione or testosterone) have been developped. 4-Hydroxyandrostenedione (4-OHA) [Citation5,Citation6] was the first aromatase steroidal inhibitor to become available, and had high enzyme selectivity. But it had to be administered as a parental formulation due to its poor oral bioavailability. Other steroidal inhibitors, in addition to 4-OHA, have been also shown to cause inactivation of aromatase Citation7-11. One of these, exemestane [Citation11], has proved to be orally effective in postmenopausal women and reached the market in 1999. Steroidal inhibitors bind either very tightly or irreversibly to aromatase and then cause its inactivation [Citation6,Citation12].
Among the non-steroidal inhibitors, the most studied compounds were aminoglutethimide [AG, 3-(4-aminophenyl)-3-ethylpiperidine-2,6-dione] [Citation13], 4-substituted anilines Citation14-16, imidazole antifungals and analogs[Citation17,Citation18]. AG, the first commercially available non-steroidal inhibitor, was launched in 1981. However, the use of AG [Citation19] has been limited due to (i) its lack of specificity - AG inhibits other cytochrome P450 enzymes such as P450scc and, P450c11 - and (ii) its intrinsic toxicity - main side-effects are lethargy, dizziness and skin rashes. Investigations of many research teams have resulted notably in the discovery of several heterocyclic azoles Citation20-23 such as fadrozole, anastrozole, letrozole, vorozole, YM-511 and MPV-2213ad (see for structures).
Figure 1 Structures of nonsteroidal inhibitors.
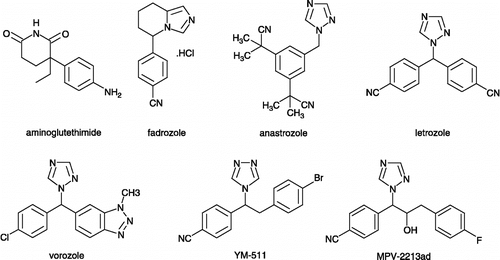
As shown in , a major advance in potency is observed for these new non-steroidal aromatase inhibitors. The selectivity profile is also increased for fadrozole [Citation24], vorozole [Citation5], letrozole [Citation26], anastrozole [Citation27], YM-511 [Citation28] and MPV-2213ad [Citation29]. About their mechanism of action, it can be noted that they perturb the catalytic properties of the heme prosthetic group of aromatase [Citation30].
Table I. Inhibition of aromatase by major nonsteroidal inhibitors.
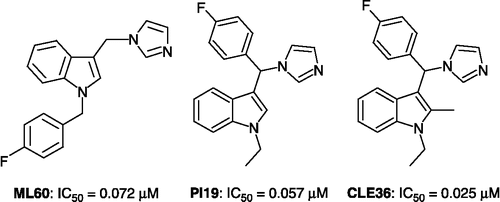
In previous works Citation31-33, we initially prepared and evaluated 3-azolylmethylindoles and 3-(α-azolylbenzyl)indoles as potential aromatase inhibitors. 1-4-Fluorobenzyl-3-(1H-imidazol-1-ylmethyl)-1H-indole ML60, 1-ethyl-3-[(1H-imidazol-1-yl)(4-fluorophenyl)methyl]-1H-indole PI19 and 1-ethyl-3-[(1H-imidazol-1-yl)(4-fluorophenyl)methyl]-2-methyl-1H-indole CLE36 (see for structures) proved to be the most potent agents in inhibiting aromatization of androgens (androstenedione and, testosterone).
Figure 2 Structures and aromatase inhibitory activities of compounds ML60, PI19 and CLE36.
The present paper describes further structural optimizations in azolylmethyl-substituted indole series, namely the development of 1-substituted-3-(azolylmethyl)-1H-indoles and 1-substituted-3-(α-azolylbenzyl)-1H-indoles. Synthesis and structure-activity studies on the in vitro activity (inhibition of P450 arom) of compounds 11–18 and 30–37 are described in this paper. The most potent compounds were further evaluated for selectivity (inhibition of 17α-hydroxylase/C17,20-lyase).
Materials and methods
Chemistry
Instrumentation
Chemicals were purchased from Sigma-Aldrich. Compounds 3 and 20 were prepared according to reported methods [Citation34,Citation33]. Evaporations of final product solutions were done under vacuum with a rotatory evaporator. Column chromatography (CC) was carried out by using silica gel (silica gel 60, 63–200 mesh, E. Merck). Melting points were determined on a Tottoli-Büchi apparatus and are uncorrected. IR spectra (KBr or NaCl) were recorded as either neat (liquid or oil) or a KBr pellet (solids) on a Perkin-Elmer Paragon 1000 PC spectrophotometer. 1H NMR spectra were recorded on a Bruker AC 250 (250 MHz) spectrometer, using DMSO d6 as solvent. Chemical shifts are expressed in ppm (δ) downfield from internal TMS. Coupling constants J are expressed in Hz. Signals of protons exchangeable with D2O are denoted by an asterisk (*). EI MS spectra were obtained on a Hewlett-Packard HP 5989A spectrometer (250°C, 70 eV).
Synthesis of N-substituted 1H-3-indolecarbaldehydes 3–6
A mixture of 1H-indole-3-carbaldehyde 2 (3.4 mmol) and K2CO3 (17 mmol) in dry acetone (15 mL) was stirred at room temperarure for 1 h. The corresponding benzyl chloride (4.1 mmol) was added and the reaction mixture was heated to reflux for 5 h. After filtration, the filtrate was evaporated. Water was added to the residue and the solution was extracted three times with CH2Cl2. The combined organic layers were dried (Na2SO4) and the solvent was evaporated under reduced pressure. The crude product was purified by trituration to give the target compound.
1-(2-Chlorobenzyl)-1H-3-indolecarbaldehyde (3)
This compound was prepared from precursor 2. Yield: 80%, brown crystals, mp 90–91°C (isopropyl ether). IR (KBr, cm− 1) 1656 (CO); 1H NMR (DMSO d6) δ 5.68 (s, 2H, CH2), 7.0 (dd, J = 7.5, 1.6 Hz, 1H, H-6′), 7.30–7.44 (m, 4H, H-5, H-6, H-4′, H-5′), 7.54–7.61 (m, 2H, H-7, H-3′), 8.16–8.21 (m, 1H, H-4), 8.41 (s, 1H, H-2), 9.99 (s, 1H, CHO).
