Abstract
A quantitative structure–activity relationship analysis was conducted on two different series of pyridinylguanidines acting as inhibitors of urokinase-type plasminogen activator using QuaSAR descriptors of molecular modeling software MOE. Multiple linear regression analysis following a stepwise scheme was employed to generate QSARs that relate molecular descriptors to uPA inhibitory activity data of the title compounds. Among the several QSARs generated by MLR analysis, the best models were selected on the basis of their statistical significance and predictive potential. The interpretation of the selected QSAR models suggest that uPA inhibitory activity of compounds in series 1 is influenced by their molecular shape, molecular flexibility and halogen atoms in the molecule whereas the uPA inhibitory potency of compounds in series 2 is dependent on molecular lipophilicity, number of double bonds and spatial orientation of bulky substituents in the molecule.
Keywords::
Introduction
Metastasis is the cause of most cancer-related deaths. The proteolytic degradation of the extracellular matrix is recognized as a mechanism that plays an important role in the metastatic process. Proteolytic enzymes are required to mediate tumor cell invasion into adjacent tissues and initiate the metastatic process [Citation1]. Urokinase plasminogen activator (urokinase, uPA) a trypsin-like serine protease has been implicated in many cellular processes such as tumorigenesis, cell proliferation and migration, cell adhesion, angiogenesis and metastasis [Citation2–4]. The primary role of uPA is to convert plasminogen into its active form plasmin [Citation5]. Plasmin, a serine protease, is involved in the degradation of several extracellular matrix components, the key stage in the process of angiogenesis and thus contributes to the tumor cell survival and metastatic properties of cancer cells [Citation6]. Clinical studies have demonstrated that increased expression of uPA in tumour tissues is highly correlated with tumor cell migration, invasion, proliferation, progression and metastasis [Citation7]. Additionally, it has also been reported that uPA activity is increased in metastatic tumors compared with primary tumors in experimental animals [Citation8]. The pivotal role of uPA in tumor angiogenesis and metastases makes it a potentially attractive target for preventing metastasis of tumors.
Several small molecules have been identified as potent inhibitors of uPA such as benzamidines, phenylguanidines, acylguanidines and bisbenzamidines [Citation6]. However, these compounds also inhibit other related serine proteases such as tissue plasminogen activator, plasmin and thrombin, which play a significant role in the fibrinolytic cascade [Citation6]. In order to circumvent these unwanted adverse effects arising out of inhibition of homeostatic enzymes, efforts were devoted to develop inhibitors with high degree of selectivity towards uPA. Recently, Barber et al. [Citation9,Citation10] reported two series of pyridinylguanidines with selective uPA inhibitory activity. In view of further progression in the development of such inhibitors, the present study attempts to explore QSAR of both the series of uPA inhibitory pyridinylguanidines reported by Barber et al. QSAR is a powerful lead-compound optimization technique, which quantitatively relates variations in biological activity to changes in molecular properties (descriptors). In other words, it attempts to link activity data with descriptors chosen via identification of the “rules” that can be further used to guide chemical synthesis when new chemical entities are developed. In the past, many quantitative structure activity relationship studies were performed on potential uPA inhibitors to explore the structural features determining the binding affinity and selectivity for the enzyme. Yang et al. [Citation11] investigated the features contributing to selective inhibition of urokinase using Hansch analysis of a series of phenyl guanidines exhibiting inhibitory activity against six serine proteinases. Bhongade et al. [Citation12,Citation13] reported comparative molecular field analysis and comparative molecular similarity indices analysis of uPA inhibitory indole/benzimidazole-5-carboxamidines. Related to the foregoing and in continuation of our efforts focused on the design and development of novel and potent protease inhibitors targeting cancer, the present work strives to examine the applicability of QSAR approach to the series of pyridinyl guanidines reported by Barber et al. [Citation9,Citation10] and gain some fundamental understanding on factors influencing uPA binding affinity of these molecules.
Materials and methods
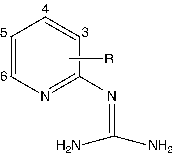
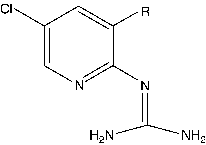
The dataset investigated for QSAR study includes two series of pyridinyl guanidines; series 1 and series 2. Series 1 comprises of 16 compounds whereas series 2 comprises of 19 compounds. The structure of molecules in Series 1 and Series 2 are presented in and respectively. The uPA inhibitory activity data of compounds in both the series have been reported as IC50 values, where IC50 refers to the experimentally determined micro-molar concentration of the compounds in the selected series required to inhibit the enzyme by 50%. The biological activity values [IC50 (μM)] were transformed into their molar scale and subsequently converted to negative logarithmic scale and then used as the response variable for the QSAR analysis.
