Abstract
Quinazolinone derivatives have been studied as both in vitro and in vivo inhibitors of aspartate transcarbamylase (ATCase). In vitro treatment of mammalian ATCase with four compounds revealed that they inhibited enzyme activity and that 2-phenyl-1,3-4(H)benzothiazin-4-thione was the most potent one. This compound acts as a noncompetitive inhibitor towards both aspartate and carbamoyl phosphate. The values of the inhibition constant (Ki) indicate that this compound exerts a potent inhibitory effect upon ATCase activity. Moreover, in vivo treatment with different doses of these derivatives showed also an inhibitory effect upon ATCase, the relative activity being decreased by 40%–58% with a 1 mg dose. These data support the inhibition of ATCase by quinazolinone derivatives as a new type of inhibitor for the enzyme.
Introduction
Quinazolinone derivatives are structurally related to pyrimidines as a continuation of our own interest in the synthesis and modification of these derivatives with pharmacological activities Citation1-14. They possess a wide range of biological properties as some derivatives act as anti-inflammatory agents by inhibiting the cyclooxygenase II [Citation15]. Also, some derivatives reduce prostaglandin levels and significantly lower protein concentration and polymorphonuclear leukocytes number [Citation16]. Quinazolinone derivatives act as anticancer agents by inhibiting some enzymes involved in tumor progress as tyrosine kinase [Citation17], poly (ADP-ribose) polymerase [Citation18,Citation19] and DNA topoisomerases [Citation20]. Also, quinazolinones have antiviral effect by inhibiting the human immunodeficiency virus -1 reverse transcriptase [Citation21,Citation22].
Aspartate transcarbamylase (abbreviated as ATCase) catalyzes the second reaction in pyrimidine biosynthesis, which is the carbamylation of the α-amino group of l-aspartate by carbamyl phosphate (abbreviated CP) to yield carbamyl aspartate and inorganic phosphate Citation23-25. In eukaryotes, five of six enzymes that involved in pyrimidine biosynthesis are clustered in two complexes whereas the corresponding bacterial enzymes appear to be unassociated. For example, the initial steps in mammalian pyrimidine biosynthesis are catalyzed by a multifunctional protein called CAD, that has carbamyl phosphate synthetase II, ATCase and dihydroorotase activities [Citation26,Citation27]. The multifunctional protein, CAD, is comprised of a single 243-kDa polypeptide that is organized into separate functional domains [Citation26,Citation27]. It is well known that CAD has a major role in the regulation of de novo pyrimidine biosynthesis [Citation27,Citation28]. In a previous study, it was found that hepatic ATCase is inhibited by a phenobarbitol derivative as a particularly related compound to pyrimidine [Citation29]. Taken together, the current study is undertaken to look for another structurally related compound to pyrimidines as a potent inhibitor to mammalian ATCase. Based on the reported biological importance of quinazolinone derivatives, these compounds were subjected to both in vitro and in vivo characterization of the mammalian enzyme.
Materials and methods
Animals
Eight-week-old male mice of Swiss albino strain weighing approximately 20 g were from the animal house, Faculty of Medicine, Alexandria University. Mice were housed with available food and water and 12 h photoperiod under conventional conditions.
Materials
Dilithium CP, carbamyl aspartate, antipyrine, diacetyl-monoxime and sephadex G-25 were purchased from Sigma Chemical Co. (St. Louis, Mo, USA). Folin-Ciocalteu's phenol reagent, l-aspartic acid and mercaptoethanol were purchased from Merck, Hohenbrunn. Bovine serum albumin (Fract V) was purchased from Fisher Biotech (Fisher Scientific Fair Lawn, N. J., USA).