1-(2-Fluorobenzyl)-1H-3-indolecarbaldehyde (4)
This compound was prepared from precursor 2. Yield: 88%, brown crystals, mp 95–97°C (diethyl ether). IR (KBr, cm− 1) 1665 (CO); 1H NMR (DMSO d6) δ 5.65 (s, 2H, CH2), 7.63–7.66 (s, 1H, H-7), 7.17–7.46 (m, 6H, H-5, H-6, H-3′, H-4′, H-5′, H-6′), 8.15-8.18 (m, 1H, H-4), 8.44 (s, 1H, H-2), 9.99 (s, 1H, CHO).
1-(3-Fluorobenzyl)-1H-3-indolecarbaldehyde (5)
This compound was prepared from precursor 2. Yield: 87%, brown solid, mp 105–107°C (isopropyl ether). IR (KBr, cm− 1) 1664 (CO); 1H NMR (DMSO d6) δ 5.61 (s, 2H, CH2), 7.14–7.35 (m, 5H, H-5, H-6, H-2′, H-4′, H-6′), 7.42 (ddd, J = 7.9, 7.9, 6.1 Hz, 1H, H-5′), 7.62–7.66 (m, 1H, H-7), 8.14–8.18 (m, 1H, H-4), 8.53 (s, 1H, H-2), 9.99 (s, 1H, CHO).
1-(4-Cyanobenzyl)-1H-3-indolecarbaldehyde (6)
This compound was prepared from precursor 2. Yield: 85%, brown solid, mp 153–155°C (isopropyl ether). IR (KBr, cm− 1) 2230 (CN), 1658 (CO); 1H NMR (DMSO d6) δ 5.71 (s, 2H, CH2), 7.26–7.34 (m, 2H, H-5, H-6), 7.47 (d, J = 8.2 Hz, 2H, H-2′, H-6′), 7.56–7.60 (m, 1H, H-7), 7.86 (d, J = 8.2 Hz, 2H, H-3′, H-5′), 8.15–8.18 (m, 1H, H-4), 8.54 (s, 1H, H-2), 9.99 (s, 1H, CHO).
Synthesis of 3-aroylindoles 21–23
To a magnetically stirred suspension of AlCl3 (10 mmol) in CH2Cl2 (20 mL) at 25°C was added 4-fluorobenzoyl chloride (10 mmol) and the mixture was stirred at the same temperature for 1 h. A solution of 5-bromo-1H-indole 19 or 5-bromo-1-ethyl-1H-indole 20 (10 mmol) in CH2Cl2 (10 mL) was added dropwise to the mixture at 25°C. After stirring for 24 h at 25°C, the reaction mixture was poured into ethyl acetate and ice water. The organic layer was separated, washed with water, and dried over Na2SO4. After evaporation of solvent, the crystalline residue was recrystallized in the appropriate solvent.
5-Bromo-3-(4-fluorobenzoyl)-1H-indole (21)
This compound was prepared from precursor 19. Yield: 45%, fine white crystals, mp 250°C (ethyl acetate, decomposition). IR (KBr, cm− 1) 3157 (NH), 1596 (CO); 1H NMR (DMSO d6) δ 7.40 (dd, J = 8.9, 8.9 Hz, 2H, H-3″, H-5″), 7.44 (dd, J = 1.7, 8.6 Hz, 1H, H-6), 7.54 (d, J = 8.6 Hz, 1H, H-7), 7.92 (dd, J = 5.7, 8.7 Hz, 2H, H-2″, H-6″), 8.09 (d, J = 3.1 Hz, 1H, H-2), 8.43 (d, J = 1.7 Hz, 1H, H-4), 12.35 (s*, 1H, NH).
5-Bromo-1-ethyl-3-(4-fluorobenzoyl)-1H-indole (22)
This compound was prepared from precursor 20. Yield: 80%, brown crystals, mp 155–157°C (diisopropyl ether/MeOH). IR (KBr, cm− 1) 1616 (CO); 1H NMR (DMSO d6) δ 1.42 (t, J = 7.2 Hz, 3H, CH3), 4.36 (q, J = 7.2 Hz, 2H, CH2), 7.42 (dd, J = 8.8, 8.8 Hz, 2H, H-3″, H-5″), 7.50 (dd, J = 2.0, 8.7 Hz, 1H, H-6), 7.69 (d, J = 8.7 Hz, 1H, H-7), 7.93 (dd, J = 5.6, 8.8 Hz, 2H, H-2″, H-6″), 8.19 (s, 1H, H-2), 8.45 (d, J = 2.0 Hz, 1H, H-4).
5-Bromo-3-(2,4-dichlorobenzoyl)-1-ethyl-1H-indole (23)
This compound was prepared from precursor 20. Yield: 91%, pink solid, mp 147–149°C (diisopropyl ether/MeOH). IR (KBr, cm− 1) 1630 (CO). 1H NMR (DMSO d6) δ 1.57 (t, J = 7.2 Hz, 3H, CH3), 4.48 (q, J = 7.2 Hz, 2H, CH2), 7.60 (dd, J = 1.8, 8.9 Hz, 1H, H-6), 7.62 (dd, J = 1.8, 8.2 Hz, 1H, H-5″), 7.68 (d, J = 8.2 Hz, 1H, H-6″), 7.72 (d, J = 8.9 Hz, 1H, H-7), 7.74 (d, J = 1.8 Hz, 1H, H-3″), 7.96 (s, 1H, H-2), 8.64 (d, J = 1.8 Hz, 1H, H-4).
Synthesis of N-substituted 3-aroylindoles 24 and 25
A mixture of 5-bromo-3-(4-fluorobenzoyl)-1H-indole 21 (2.50 mmol) and Cs2CO3 (5 mmol) in acetonitrile (15 mL) was heated to reflux for 2 h. 2-Chlorobenzyl chloride or 4-fluorobenzyl chloride (2.80 mmol) was added and the reaction mixture was heated to reflux for 1 h. Then, it was filtered and the filtrate was evaporated. To the residue was added water and the solution was extracted three times with CH2Cl2. The combined organic layers were dried (Na2SO4) and the solvent was evaporated under reduced pressure. The crude product was triturated from isopropyl ether to give 24 or 25.