Synthesis of compounds 1, 2, 3 and 4
Compounds 1, 2, 3 and 4 () were prepared according to the methods described previously Citation30-35. The structure and purity of compounds 1, 2, 3 and 4 were confirmed using different methods. The difference in melting points of these compounds (238, 123, 248 and 228°C, respectively) and the obtained data of the IR spectrum for each examined compound is consistent with the proposed structure. Further evidence of the examined compounds was obtained from 1H-NMR, 13C-NMR, 13C-NMR DEPT and mass spectra. 1H-NMR spectral data: compound 1, quinazolinone protons at δ 8.07, 7.78, 7.55 and 7.66 ppm corresponding to H-5, H-6, H-7 and H-8 of benzene moiety, while NH proton resonates at down field ca 12.2 ppm disappeared by addition of D2O; compound 2, H-5, H-6, H-7 and H-8 protons at 8.8, 8.1, 7.83 and 7.92 ppm, respectively; compound 3, the absence of H-6 and H-8 protons, while the phenyl group resonates at 7.4–7.7 ppm region and the two protons of NH2 resonate at 3.3 ppm disappear by D2O addition; compound 4, H-5 and H-7 at 8.4 and 8.3 ppm, respectively. The methyl group at position 2- resonates at 2.6 ppm as singlet corresponding to three protons. 13C-NMR spectral data showed the chemical shifts of C-2, C-4, C-4a, C-5, C-6, C-7, C-8 and C-8a of quinazolinone nucleus, while the assignment of substitution moiety (methyl group in compounds 1 and 4) could be seen at δ = 22.24 ppm and 22.8 ppm, respectively. On the other hand, the phenyl group at position 2- of the quinazolinone nucleus is shown in compounds 2 and 3. 13C-NMR DEPT spectra: compound 1, the disappearance of quaternary carbons at C-2, C-4, C-4a and C-8a; compound 2, the disappearance of quaternary carbons at C-2, C-4, C-4a and C-8a; compound 3, the disappearance of quaternary carbons at C-2, C-4, C-4a, C-6, C-8 and C-8a as well as C-1 of phenyl ring substitution at position 2- of the quinazolinone nucleus; compound 4; the disappearance of carbons C-2, C-4, C-4a, C-6, C-8 and C-8a. Mass spectra: compound 1, its molecular ion peak at 160 (48%) that has peak at m/z 42 (100%) in EI technique and M + 1 m/z 161 (100%) in CI technique; compound 2, its molecular ion peak as a base peak 225 (100%) in EI technique and M + 1 m/z 226 (100%) as base peak in CI technique; compound 3, its molecular ion peak m/z 395 (30%) and its base peak at m/z 105 (100%) in EI technique. M + 1 m/z 396 (57%) and base peak at m/z 382 (100%) in CI technique; compound 4, its molecular ion peak at m/z 332 (100%) as its base peak in EI technique and M + 2 m/z 334 (78%) as appears at m/z 161 (100%).
Figure 1 The quinazolinone derivatives 1, 2, 3 and 4 tested for the inhibition of ATCase.
Isolation and purification of ATCase
Isolation and purification were done as described previously [Citation36] with some modifications [Citation29].
Enzyme assay
The activity of ATCase was assayed as described previously [Citation37] with some modifications [Citation29]. Briefly, the carbamyl aspartate production was determined in a system containing 40 mM sodium phosphate buffer, pH 8.2, 12.5 mM aspartate, 10–50 μg enzyme and 3.6 mM dilithium CP at 30°C for about 5 min in a final volume of 1.0 mL. The reaction was allowed to proceed at 30°C for 30 min and stopped by the addition of 1.0 mL of 2% HClO4 followed by protein removal by centrifugation. The color reagent was prepared immediately before use by mixing two parts of antipyrine-H2SO4 reagent with one part of diacetylmonoxime reagent and 3.0 mL of the color reagent were added to each assay tube. The reaction tubes were capped with marbles, covered with aluminum foil and stored in a dark place at room temperature for 24 h. The tubes were then incubated in a 45°C water bath for 70 min, cooled in cold water and measured for absorbance at 466 nm versus blank (zero time incubation). The production of carbamyl aspartate is measured by a standard curve.
In vitro studies
Determination of the IC50 of compounds 1, 2, 3 and 4
ATCase activity towards compounds 1, 2, 3 and 4 was carried out using 0.1 mL of different concentrations of these compounds (0.05, 0.1, 0.15, 0.2, 0.25 and 0.3 mM in ethanol). Into each tube, the assay mixture was added in a final concentration as mentioned above. Each tube was incubated at 30°C for 30 min and the reaction was then stopped by the addition of 1 mL of 2% HClO4. Parallel control experiments, containing 0.1 mL of ethanol instead of compounds 1, 2, 3 or 4 were run. The color reaction was developed as mentioned above.
Detection of the reversibility of binding of compounds 2 and 3 to ATCase
The reversibility of binding of each compound to ATCase was studied as described before for other enzymes [Citation38,Citation39]. Briefly, 1.0 mL of 0.2 mM of each compound was added to 1.0 mL of the enzyme for 60 min at room temperature and the mixture was then put in a cellophane bag. Dialysis was allowed to proceed for 12 h by keeping the cellophane bag submerged in 10 mM sodium phosphate buffer, pH 7.4 with 5 changes. A parallel control was also performed by adding 1.0 ml of ethanol to 1.0 ml of the enzyme. From each cellophane bag, 0.2 ml of the mixture was withdrawn and assayed as mentioned above in sodium phosphate buffer, pH 8.2.
Determination of the kinetic parameters and the inhibition constant (Ki)
The effect of Asp concentration was carried out in a reaction mixture containing different concentrations of Asp (02.0–12.5 mM), 3.6 mM dilithium CP at pH 8.2 and 30°C for 5 min the reactions proceeded in the absence and presence of each compound (0.05, 0.1, 0.15 and 0.2 mM). Also, the effect of CP concentration was carried using 12.5 mM Asp and different concentrations (1.0–3.6 mM) of dilithium CP at pH 8.2 and 30°C for 5 min. One ml of the assay mixture was withdrawn and added to 1.0 mL of 2% HClO4 at 5 min time interval. The denatured protein was removed by centrifugation. The color reaction was developed and absorbance read at 466 nm. The kinetic parameters of ATCase (Km and Vmax) and the inhibition constant (Ki) were determined from Lineweaver Burk plot in which 1/v was plotted against 1/[S] [Citation40]. Ki values were calculated by taking the slops of the linear part of the plot. Also Dixon plot in which 1/v is plotted against different concentrations of the inhibitor [I] was used to calculate Ki value [Citation41].