5-Bromo-1-(2-chlorobenzyl)-3-(4-fluorobenzoyl)-1H-indole (24)
This compound was prepared from precursor 21. Yield: 98%, brown solid (isopropyl ether/CH2Cl2), mp 150–151°C. IR (KBr, cm− 1) 1632 (CO); 1H NMR (DMSO d6) δ 5.70 (s, 2H, CH2), 6.83 (d, J = 7.3 Hz, 1H, H-6′), 7.25–7.38 (m, 2H, H-4′, H-5′), 7.37–7.52 (m, 4H, H-6, H-7, H-3″, H-5″), 7.56 (d, J = 7.6 Hz, 1H, H-3′), 7.94 (dd, J = 5.8 Hz, 8.4 Hz, 2H, H-2″, H-6″), 8.33 (s, 1H, H-2), 8.48 (s, 1H, H-4).
5-Bromo-3-(4-fluorobenzoyl)-1-(4-fluorobenzyl)-1H-indole (25)
This compound was prepared from precursor 21. Yield: 93%, white solid, mp 150–152°C (isopropyl ether). IR (KBr, cm− 1) 1629 (CO). 1H NMR (DMSO d6) δ 5.58 (s, 2H, CH2), 7.19 (dd, J = 8.8, 8.8 Hz, 2H, H-3′, H-5′), 7.38–7.43 (m, 2H, H-2′, H-6′), 7.43 (dd, J = 8.8, 8.8 Hz, 2H, H-3″, H-5″), 7.45 (dd, J = 1.9, 8.8 Hz, 1H, H-6), 7.59 (d, J = 8.8 Hz, 1H, H-7), 7.95 (dd, J = 5.6, 8.8 Hz, 2H, H-2″, H-6″), 8.42 (s, 1H, H-2), 8.45 (d, J = 1.9 Hz, 1H, H-4).
Synthesis of carbinol derivatives 7–10 and 26–29
A solution of sodium borohydride (12.6 mmol) in methanol (10 mL) was added dropwise to a solution of the appropriate indole-3-carbaldehyde 3–6 or ketone derivative 22–25 (4.2 mmol) in methanol (10 mL). The reaction mixture was stirred at room temperature for 1 h. Water (30 mL) was added and the solution was extracted three times with diethyl ether. The combined organic layers were dried (Na2SO4) and the solvent was carefully evaporated leaving the desired crude product.
1-(2-Chlorobenzyl)-3-hydroxymethyl-1H-indole (7)
This compound was prepared from precursor 3. Yield: 82%, white solid, mp 85–87°C (isopropyl ether). IR (KBr, cm− 1) 3380 (OH); 1H NMR (DMSO d6) δ 4.70 (d, J = 5.4 Hz, 2H, CH2), 4.89 (t*, J = 5.4 Hz, 1H, OH), 5.51 (s, 1H, CH2), 6.76 (dd, J = 7.5, 1.6 Hz, 1H, H-6′), 7.05–7.18 (m, 2H, H-5, H-6), 7.25 (ddd, J = 7.5, 7.5, 1.3 Hz, 1H, H-5′), 7.31–7.41 (m, 3H, H-2, H-7, H-4′), 7.55 (dd, J = 7.8, 1.3 Hz, 1H, H-3′), 7.68 (m, 1H, H-4).
1-(2-Fluorobenzyl)-3-hydroxymethyl-1H-indole (8)
This compound was prepared from precursor 4. Yield: 88%, yellow oil. IR (NaCl, cm− 1) 3390 (OH). 1H NMR (DMSO d6) δ 4.70 (d, J = 5.4 Hz, 2H, CH2), 4.91 (t*, J = 5.4 Hz, 1H, OH), 5.47 (s, 2H, CH2), 7.05–7.38 (m, 6H, H-5, H-6, H-3′, H-4′, H-5′, H-6′), 7.39 (s, 1H, H-2), 7.46–7.52 (m, 1H, H-7), 7.66–7.69 (m, 1H, H-4).
1-(3-Fluorobenzyl)-3-hydroxymethyl-1H-indole (9)
This compound was prepared from precursor 5. Yield: 66%, white solid, mp 63–65°C (isopropyl and diethyl ethers, cyclohexane). IR (KBr, cm− 1) 3388 (OH); 1H NMR (DMSO d6) δ 4.68 (d, J = 5.4 Hz, 2H, CH2), 4.86 (t*, J = 5.4 Hz, 1H, OH), 5.43 (s, 2H, CH2), 7.03–7.17 (m, 5H, H-5, H-6, H-2′, H-4′, H-6′), 7.38 (ddd, J = 8.0, 8.0, 6.1 Hz, 1H, H-5′), 7.45 (s, 1H, H-2), 7.45–7.48 (m, 1H, H-7), 7.64–7.67 (m, 1H, H-4).
1-(4-Cyanobenzyl)-3-hydroxymethyl-1H-indole (10)
This compound was prepared from precursor 6. Yield: 93%, white solid, mp 60–62°C (isopropyl ether). IR (KBr, cm− 1) 3560 (OH), 2220 (CN); 1H NMR (DMSO d6) δ 4.68 (d, J = 5.4 Hz, 2H, CH2), 4.88 (t*, J = 5.4 Hz, 1H, OH), 5.53 (s, 2H, CH2), 7.03–7.16 (m, 2H, H-5, H-6), 7.35 (d, J = 8.2 Hz, 2H, H-2′, H-6′), 7.42 (d, J = 7.9 Hz, 1H, H-7), 7.45 (s, 1H, H-2), 7.66 (d, J = 7.4 Hz, 1H, H-4), 7.81 (d, J = 8.2 Hz, 2H, H-3′, H-5′).
5-Bromo-1-ethyl-3-[(4-fluorophenyl)(hydroxy)methyl]-1H-indole (26)
This compound was prepared from precursor 22. Yield: 88%, white solid, mp 162–164°C (isopropyl ether). IR (KBr, cm− 1) 3164 (OH). 1H NMR (DMSO d6) δ 1.34 (t, J = 7.2 Hz, 3H, CH3), 4.18 (q, J = 7.2 Hz, 2H, CH2), 5.80 (d*, J = 4.5 Hz, 1H, OH), 5.97 (d, J = 4.5 Hz, 1H, CH), 7.18 (dd, J = 8.8, 8.8 Hz, 2H, H-3″, H-5″), 7.24 (dd, J = 1.9, 8.6 Hz, 1H, H-6), 7.25 (s, 1H, H-2), 7.45 (d, J = 8.6 Hz, 1H, H-7), 7.50 (dd, J = 5.7, 8.8 Hz, 2H, H-2″, H-6″), 7.66 (d, J = 1.9 Hz, 1H, H-4).