In vivo studies
LD50 of quinazolinone derivatives
LD50 can be determined as described previously [Citation42]. Thirty mice divided into 6 groups and each group includes five mice. Different doses of compounds 1, 2, 3 and 4 were prepared in very small amount of ethanol, completed to 0.2 mL with corn oil and administrated by oral injection using gavage for two days. LD50 can be calculated from proportionate distance (P.D) where LD50 is the dose above 50% + P.D. Animal work was conducted in accordance with the institutions protocol.
Effect of quinazolinone derivatives on the activity of ATCase
In vivo effect of compounds 1, 2, 3 and 4 on ATCase was studied as described previously for other enzymes [Citation43]. Briefly, mice were allocated randomly into six groups and each group contained five mice. Each group was treated with specific dose of compounds 1, 2, 3 and 4 (200, 400, 600, 800 and 1000 μg in 0.2 mL of corn oil/day) for two days using gavage. The vehicle-treated group (control) was incubated with 0.2 mL of corn oil per day for two days. Mice were sacrificed after 24 h from the final treatment and their livers were removed and prepared for the enzyme assay. The specific and relative activities were estimated.
Protein assay
Protein concentration was determined using bovine serum albumin as a standard [Citation44].
Statistical analyses
Data analyses involved estimation of means, mean standard errors ( ± SEs) and probabilities (p values) for each of the groups. Student's t-test was used to determine statistical differences among the groups. Statistical significance was identified at p < 0.05.
All experiments were run on three occasions for reproducibility and all assays were done in triplicate.
Results
In vitro treatment of ATCase
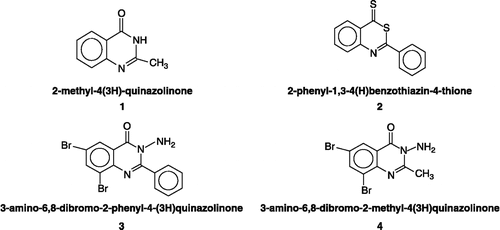
As illustrated in (A, right and left panels; B, right and left panels), the values of relative activities of ATCase reveal that the enzyme is inhibited by compounds 1, 2, 3 and 4. The panels of (A and B) show that ATCase is inhibited by different compounds in a concentration-dependent manner. The observed IC50 values are 0.20, 0.15, 0.18 and 0.35 mM, respectively as shown in . The data imply that ATCase is inhibited by these compounds and that compounds 2 and 3 are the most potent.
Figure 2 In vitro inhibition of ATCase by quinazolinone derivatives. (A): compounds 1 (left panel) and 2 (right panel). (B): compounds 3 (left panel) and 4 (right panel). The relative activity is the activity % of control and assays were carried out at pH 8.2 and 30°C.
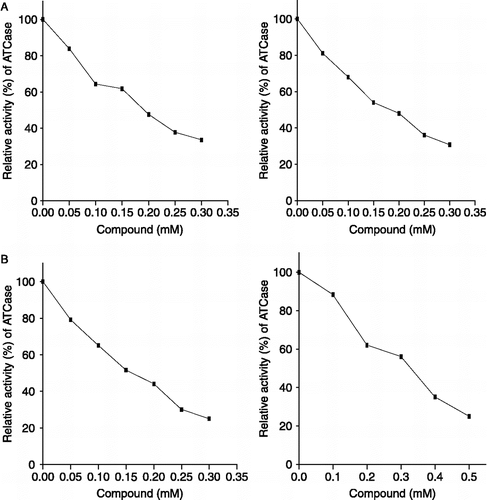
Figure 3 IC50 of the tested quinazolinone derivatives 1, 2, 3 and 4 for ATCase inhibition.
Reversibility of compound 2 binding was readily realized when the full activity of the enzyme incubated for 60 min with this compound and was recovered after its dialysis for 12 hours at 4°C. This reversible binding of compound 2 is likewise that reported for 3. As shown in , the dialysis recovered the full activity of ATCase incubated with compounds 2 or 3.
Table I. The reversibility of the binding of compounds 2 and 3 to ATCase. The enzyme was incubated in vitro with each compound for 60 min followed by dialysis for 12 h at pH 7.4. ATCase activity was then measured for the treated enzyme at 30°C and compared to control.
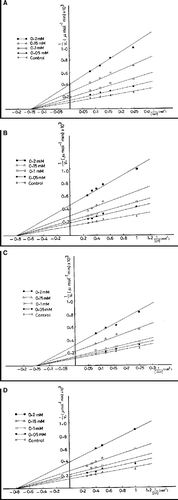
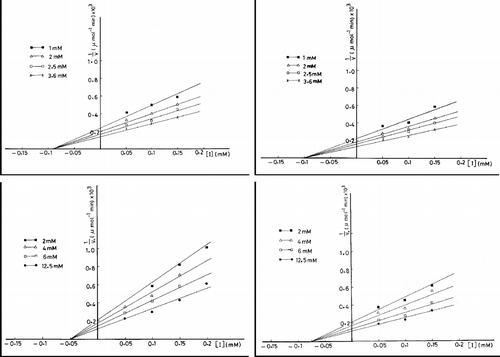
According to Lineweaver Burk plot, different concentrations of compound 2 (0.05, 0.10, 0.15 and 0.20 mM) yielded an inhibition pattern in which the lines intersected at the X-axis and Vmax of the enzyme towards Asp was decreased and the observed Km value is unchanged (, panel A). Accordingly, the type of inhibition is a noncompetitive type. The observed values of Km and dissociation constant of the enzyme inhibitor complex, Ki, were found to be 6.70 mM and 6.6 × 10− 5 M, respectively. Similarly, the same inhibition pattern of the noncompetitive type is obtained when CP is subjected as a substrate. The observed Km value of the enzyme is 1.25 mM and that of Ki value for compound 2 was calculated as 9x10− 5 M (, panel B). On treating the enzyme by different concentrations of compound 3 (0.05, 0.10, 0.15 and 0.20 mM), Vmax of the enzyme towards Asp was decreased. The observed Ki value is 8.6 × 10− 5 M. Again, the inhibition pattern is a noncompetitive type since a comparison between the control and treated enzyme with compound 3 showed that there is no change in Km value that is 6.70 mM in both cases (, panel C). On using CP as a substrate, the treated enzyme with compound 3 showed a decreased Vmax and unchanged Km that is equal to 1.25 mM. The observed Ki was found to be 1.0 × 10− 4 M (, panel D).
Figure 4 Lineweaver Burk plots of ATCase. The enzyme is assayed in the absence and presence of compound 2 (A and B) or compound 3 (C and D) at varying concentrations of Asp (A and C) and CP (B and D).
However, the observed Ki value determined by Dixon plots that is made by plotting 1/v against different concentrations of compounds 2 and 3, were nearby to those obtained by the reciprocal plots (). In the inhibition of the enzyme by compound 2 (left panels), Ki values are 5.0 × 10− 5 and 9.0 × 10− 5 M when Asp (lower panel) and CP (upper panel) are subjected as substrates, respectively. These values for the inhibition by compound 3 (right panels) are 7.5 × 10− 5 and 1.0 × 10− 4 M in the enzyme reaction towards Asp (lower panel) and CP (upper panel), respectively.
Figure 5 Dixon plots of ATCase inhibition. The enzyme is assayed at varying concentrations of CP in presence of different concentrations of compound 2 (upper panel, left) and 3 (upper panel, right) and varying concentrations of Asp in presence of different concentrations of compound 2 (lower panel, left) and 3 (lower panel, right).
In vivo treatment of ATCase
The LD50 for each compound was calculated according to that demonstrated previously [Citation25]. The observed LD50 values of compounds 1, 2, 3 and 4 are 2.60, 1.50, 2.10 and 1.36 mg /20 g body weight, respectively. Furthermore, the treated animals with different doses of compounds 1, 2, 3 and 4 for two days showed an inhibitory effect upon ATCase. The measured specific activities of the hepatic enzyme by the in vivo-treated animals are illustrated in . The corresponding relative activities are 52.50, 42.20, 47.90 and 60.00% by 1.0 mg treatment of compounds 1, 2, 3 and 4, respectively. Higher specific activities are observed at lower concentrations of the four compounds. The specific activities obtained at the different doses of compounds 1, 2, 3 and 4 are all much lower than that of control (0.040 ± 0.0008, 0.045 ± 0.0007, 0.048 ± 0.0017 and 0.040 ± 0.0016 μmol/30 min/mg protein, respectively). The corresponding relative activities of ATCase at doses of 200, 400, 600 and 800 mg are 85.0, 75.0, 70.0 and 62.5%, respectively by compound 1; 80.0, 75.5, 64.4 and 48.9%, respectively by compound 2; 75.0, 72.9, 68.8 and 56.3%, respectively by compound 3; 90.0, 72.6, 67.5 and 62.5%, respectively for compound 4. These results emphasize that in vivo inhibition of ATCase by compounds 1, 2, 3 and 4 occurs in a dose-dependent manner.
Table II. Specific activities of in vivo treatment of ATCase by quinazolinone derivatives (compounds 1–4). Mice were treated with the specified dose twice for two days, sacrificed and their livers were removed and prepared for the enzyme assay. The vehicle-treated group (control) was treated with corn oil. Assays were carried out at pH 8.2 at 30°C. Values of specific activity are expressed as mean±standard error (SE).