5-Bromo-3-[(2,4-dichlorophenyl)(hydroxy)methyl]-1-ethyl-1H-indole (27)
This compound was prepared from precursor 23. Yield: 78%, white solid, mp 120–122°C (isopropyl ether). IR (KBr, cm− 1) 3232 (OH). 1H NMR (DMSO d6) δ 0.85 (t, J = 7.0 Hz, 3H, CH3), 4.16 (q, J = 7.0 Hz, 2H, CH2), 6.01 (d*, J = 4.9 Hz, 1H, OH), 6.20 (d, J = 4.9 Hz, 1H, CH), 7.06 (s, 1H, H-2), 7.28 (dd, J = 1.9, 8.7 Hz, 1H, H-6), 7.47 (d, J = 8.7 Hz, 1H, H-7), 7.55 (dd, J = 8.3 Hz, 1H, H-5″), 7.60 (d, J = 2.1 Hz, 1H, H-3″), 7.78 (d, J = 1.9 Hz, 1H, H-4), 7.84 (d, J = 8.3 Hz, 1H, H-6″).
5-Bromo-1-(2-chlorobenzyl)-3-[(4-fluorophenyl)(hydroxy)methyl]-1H-indole (28)
This compound was prepared from precursor 24. Yield: 93%, brown solid, mp 101–103°C (isopropyl ether). IR (KBr, cm− 1) 3274 (OH); 1H NMR (DMSO d6) δ 5.51 (s, 2H, CH2), 5.85 (d*, J = 4.5 Hz, 1H, OH), 5.99 (d, J = 4.5 Hz, 1H, CH), 6.75 (d, J = 7.3 Hz, 1H, H-6′), 7.15-7.55 (m, 8H, H-2, H-6, H-7, H-3′, H-4′, H-5′, H-3″, H-5″), 7.50 (dd, J = 5.9, 7.2 Hz, 2H, H-2″, H-6″), 7.67 (d, J = 1.7 Hz, 1H, H-4).
5-Bromo-1-(4-fluorobenzyl)-3-[(4-fluorophenyl)(hydroxy)methyl]-1H-indole (29)
This compound was prepared from precursor 25. Yield: 95%, white solid, mp 125–127°C (cyclohexane). IR (KBr, cm− 1) 3114 (OH); 1H NMR (DMSO d6) δ 5.40 (s, 2H, CH2), 5.85 (d*, J = 4.5 Hz, 1H, OH), 5.99 (d, J = 4.5 Hz, 1H, CH), 7.14–7.24 (m, 5H, H-6, H-3′, H-5′, H-3″, H-5″), 7.28 (dd, J = 5.9 Hz, 8.6 Hz, 2H, H-2′, H-6′), 7.38 (s, 1H, H-2), 7.45 (d, J = 8.8 Hz, 1H, H-7), 7.51 (dd, J = 5.8, 8.5 Hz, 2H, H-2″, H-6″), 7.67 (d, J = 1.7 Hz, 1H, H-4).
Synthesis of imidazole derivatives 11–14, 30, 31, 34 and 35
A solution of the appropriate carbinol derivative 7–10 or 26–29 (3.70 mmol) and CDI (3.70 mmol) in dry THF (20 mL) was stirred at room temperature for 3 h. The reaction mixture was partitioned between H2O and diethyl ether and extracted three times with diethyl ether. The combined organic layers were dried (Na2SO4) and concentrated. The residue was purified by CC (CH2Cl2:EtOH = 19:1).
1-(2-Chlorobenzyl)-3-(1H-imidazol-1-ylmethyl)-1H-indole (11)
This compound was prepared from precursor 7. Yield: 42%, white solid, mp 45–47°C (isopropyl ether). 1H NMR (DMSO d6) δ 5.38 (s, 2H, CH2), 5.54 (s, 2H, CH2), 6.80 (dd, J = 7.5, 1.8 Hz, 1H, H-6′), 6.88 (s, 1H, Himide), 7.06–7.20 (m, 2H, H-5, H-6), 7.20 (s, 1H, Himide), 7.28 (m, 1H, H-5′), 7.35 (m, 1H, H-4′), 7.43 (d, J = 8.0 Hz, 1H, H-7), 7.55 (dd, J = 7.8, 1.3 Hz, 1H, H-3′), 7.59 (s, 1H, H-2), 7.62 (d, J = 7.7 Hz, 1H, H-4), 7.80 (s, 1H, Himide).
1-(2-Fluorobenzyl)-3-(1H-imidazol-1-ylmethyl)-1H-indole (12)
This compound was prepared from precursor 8. Yield: 42%, white solid, mp 65–68°C (isopropyl ether). 1H NMR (DMSO d6) δ 5.38 (s, 2H, CH2), 5.51 (s, 2H, CH2), 6.91 (s, 1H, Himide), 7.06–7.41 (m, 6H, H-5, H-6, H-3′, H-4′, H-5′, H-6′), 7.21 (s, 1H, Himide), 7.53 (d, J = 8.1 Hz, 1H, H-7), 7.61 (s, 1H, H-2), 7.63 (d, J = 7.7 Hz, 1H, H-4), 7.82 (s, 1H, Himide).
1-(3-Fluorobenzyl)-3-(1H-imidazol-1-ylmethyl)-1H-indole (13)
This compound was prepared from precursor 9. Yield: 92%, white solid, mp 62–65°C (cyclohexane). 1H NMR (DMSO d6) δ 5.37 (s, 2H, CH2), 5.47 (s, 2H, CH2), 6.88 (s, 1H, Himide), 7.04–7.16 (m, 5H, H-5, H-6, H-2′, H-4′, H-6′), 7.20 (s, 1H, Himide), 7.34–7.43 (m, 1H, H-5′), 7.49 (d, J = 8.2 Hz, 1H, H-7), 7.60 (d, J = 7.7 Hz, 1H, H-4), 7.66 (s, 1H, H-2), 7.81 (s, 1H, Himide); IR (KBr, cm− 1) 1086.
1-(4-Cyanobenzyl)-3-(1H-imidazol-1-ylmethyl)-1H-indole (14)
This compound was prepared from precursor 10. Yield: 60%, white solid, mp 102–104°C (isopropyl ether). IR (KBr, cm− 1) 2229 (CN); 1H NMR (DMSO d6) δ 5.38 (s, 2H, CH2), 5.57 (s, 2H, CH2), 6.89 (s, 1H, Himide), 7.04–7.16 (m, 2H, H-5, H-6), 7.21 (s, 1H, Himide), 7.36 (d, J = 8.2 Hz, 2H, H-2′, H-6′), 7.45 (d, J = 8.1 Hz, 1H, H-7), 7.60 (d, J = 7.6 Hz, 1H, H-4), 7.65 (s, 1H, H-2), 7.81 (s, 1H, Himide), 7.83 (d, J = 8.2 Hz, 2H, H-3′, H-5′).
5-Bromo-1-ethyl-3-[(4-fluorophenyl)(1H-imidazol-1-yl)methyl]-1H-indole (30)
This compound was prepared from precursor 26. Yield: 71%, white solid, mp 143–145°C (isopropyl ether). 1H NMR (DMSO d6) δ 1.33 (t, J = 7.2 Hz, 3H, CH3), 4.22 (q, J = 7.2 Hz, 2H, CH2), 6.97 (s, 1H, Himide), 7.12 (s, 1H, CH), 7.14 (s, 1H, H-2), 7.20 (s, 1H, Himide), 7.24–7.30 (m, 2H, H-3″, H-5″), 7.30–7.34 (m, 4H, H-4, H-6, H-2″, H-6″), 7.54 (d, J = 8.6 Hz, 1H, H-7), 7.75 (s, 1H, Himide).
5-Bromo-3-[(2,4-dichlorophenyl)(1H-imidazol-1-yl)methyl]-1-ethyl-1H-indole (31)
This compound was prepared from precursor 27. Yield: 66%, white solid, mp 138–140°C (isopropyl ether/EtOH). 1H NMR (DMSO d6) δ 1.32 (t, J = 7.1 Hz, 3H, CH3), 4.21 (q, J = 7.1 Hz, 2H, CH2), 6.91 (d, J = 8.4 Hz, 1H, H-6″), 6.99 (s, 1H, Himide), 7.07 (s, 1H, H-2), 7.15 (s, 1H, Himide), 7.27 (s, 1H, CH), 7.34 (dd, J = 8.8, 8.8 Hz, 1H, H-6), 7.49 (d, J = 1.7 Hz, 1H, H-4), 7.52 (dd, J = 2.1, 8.4 Hz, 1H, H-5″), 7.56 (d, J = 8.8 Hz, 1H, H-7), 7.71 (s, 1H, Himide), 7.75 (d, J = 2.1 Hz, 1H, H-3″).
5-Bromo-1-(2-chlorobenzyl)-3-[(4-fluorophenyl)(1H-imidazol-1-yl)methyl]-1H-indole (34)
This compound was prepared from precursor 28. Yield: 71%, white crystals, mp 145°C (isopropyl ether/MeOH, decom position). 1H NMR (base) (DMSO d6) δ 5.55 (s, 2H, CH2), 6.70 (d, J = 7.1 Hz, 1H, H6′), 6.99 (s, 1H, Himide), 7.21 (s, 1H, Himide), 7.17–7.42 (m, 11H, H-2, H-4, H-6, H-7, H-4′, H-5′, H-2″, H-3″, H-5″, H-6″, CH), 7.53 (d, J = 7.6 Hz, 1H, H-3′), 7.77 (s, 1H, Himide).
5-Bromo-1-(4-fluorobenzyl)-3-[(4-fluorophenyl)(1H-imidazol-1-yl)methyl]-1H-indole (35)
This compound was prepared from precursor 29. Yield: 80%, white solid, mp 140–142°C (cyclohexane/diethyl ether). 1H NMR (DMSO d6) δ 5.50 (s, 2H, CH2), 6.99 (s, 1H, Himide), 7.15–7.33 (m, 12H, H-2, H-4, H-6, H-2′, H-3′, H-5′, H-6′, H-2″ H-3″ H-5″ H-6″, CH), 7.21 (s, 1H, Himide), 7.47 (d, J = 8.7 Hz, 1H, H-7), 7.77 (s, 1H, Himide).
Synthesis of triazole derivatives 15–18, 32, 33, 36 and 37
Thionyl chloride (11.8 mmol) was dropped into an ice-cooled solution of 1H-1,2,4-triazole (47.2 mmol) in dry acetonitrile (30 mL). The mixture was stirred at room temperature for 1 h, then filtered. This solution was added dropwise to a solution of the appropriate carbinol derivative 7–10 or 26–29 (3.0 mmol) in dry acetonitrile (10 mL). After addition, the mixture was stirred at room temperature for 2 h, then filtered and concentrated. The residue was dissolved in CH2Cl2. The organic solution was washed with brine, dried over Na2SO4, filtered, and evaporated to provide the crude product. This mixture was purified by CC (CH2Cl2:EtOH = 19:1). In the case of compounds 15 and 16, appropriate eluates were collected and evaporated to give the corresponding regioisomers.
1-(2-Chlorobenzyl)-3-(1H-1,2,4-triazol-1-ylmethyl)-1H-indole (15a)
This compound was prepared from precursor 7. Yield: 74%, pale yellow crystals, mp 65–66°C (cyclohexane/CH2Cl2). 1H NMR (DMSO d6) δ 5.54 (s, 2H, CH2), 5.60 (s, 2H, CH2), 6.81 (d, J = 7.5 Hz, 1H, H-6′), 7.07–7.12 (m, 1H, H-5), 7.15–7.20 (m, 1H, H-6), 7.24–7.30 (m, 1H, H-5′), 7.33–7.38 (m, 1H, H-4′), 7.43 (d, J = 8.1 Hz, 1H, H-7), 7.55 (d, J = 7.8 Hz, 1H, H-3′), 7.60 (s, 1H, H-2), 7.67 (d, J = 7.7 Hz, 1H, H-4), 7.97 (s, 1H, Htriazole), 8.64 (s, 1H, Htriazole).
1-(2-Chlorobenzyl)-3-(4H-1,2,4-triazol-4-ylmethyl)-1H-indole (15b)
This compound was prepared from precursor 7. Yield: 6%, white solid, mp 130°C (diisopropyl ether). 1H NMR (DMSO d6) δ 5.47 (s, 1H, CH2), 5.55 (s, 1H, CH2), 6.83 (d, J = 7.1 Hz, 1H, H-6′), 7.08–7.22 (m, 2H, H-5, H-6), 7.25–7.31 (m, 1H, H-5′), 7.33–7.39 (m, 1H, H-4′), 7.44 (d, J = 8.1 Hz, 1H, H-7), 7.55 (d, J = 7.8 Hz, 1H, H-3′), 7.62 (s, 1H, H-2), 7.66 (d, J = 7.6 Hz, 1H, H-4), 8.62 (s, 2H, Htriazole).