Discussion
CP Synthetase II, ATCase and dihydroorotase exist as a multienzyme complex and still associated following ammonium sulphate fractionation and hydroxylapatite chromatography [Citation45,Citation46]. The reaction of ATCase enzyme is not affected by the presence of the two other enzymes. The extremely small amount of dihydroorotate obtained when the CAD protein is incubated with only the substrates of ATCase is due to the fact that the equilibrium constant of dihydroorotase is much in favor of the reverse reaction towards the reduction of carbamyl aspartate. The equilibrium between carbamyl aspartate and dihydroorotate is a pH-dependent with maximal rate of cyclization of carbamyl aspartate to dihydroorotate at pH 4.4. It decreases to a very low rate at alkaline range [Citation47]. Therefore, ATCase reaction carried out at pH 8.2 indicates that the third step reaction of CAD mainly proceeds towards the carbamyl aspartate formation.
The regulation of the ATCase reaction should be critical for the pyrimidine nucleotide biosynthesis, which might provide a major part of the pyrimidine supply in vivo. ATCase in E. Coli has been shown to be subject to feedback inhibition by the pyrimidines specially cytosine nucleotides such as CDP, CTP, and dCTP which are the best inhibitors of the bacterial enzyme among the tested pyrimidine derivatives [Citation48]. The mammalian enzyme is different since thymidine is the most potent inhibitor of this enzyme [Citation49]. As reported previously, the concentration of cytidine triphosphate that is required to exert 50% inhibition upon mouse hepatic ATCase is higher than that required of thymidine [Citation29]. A comparison of the effect of structure of the examined compounds with their inhibitory potency revealed that the four tested compounds 1, 2, 3 and 4 are structurally related to pyrimidines, especially to thymidine more than cytidine triphosphate.
The purpose of this study is to investigate the effect of compounds 1, 2, 3 and 4 on ATCase. The measured enzyme in the present work was inhibited in both in vitro and in vivo by the four compounds. This inhibition is valuable in the establishment of new inhibitors for this enzyme. The in vitro results showed that the four compounds were potent inhibitors of mouse hepatic ATCase. The inhibition occurred in a concentration-dependent manner indicating that there is a direct interaction between the enzyme and the four tested compounds.
The 50% reduction of the activity of ATCase enzyme was achieved at a concentration of compounds 1, 2, 3 and 4 equal to 0.24, 0.15, 0.18 and 0.35 mM, respectively. As reported previously [Citation29], the IC50 value of thymidine was 0.6 mM. In this respect, the four tested compounds in the present study are more potent inhibitors than thymidine. Compounds 2 and 3 have more inhibitory effect than compounds 1 and 4 (). This may be attributed to the presence of phenyl group at position 2 of the latter two compounds.
Compound 2 is considered to be the most potent inhibitor due to the presence of phenyl group at position 2- (electron attracting group) that enhances the activation of sulfur atoms at positions 3- and 4-. Therefore, this compound may react with a sulfhydryl group of the enzyme. The presence of sulfhydryl group was demonstrated previously in the enzyme from E. coli [Citation50]. Each catalytic chain of the trimeric catalytic subunit of ATCase from E. coli contains free sulfhydryl group and no disulfides [Citation50]. It was reported that the mammalian ATCase domain and the E. coli ATCase catalytic chain have the same tertiary fold [Citation51]. It was inferred that a sulfhydryl group is intimately connected with enzymatic activity in rat liver [Citation49].Thus, the binding between compound 2 and the enzyme to form disulfide may affect the conformation of the catalytic site that is responsible for catalysis and decreases the enzyme activity.
The inhibitory effect of compound 3 is due to the presence of phenyl group at carbon 2, which enhances the acidic character of the amino group at carbon 3. Accordingly, this may facilitate the approach between compound 3 and the enzyme surface that mainly has positively charged groups. The existence and role of the positively charged groups was demonstrated previously [Citation28]. In fact, the basic amino acids represent 11% of the total amino acids of ATCase. Also, the acidic amino acids, which are about 60%, have to be amidated to account for the isoelectric point of ATCase (9.4).
In addition, the measured Ki values for the inhibition of ATCase by compounds 2 and 3 revealed that the inhibitory capacity of compound 3 is lower than that of compound 2. This may attributed to the absence of sulfur atoms in compound 3. Moreover, compounds 1 and 4 have lesser effect due to the presence of methyl group at position 2- which increases electron density over hetero atoms in these compounds by positive inductive effect (R is electron donating group). So, these compounds exhibit basic nature allowing decreasing their binding to the enzyme. The kinetic studies revealed that the mammalian hepatic ATCase is devoid of the characteristic sigmoidal dependence of activity on the substrates (CP or Asp) concentration. However, the kinetic behavior of ATCase in the present study did not display sigmoidity and obeys Michaelis–Menten kinetics. This behavior agrees well with those obtained in the previous studies [Citation49,Citation50].