1-(2-Fluorobenzyl)-3-(1H-1,2,4-triazol-1-ylmethyl)-1H-indole (16a)
This compound was prepared from precursor 8. Yield: 29%, brown solid, mp 121–123°C (diisopropyl ether/CH2Cl2). 1H NMR (DMSO d6) δ 5.50 (s, 2H, CH2), 5.58 (s, 2H, CH2), 7.05–7.42 (m, 6H, H-5, H-6, H-3′, H-4′, H-5′, H-6′), 7.52 (d, J = 8.2 Hz, 1H, H-7), 7.61 (s, 1H, H-2), 7.65 (d, J = 7.7 Hz, 1H, H-4), 7.96 (s, 1H, Htriazole), 8.64 (s, 1H, Htriazole).
1-(2-Fluorobenzyl)-3-(4H-1,2,4-triazol-4-ylmethyl)-1H-indole (16b)
This compound was prepared from precursor 8. Yield: 24%, yellow crystals, mp 118–120°C (diisopropyl ether/CH2Cl2). 1H NMR (DMSO d6) δ 5.46 (s, 2H, CH2), 5.51 (s, 2H, CH2), 7.07–7.39 (m, 6H, H-5, H-6, H-3′, H-4′, H-5′, H-6′), 7.53 (d, J = 8.0 Hz, 1H, H-7), 7.64 (s, 1H, H-2), 7.65 (d, J = 7.8 Hz, 1H, H-4), 8.62 (s, 2H, Htriazole).
1-(3-Fluorobenzyl)-3-(1H-1,2,4-triazol-1-ylmethyl)-1H-indole (17)
This compound was prepared from precursor 9. Yield: 65%, white solid, mp 135–137°C (cyclohexane). 1H NMR (DMSO d6) δ 5.48 (s, 2H, CH2), 5.59 (s, 2H, CH2), 7.05–7.20 (m, 5H, H-5, H-6, H-2′, H-4′, H-6′), 7.35–7.43 (m, 1H, H-5′), 7.50 (d, 1H, H-7, J = 8.0 Hz), 7.65 (d, 1H, H-4, J = 7.60 Hz), 7.68 (s, 1H, H-2), 7.96 (s, 1H, Htriazole), 8.65 (s, 1H, Htriazole).
1-(4-Cyanobenzyl)-3-(1H-1,2,4-triazol-1-ylmethyl)-1H-indole (18)
This compound was prepared from precursor 10. Yield: 63%, white solid, mp 104–106°C (isopropyl ether). IR (KBr, cm− 1) 2226; 1H NMR (DMSO d6) δ 5.58 (s, 2H, CH2), 5.60 (s, 2H, CH2), 7.08–7.17 (m, 2H, H-5, H-6), 7.37 (d, J = 8.1 Hz, 2H, H-2′, H-6′), 7.45 (d, J = 7.9 Hz, 1H, H-7), 7.64–7.68 (m, 2H, H-2, H-4), 7.83 (d, J = 8.1 Hz, 2H, H3′, H-5′), 7.97 (s, 1H, Htriazole), 8.65 (s, 1H, Htriazole).
5-Bromo-1-ethyl-3-[(4-fluorophenyl)(1H-1,2,4-triazol-1-yl)methyl]-1H-indole (32)
This compound was prepared from precursor 26. Yield: 67%, white solid, mp 126–128°C (isopropyl ether/EtOH). 1H NMR (DMSO d6) δ 1.33 (t, J = 7.20 Hz, 3H, CH3), 4.22 (q, J = 7.20 Hz, 2H, CH2), 7.22–7.27 (m, 3H, H-2, H-3″, H-5″), 7.28–7.35 (m, 2H, H-6, CH), 7.37–7.42 (m, 2H, H-2″, H-6″), 7.43 (d, J = 1.50 Hz, 1H, H-4), 7.54 (d, J = 8.70 Hz, 1H, H-7), 8.08 (s, 1H, Htriazole), 8.67 (s, 1H, Htriazole).
5-Bromo-3-[(2,4-dichlorophenyl)(1H-1,2,4-triazol-1-yl)methyl]-1-ethyl-1H-indole (33)
This compound was prepared from precursor 27. Yield: 30%, white solid, mp 124–126°C (acetonitrile). 1H NMR (DMSO d6) δ 1.33 (t, J = 7.2 Hz, 3H, CH3), 4.22 (q, J = 7.2 Hz, 2H, CH2), 7.18 (s, 1H, H-2), 7.22 (d, J = 8.5 Hz, 1H, H-6″), 7.33 (dd, J = 1.8, 8.7 Hz, 1H, H-6), 7.48 (s, 1H, CH), 7.49–7.57 (m, 3H, H-4, H-7, H-5″), 7.76 (d, J = 2.0 Hz, 1H, H-3″), 8.11 (s, 1H, Htriazole), 8.69 (s, 1H, Htriazole).
5-Bromo-1-(2-chlorobenzyl)-3-[(4-fluorophenyl)(1H-1,2,4-triazol-1-yl)methyl]-1H-indole (36)
This compound was prepared from precursor 28. Yield: 70%, pale yellow crystals, mp 145–147°C (MeOH/CH2Cl2). 1H NMR (DMSO d6) δ 5.56 (s, 2H, CH2), 6.73 (d, J = 6.5 Hz, 1H, H-6′), 7.23-7.47 (m, 11H, H-2, H-4, H-6, H-7, H-4′, H-5′, H-2″, H-3″, H-5″, H-6″, CH), 7.53 (d, J = 7.6 Hz, 1H, H-3′), 8.10 (s, 1H, Htriazole), 8.71 (s, 1H, Htriazole).
5-Bromo-1-(4-fluorobenzyl)-3-[(4-fluorophenyl)(1H-1,2,4-triazol-1-yl)methyl]-1H-indole (37)
This compound was prepared from precursor 29. Yield: 85%, white solid, mp 163–165°C (cyclohexane). 1H NMR (DMSO d6) δ 5.45 (s, 2H, CH2), 7.17 (dd, J = 8.7, 8.7 Hz, 2H, H-3′, H-5′), 7.24–7.30 (m, 4H, H-2′, H-6′, H-3″, H-5″), 7.28 (dd, J = 1.8, 8.7 Hz, 1H, H-6), 7.38–7.45 (m, 5H, H-2, H-4, H-2″, H-6″, CH), 7.48 (d, J = 8.6 Hz, 1H, H-7), 8.09 (s, 1H, Htriazole), 8.69 (s, 1H, Htriazole).
Pharmacology
Inhibition of human placental aromatase (in Vitro)
The compounds were tested for aromatase inhibitory activity according to the procedure of Thompson and Siiteri [Citation35]. The microsomal fraction of freshly delivered human term placenta provided the source of the aromatase enzyme and [1β,2β-3H]testosterone was used as substrate [Citation36].
Inhibition of rat testicular P450 17α (in Vitro)
Rat testicular microsomes were used as source of the enzyme, and nonlabeled progesterone served as substrate. The separation of the steroids was accomplished using the procedure recently described by Sergejew and Hartmann [Citation37].
Results and discussion
Chemistry
3-(Azolylmethyl)-1H-indoles incorporating differently substituted R groups were synthezised from indole-3-carbaldehyde 2 (see Scheme ). N-Substitution of indole-3-carbaldehyde 2 with benzyl chlorides, using potassium carbonate in acetone, afforded the N-substituted indole derivatives 3–6. Reduction of the carbonyl group was carried out by using sodium borohydride [Citation38] in methanol and provided 7–10. The synthesis of imidazolyl-substituted compounds 11–14 was achieved by the direct reaction of the carbinols 7–10 with 1,1′-carbonyldiimidazole (CDI) in THF [Citation39], via decarboxylation of the carbamate intermediate. The triazolyl-substituted compounds 15–18 were prepared by the direct reaction of the corresponding carbinols with sulfinylditriazole (SDT) [Citation40], which was readily generated from triazole and SOCl2 in acetonitrile.
Scheme 1 Synthetic route to 3-azolylmethyl-1H-indoles 11–14. Reagents: (a) see reference [Citation34]; (b) K2CO3, acetone, RCl; (c) NaBH4, methanol; (d) CDI, THF; (e) SDT, CH3CN.
![Scheme 1 Synthetic route to 3-azolylmethyl-1H-indoles 11–14. Reagents: (a) see reference [Citation34]; (b) K2CO3, acetone, RCl; (c) NaBH4, methanol; (d) CDI, THF; (e) SDT, CH3CN.](/cms/asset/5ee80dc2-41e5-4b46-b550-ab7c02ef47ed/ienz_a_265107_f0003_b.gif)
3-(α-Azolylbenzyl)-1H-indoles were obtained by two synthetic routes as depicted in Scheme . A Friedel Crafts procedure [Citation41,Citation42], using AlCl3 catalysis, was employed to give the N-ethyl-3-aroylindole derivatives 22 and 23. A direct acylation of 5-bromo-1H-indole 19 was attempted, with 4-fluorobenzoyl chloride; it gave 3-acylindole derivative 21, in moderate yield. N-Substitution of 21 was achieved with benzyl chlorides, using Cs2CO3 in acetonitrile, and gave 3-aroyl-1-benzyl-1H-indoles 24 and 25 in excellent yields. Then the corresponding alcohols 28 and 29 were obtained by using NaBH4 in methanol. Introduction of the imidazole and triazole moieties was achieved with CDI and SDT, leading to compounds 30–37.
Scheme 2 Synthetic route to 3-(azolylbenzyl)-1H-indoles 30–37. Reagents: (a) see reference [Citation33]; (b) AlCl3, CH2Cl2, ArCOCl; (c) CsCO3, CH3CN, R1Cl; (d) NaBH4, methanol; (e) CDI, THF; (f) SDT, CH3CN.
![Scheme 2 Synthetic route to 3-(azolylbenzyl)-1H-indoles 30–37. Reagents: (a) see reference [Citation33]; (b) AlCl3, CH2Cl2, ArCOCl; (c) CsCO3, CH3CN, R1Cl; (d) NaBH4, methanol; (e) CDI, THF; (f) SDT, CH3CN.](/cms/asset/510b5911-bc41-4fd3-95f4-13c39c3b7ce6/ienz_a_265107_f0004_b.gif)
Biological results
The ability of compounds under investigation to inhibit aromatase was investigated. The IC50 values and the potencies of the compounds, relative to AG, are given in Tables and . The reference standard used was aminoglutethimide, with an IC50 value of 18.5 μM.
Table II. Inhibition of P450 arom and P450 17α by 3-(azolylmethyl)-1H-indoles 11–18.
Table III. Inhibition of P450 arom and P450 17α by 3-(α-azolylbenzyl)-1H-indoles 30–37.
In the sub-series of 3-(azolylmethyl)-1H-indoles, all imidazole derivatives 11–14 were more active than their triazole analogues 15–18. Triazol-4-yl derivatives 15b and 16b showed the lowest inhibitory potencies (IC50 > 3.5 μM). The presence of a chloro substituent in the ortho position of the N-benzyl moiety gave a positive result (compare compound 11 with respect to compound 12). The presence of a fluorine atom at position 3 of the N-benzyl substituent improved the antiaromatase activity: compound 13 is 1.5 times more active than parent compound 12. Imidazole derivative 14 in which the benzyl moiety was substituted by a 4-cyano group, was the most active, with an IC50 value of 0.050 μM. All compounds of this sub-series did not exhibit any significant inhibitory activity on rat testicular P450 17α. The highest percentage inhibition value found was 70 with compound 13 at the concentration of 2.5 μM. IC50 value was only calculated in the case of percentage inhibition value superior to 85%.
shows the in vitro data of the antiaromatase activity of 3-(α-azolylbenzyl)-1H-indoles 30–37. The N-ethyl derivative 30 showed an IC50 value of 0.052 μM and this imidazole derivative was the best inhibitor of this sub-series. Further structure-activity study regarding the presence of a 2,4-dichlorophenyl substituent did not improve activity (compound 30 compared with 31). The replacement of the N-ethyl chain of compound 30 by the N-halobenzyl moiety (2-chloro or 4-fluoro) gave negative results for compounds 32 and 33. The triazole derivative 34 was 19 times less active than its imidazole analogue 30. Other triazole analogues 35–37 were less active than parent molecules 31–33. As found with the first sub-series, all 3-(α-azolylbenzyl)-1H-indoles 30–37 were poor P450 17α inhibitors, with no significant percentage inhibition values ( < 3.7).