As illustrated in “Results”, compounds 2 and 3 act as non-competitive inhibitors with respect to CP and Asp. These results are consistent with those previously obtained [Citation29] in which hepatic mammalian ATCase was inhibited by phenobarbital-p-nitrohydrazone in a non-competitive type, whereas the inhibition of hepatic mammalian ATCase by thymidine is a competitive one [Citation49].
It had been noted previously that Ki values of thymidine is 5.86 × 10− 4 and 8.58 × 10− 4 M for the enzyme towards CP and Asp, respectively [Citation29]. A comparison of the Ki values of compounds 2 and 3 in the present study () with that of thymidine reveals that those compounds are more potent inhibitors than it. The inhibition of ATCase by compound 2 can be described as potent according to its Ki value that is of the magnitude of 10− 5.
In regard to Km of ATCase towards CP and Asp, the observed values were calculated to be 1.25 and 6.7 mM, respectively. These values are coincident to that reported previously [Citation29]. In addition, Lineweaver-Burk plot for the inhibitory effect of compounds 2 and 3 is confirmed by Dixon plot which also suggested non-competitive inhibition of the enzyme by the two compounds.
The removal of the inhibitor by dialysis restores full enzymatic activity. The data reported for dialysis experiment () indicate that both the dialyzed control and treated enzyme with compounds 2 or 3 showed nearly equal values of relative activity. Consequently, compounds 2 and 3 have been found to be reversible inhibitors of ATCase enzyme like that of thymidine as described previously [Citation49], whereas the inhibition by thymidine is a reversible type. Also, this is in agreement with that described previously [Citation29], whereas phenobarbital p-nitrohydrazone is a reversible inhibitor for mammalian ATCase.
In vivo effect of compounds 1, 2, 3 and 4 was also investigated and the most impressive results of the compounds inhibition were obtained after two successive days of treatment. The data showed in indicated that the rate of enzymatic reaction was decreased below the control upon in vivo treatment with 1.0 mg of each compound in a dose-dependent manner. Compound 2 is the most potent inhibitor in this regard. Therefore, compounds 2 and 3 may bind the enzyme reversibly as observed from the in vitro studies. These observations suggest that completely different inhibition mechanisms seem to be operative as reported earlier [Citation52].
In conclusion, compounds 2 and 3 as quinazolinone derivatives are inhibitors of hepatic mammalian ATCase, which exert a stronger effect than do some pyrimidines such as thymidine. This was supported by Ki values and in vivo studies. The inhibition of ATCase by compounds 2 and 3 is due to the reversible binding to these compounds.
Acknowledgements
We thank Dr. Fatma El-Rashidy and Dr. Mohamed El-Kersh, Department of Biochemistry, Faculty of Science, Alexandria University, Alexandria, Egypt for their helpful assistance.
References
- Abdel-Megeed MF, El-Hiti GA, Saleh MA, Abdo MA, Awadalla SE. A convenient synthesis of novel hydrazones and osazones of 2-hydrazino-3-phenyl-4(3H)quinazolinone. Rev Roum Chim 1999; 44(1)67–76
- Abdel-Megeed MF, El-Hiti GA, Abdo MA, Saleh MA. A convenient synthesis of 3-(aminophenyl)-4(3H)quinazolinone derivatives. Rev Roum Chim 2002; 43(6)545–554
- El-Hiti GA, Abdel-Megeed MF, Mahmoud YA-G. Synthesis and antimicrobial activities of novel sugar (2-phenylquinazolin-4-yl) hydrazones and their osazones. Indian J Chem 2000; 39(5)368–376
- Saleh MA, Abdel-Megeed MF, Abdo MA, Shokr AM. Synthesis of aldohydrosugar (4-oxoquinazolin-2-yl) hydrazones and their transformation into 1-(alditol-1-yl)-1,2,4-triazolo [4,3-a]quinazolin-5(4H)-ones. J Heterocycl Chem 2003; 40: 85–92
- Smith K, El-Hiti GA, Abdo MA, Abdel-Megeed MF. Regiospecific electrophilic substitution of aminoquinazolinones directed lithiation of 3-(pivaloylamino)- and 3-(acetylamino)-2-methylquinazolin-4(3H)-ones. J Chem Soc Perkin Trans 1995; 1: 1029–1033