In order to evaluate the in vivo efficacy, the effect of select 3-(azolylmethyl)-1H-indoles and 3-(α-azolylbenzyl)-1H-indoles on the androgen-stimulated uterine growth will be determined. In juvenile female rats, androstedione treatment strongly stimulates uterine weight. This effect is due to ovarian aromatization of the androgen and can be dose-dependently antagonized by aromatase inhibitors such as compounds 14 and 30. Thus the antiuterotrophic activity could give further biological data to continue our research. Furthermore we are doing molecular modelling studies on compound 14 and 30 to better understand their interactions in the active site of P450 arom and then to be able to design new non steroidal aromatase inhibitors.
References
- Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD, Mendelson CR, Bulun SE. Endocr Rev 1994; 15: 342–355
- Kendall A, Folkerd EJ, Dowsett M. J Steroid Biochem Mol Biol 2007; 103: 99–109
- Mokbel K. Int J Clin Oncol 2002; 7: 279–283
- Miller WR. Semin Oncol 2003; 30(Suppl. 14)3–11
- Wiseman LR, McTavish D. Drugs 1993; 45: 66–84
- Jordan VC, Brodie AMH. Steroids 2007; 72: 7–25
- Lombardi P. Curr Pharmaceut Des 1995; 1: 23–50
- Grau A, Castaner J. Drugs Fut 1996; 21: 1028–1031
- Numazawa M, Oshibe M, Yamaguchi S, Tachibana M. J Med Chem 1996; 39: 1033–1038
- Numazawa M, Kamiyama T, Tachibana M, Oshibe M. J Med Chem 1996; 39: 2245–2252
- Johannessen DC, Engan T, Di Salle E, Zurlo MG, Paolini J, Ornati G, Piscitelli G, Kvinnsland S, Lonning PE. Clin Cancer Res 1997; 3: 1101–1108
- Brueggemeier RW. Breast Cancer Res Treat 1994; 30: 31–42
- Brodie A, Brodie HB, Callard G, Robinson C, Roselli C, Santen R. J Steroid Biochem Mol Biol 1993; 44: 321–696
- Foster AB, Jarman M, Leung CS, Rowlands MG, Taylor GN, Plevey RG, Sampson P. J Med Chem 1985; 28: 200–204
- Daly MJ, Jones GW, Nicholls PJ, Smith HJ, Rowlands MG, Bunnett MA. J Med Chem 1986; 29: 520–523
- Hartmann RW, Batzl C, Pongratz TM, Mannschreck A. J Med Chem 1992; 35: 2210–2214
- Bossche HV. J Steroid Biochem Mol Biol 1992; 43: 1003–1021
- Karjalainen A, Kalapudas A, Södervall M, Pelkonen O, Lammintausta R. Eur J Pharm Sci 2000; 11: 109–131
- Santen RJ, Samojlik E, Worgul TJ. In a Comprehensive Guide to the Therapeutic Use of Aminoglutethimide. Karger, Basel 1982; 101–160
- Brodie AMH, Njar VCO. Semin Oncol 1996; 23(Supplement 9)10–20
- Okada M, Yoden T, Kawaminami E, Shimada Y, Kudoh M, Isomura Y, Shikama H, Fujikura T. Chem Pharm Bull 1996; 44: 1871–1879
- Ahokoski O, Irjala K, Huupponen R, Halonen K, Salminen E, Scheinin H. Br J Clin Pharmacol 1998; 45: 141–146
- Sorbera LA, Silvestre J, Castaner J. Drugs Fut 1998; 23: 1071–1074
- Costa LA, Kopreski MS, Demers LM, Chinchilli VM, Santen RJ, Harvey HA, Lipton A. Cancer 1999; 85: 100–103
- Goss PE, Walde D, De Coster R, Langenaeken C. Oncol 1999; 56: 114–121
- Bhatnagar AS, Häusler A, Schieweck K, Lang M, Bowman R. J Steroid Biochem Mol Biol 1990; 37: 1021–1027
- Plourde PV, Dyroff M, Dukes M. Breast Cancer Res Treat 1994; 30: 103–111
- Kudoh M, Susaki Y, Ideyama Y, Nanya T, Okada M, Shikama H, Fujikura T. J Steroid Biochem Mol Biol 1995; 54: 265–271
- Ahokoski O, Irjala K, Huhtala S, Salminen E, Scheinin H, Huupponen RA. Eur J Clin Pharmacol 1999; 55: 27–34
- Bossche HV, Moereels H, Koymans LMH. Breast Cancer Res Treat 1994; 30: 43–55
- Le Borgne M, Marchand P, Duflos M, Delevoye-Seiller B, Robert-Piessard S, Le Baut G, Hartmann RW, Palzer M. Arch Pharm Pharm Med Chem 1997; 330: 141–145
- Le Borgne M, Marchand P, Delevoye-Seiller B, Robert J-M, Le Baut G, Hartmann RW, Palzer M. Bioorg Med Chem Lett 1999; 9: 333–336
- Lézé M-P, Le Borgne M, Marchand P, Loquet D, Kogler M, Le Baut G, Palusczak A, Hartmann RW. J Enz Inhib Med Chem 2004; 19: 549–557
- James PN, Snyder HR. Org Synth 1959; 39: 30–33
- Thompson EAJr, Siiteri PK. J Biol Chem 1974; 249: 5364–5372
- Hartmann RW, Batzl C. J Med Chem 1986; 29: 1362–1369
- Sergejew T, Hartmann RW. J Enz Inhib 1994; 8: 113–122
- Cavrini V, Gatti R, Roveri P, Cesaroni MR. Arch Pharm Pharm Med Chem 1984; 317: 662–668
- Jones CD, Winter MA, Hirsch KS, Stamm N, Taylor HM, Holden HE, Davenport JD, Krumkalns EV, Suhr RG. J Med Chem 1990; 33: 416–429
- Massa S, Di Santo R, Retico A, Artico M, Simonetti N, Fabrizi G, Lamba D. Eur J Med Chem 1992; 27: 495–502
- Yamada K, Rice KC, Flippen-Anderson JL, Eissenstat MA, Ward SJ, Johnson MR, Howlett AC. J Med Chem 1996; 39: 1967–1974
- Bell MR, D'Ambra TE, Kumar V, Eissenstat MA, Herrmann JL, Jr, Wetzel JR, Rosi D, Philion RE, Daum SJ, Hlasta DJ, Kullnig RK, Ackerman JH, Haubrich DR, Luttinger DA, Baizman ER, Miller MS, Ward SJ. J Med Chem 1991; 34: 1099–1110