- Smith K, El-Hiti GA, Abdel-Megeed MF, Abdo MA. Lithiation of 3-(acylamino)-2-unsubstituted-, 3-(acylamino)-2-ethyl-and 3-(acylamino)-2-propyl)-4-(3H)quinazolinones: Convenient synthesis of more complex quinazolinones. J Org Chem 1996; 61: 647–655
- Smith K, El-Hiti GA, Abdel-Megeed MF, Abdo MA. Lithiation of 2-alkyl-3-amino and 2-alkyl-3-(methylamino)-4(3H)quinazolinones. J Org Chem 1996; 61: 656–661
- Smith K, El-Hiti GA, Abdel-Megeed MF. Non expected products from carbonylation of lithiated quinazolin-4(3H)-one derivatives. J Org Chem 2003; 39: 430–435
- Smith K, El-Hiti GA, Abdel-Megeed MF. Regioselective lithiation of chiral 3-acylamino-2-alkylquinazolinone-4(3H)-ones: Application in synthesis. Synthesis 2004; 13: 2121–2130
- El-Hiti GA, Abdel-megeed MF. Synthesis of glycosides and hydrazones of quinazolin-4(3H)-one derivatives. Heterocycles 2005; 65: 3007–3041
- Abdel-Megeed MF, Azaam MM, El-Hiti GA. 3-arylazo-2-thioxo2,3-dihydro-1H-quinazolinone-4-ones as azodisperse dyes for dying polyester fabrics. Monatsh Chem 2007; 138: 153–156
- El-Hiti GA, Abdel-Megeed MF, Zeid TM. Synthesis and reactions of some 3-aryl-2-thioxoquinazolin-4(3H)-ones. Indian J Chem 2002; 41: 1519–1522
- Saleh MA, Abdel-Megeed MF. Glcosylation of 4-aryl-1-thioxo[1,2,4]-triazolo[4,3-9]quinazolin-5(4H)-one. J Carbohydr Chem 2003; 22: 79–94
- Saleh MA, Abdel-Megeed MF, Abdo MA, Shokr AM. Synthesis and evaluation of some new glycosylthioureas containing a quinazolinone nucleus. Nucleosides Nucleotides Nucleic Acids 2002; 21: 93–106
- Kumar A, Sharma S, Archana, Bajaja K, Sharma S, Panwar H, Singh T, Srivastava VK. Some new 2,3,6-trisubstituted quinazolinones as potent anti-inflammatory, analgesic and COX-II inhibitors. Bioorg Med Chem 2003; 11: 5293–5299
- Perrine JW, Houlihan WJ, Takesue EI. Anti-inflammatory and other pharmacodynamic properties of five members of the 4-aryl-1-isopropyl-2(1H)-quinazolinone series. Arzneim Forsch 1984; 34(8)879–885
- Steinberg D. Closing in on multiple cancer targets: EGFR inhibitors, through one receptor, attack many tumor types. Scientist 2002; 16(7)29–33
- Kulcsar G, Kalai T, Osz E, Sar CP, Jeko J, Sumegi B, Hideg K. Synthesis and study of new 4-quinazolinone inhibitors of the DNA repair enzyme poly (ADP-ribose) polymerase (PARP). Arkivoc 2003; v: 121–131
- Delaney CA, Wang L, Kyle S, White AW, Calvert AH, Curtin NJ, Durkacz BW, Hostomsky Z, Newell DR. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly (adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res 2000; 6: 2860–2867
- Gupta M, Abdel-Megeed M, Hoki Y, Kohlhagen G, Paull K, Pommier Y. Eukaryotic DNA topoisomerases mediated DNA cleavage induced by a new inhibitor: NSC 665517. Mol Pharmacol 1995; 48: 658–665
- Chen H, Chen W, Gan L, Mutlib AE. Metabolism of (S)-5, 6-difluoro-4-cyclopropylethynyl-4-trifluoromethyl-3,4-dihydro-2(1H)-quinazolinone, a non-nucleoside reverse transcriptase inhibitor, in human liver microsomes: Metabolic activation and enzyme kinetics. Drug Metab Dispos 2003; 31: 122–132
- Chen H, Shockcor J, Chen W, Espina R, Gan L, Mutlib AE. Delineating novel metabolic pathways of DPC 963, a non-nucleoside reverse transcriptase inhibitor, in rats: Characterization of glutathione conjugates of postulated oxirene and benzoquinone imine intermediates by LC/MS and LC/NMR. Chem Res Toxicol 2002; 15: 388–399
- Bresnick E. Feedback inhibition by deoxyribonucleosides of aspartate transcarbamylase activity in liver preparations. Biochim Biophys Acta 1962; 61: 598–605
- Dennis PR, Krishna MV, Di Gregorio M, Chan WW. Ligand interactions at the active site of aspartate transcarbamoylase from Escherichia coli. Biochemistry 1986; 25: 1605–1611
- Monks A, Anderson LW, Strong J, Cysyk RL. Flux through the de novo pyrimidine pathway in vivo: Effect of N-phosphonacetyl-l-aspartate, a potent inhibitor of aspartate transcarbamylase. J Biol Chem 1983; 258: 13564–13569
- Serre V, Penverne B, Souciet J, Potier S, Guy H, Evans D, Vicart P, Herve G. Integrated allosteric regulation in the S. cerevisiae carbamylphosphate synthetase-aspartate transcarbamylase multifunctional protein. BMC Biochem 2004; 5: 6–20
- Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J Biol Chem 2004; 2279: 33035–33038
- Grayson DR, Evans DR. The isolation and characterization of the aspartate transcarbamylase domain of the multifunctional protein, CAD. J Biol Chem 1983; 258: 4123–4129
- Balbaa M, Yacout G, Ghonaim T, Othman D. Inhibition of aspartate transcarbamylase by a phenobarbital derivative. J Enzy Inhib 2001; 16: 259–267
- Roberts TD, Munchausen L, Shechter H. Ortho neighboring-group participation of amides in photolytic hydration of triple bonds. J Am Chem Soc 1975; 97: 3112–3117
- Cailsen K, Lawesson SO. Studies on organophosphorous compounds. Thiation of ethyl-2-acylaminobenzoates. Bull Soc Chim Belg 1979; 88: 305–311
- Bain DI, Smally RK. Synthesis of 2-substituted-4H-3,1-benzoxazin-4-ones. J Chem Soc 1968; 13: 1593–1597
- Wheeler AS, Oates WM. The bromination of anthranilic acid. J Chem Soc 1910; 32: 770–775
- Ismail MF, Shams NA, Nageib MI. Reaction of 6,8-dibrimo-2-methyl-3,1-benzoxazin-4(H)-one with some nucleophilic reagents: Synthesis of quinazolinone, tetrazole and benzimidazole derivatives. Indian J Chem 1981; 20(5)394–397
- Misra VS, Saxena VK, Srivastava R. Synthesis of some N1(2′-Alkylor aryl-6′,8′-disubstituted quinazolinone3′-ylaminoacetyl)-N3-aryl ureas and their in vivo action on caecal amoebiasis of rats. J Indian Chem Soc 1983; 60: 610–611
- Inagaki A, Tatibana M. Control of pyrimidine biosynthesis in mammalian tissues: III multiple forms of aspartate transcarbamoylase of mouse spleen. Biochim Biophys Acta 1970; 220: 491–502
- Clark JM, Switzer RL. Kinetic properties of aspartate transcarbamylase. Exprimental biochemistry. Freeman, San Francisco, USA 1977; 2nd ed: 123–128
- Balbaa M, Khalifa M, El-Sabaway M, Kandeel K. Inhibition of succinate-cytochrome c reductase by a ferromacrocyclic complex. J Enzy Inhib 2001; 16: 381–390
- Balbaa M, Abdel-Hady N, El-Rashidy F, Awad L, El-Ashry ESH, Schmidt RR. Inhibition of some hepatic lysosomal glycosidases by ethanolamines and phenyl 6-deoxy-6-(morpholin-4-yl)-β-d-glucopyranoside. Carbohydr Res 1999; 317: 100–109
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934; 56: 658–666
- Segel IH. Enzymes. Biochemical calculations2nd ed. John Wiley & Sons, New York 1976; 208–323
- Read LJ, Muench H. A simple method for estimating fifty percent endpoint. Am J Hyg 1938; 27: 493–497
- Balbaa M, Abdelhamid EME, Bassiouny K. Enhancement of lysosomal enzymes by the pyrethroids, fenvalerate and trans-cypermethrin. Jpn J Toxicol Environ Health 1998; 44: 83–91
- Ohnishi ST, Barr JK. A simplified method of quantitating protein using the biuret and phenol reagents. Anal Biochem 1978; 86: 193–200
- Hoogenraad NJ, Levine RL, Kretchmer N. Copurification of carbamoyl phosphate synthetase and aspartate transcarbamoylase from mouse spleen. Biochem Biophys Res Commun 1971; 44: 981–988
- Shoaf WT, Jones ME. Initial steps in pyrimidine synthesis in Ehrlich ascites carcinoma. Biochem Biophys Res Commun 1971; 45: 796–802
- Christopherson RI, Jones ME. Interconversion of carbamyl-l-aspartate and l-dihydroorotate by dihydroorotase from mouse Ehrlich ascites carcinoma. J Biol Chem 1979; 254: 12506–12512
- Gerhart JC, Pardee AB. The enzymology of control by feedback inhibition. J Biol Chem 1962; 237: 891–896
- Bresnick E. Inhibition by pyrimidines of aspartate transcarbamylase partially purified from rat liver. Biochim Biophys Acta 1963; 67: 425–434
- Vanaman TC, Stark GR. A study of the sulfhydryl groups of the catalytic subunit of Escherichia coli aspartate transcarbamylase: The use of enzyme-5-thio-2-nitrobenzoate mixed disulfides as intermediates in modifying enzyme sulfhydryl groups. J Biol Chem 1970; 245: 3565–3573
- Simmer JP, Kelly RE, Scully JL, Grayson DR, Rinker AG, J R, Bergh ST, Evans DR. Mammalian aspartate transcarbamylase (ATCase): Sequence of the ATCase domain and interdomain linker in the CAD multifunctional polypeptide and properties of the isolated domain. Proc Natl Acad Sci USA 1989; 86: 4382–4386
- Balbaa M, Mansour H, El-Sawy H, El-Ashry ES. Inhibition of some hepatic glycosidases by the diseco nucleoside, 4-amino 3-(d-glucopentitol-1-yl)-5-mercapto 1,2,4-triazole and its 3-methyl analog. Nucleosides Nucleotides Nucleic Acids 2002; 21: 695–708