Table I. Structure, biological activity and physicochemical descriptors of series 1
 .
.
Table II. Structure, biological activity and physicochemical descriptors of series 2
 .
.
The softwares employed for the present study are Molecular Operating Environment [Citation14] (MOE 2002.03), statistical software SYSTAT [Citation15] (Version 10.2) and inhouse validation program VALSTAT [Citation16]. All the computations were carried out on Compaq PIV workstation. The structures of the compounds in the selected series were sketched using builder module of MOE software and sketched structures were subsequently energy minimized up to root mean square gradient of 0.01 kcal/molÅ using MMFF94 force field. Conformational search of each energy-minimized structure was performed employing stochastic search routine. All the conformers generated for each structure were carefully scrutinized in conformational geometry panel and the lowest energy conformer of each structure selected for re-optimization by applying AM1 semi-empirical method as implemented in semi-empirical molecular orbital software program MOPAC (version 7) of MOE.
Molecular descriptors programmed into QuaSAR module of MOE were calculated for the geometry-optimized structures of the compounds in the series. The MOE package calculates nearly about 180 descriptors named as QuaSAR descriptors [Citation17]. The QuaSAR descriptors can be broadly classified into 2D and 3D descriptors. The two dimensional descriptors include traditional physicochemical properties, (atom counts and bond counts, mr, logP and vdw_area etc), connectivity-based topological descriptors (Kier and Hall connectivity and Kappa Shape indices; adjacency and distance matrix descriptors), pharmacophore feature descriptors (e.g. Donor, Acceptor, Polar, Positive, Negative, Hydrophobic.), partial charge descriptors based on Partial Equalization of Orbital Electronegativities method and quantum chemical descriptors such as HOMO and LUMO energies, dipole, etc. The 3D descriptors in the module include potential energy descriptors, surface area descriptors, volume descriptors, shape descriptors, and conformation-dependent charge descriptors.
The calculated descriptors were initially screened for invariant nature (constant and near constant values) to reduce the number of descriptors for statistical analysis. Further reduction in the descriptor pool was accomplished by eliminating the descriptors with no significant correlation with the dependent variable (R2 < 0.1) using QuaSAR-contingency module of MOE. QuaSAR-Contingency is a statistical application designed to assist in the selection of descriptors for QSAR.
In the first step of statistical analysis, pairwise correlation analysis of the reduced set of descriptors was established by the calculation of correlation matrix. If given pair of descriptors have a Pearson correlation greater than 0.7, then one member of the pair is randomly omitted for the QSAR study. As a resultant, the descriptor pool is further reduced to 45 descriptors for series 1 and 52 descriptors for series 2. This reduced descriptor pool was used in the generation of QSAR models.
The QSAR models were found through forward stepwise regression procedure employing statistical program SYSTAT 10.2 version. The best QSAR models were selected on the basis of the highest correlation coefficient (R2), the lowest standard error SEE, F statistics and the statistical relevance of the incorporated descriptors. Another important criterion for the model selection is low variable collinearity between the descriptors in the same model. To confirm the absence of multicollinearity in the selected correlations, variance inflation factor (VIF) values [Citation18] were calculated for each parameter in the regression. VIF value was calculated from 1/1 − R2, where R2 is the multiple correlation coefficient of one parameter's effect regressed on the remaining parameters. Although it is stated that VIF values less than 10 are statistically satisfactory, we chose a more stringent criterion (VIF < 5) for selection of QSAR models. The Z-score method was adopted for the detection of outliers. Z-Score can be defined as absolute difference between the value of the model and the activity field, divided by the square root of the mean square error of the data set. Any compound which shows a value of Z-score higher than 2.5, during generation of a particular QSAR model is considered as outlier.
Finally, the stability and predictive ability of every potential model was tested by a cross validation method following a leave-one-out scheme using inhouse program VALSTAT. The validation parameters calculated are squared cross-correlation coefficient (q2), standard deviation of sum of square of difference between predicted and observed values (SPRESS) and standard deviation of error of prediction (SDEP). The validation parameter q2 describes the stability of a regression model obtained by focusing on the sensitivity of the model to the elimination of any single data point and capacity to estimate the activity of compounds outside the training set. For a reliable QSAR model, the calculated squared cross-correlation coefficient q2 should be greater than 0.6.
Results and discussion
QSAR model for series 1
The best QSAR model generated for uPA inhibitory activity of compounds in Series 1 is given below
N = 16, R = 0.94, R2 = 0.89, SEE = 0.21, F = 33.28, P>0.000, Q2 = 0.83, SPRESS = 0.27, SDEP = 0.24.
In QSAR model given above, N is the number of data points and the figures in the parentheses are 95% confidence limits.
Model 1 produces good description for uPA inhibitory activity of pyridinyl guanidines, as it accounts for more than 80% of the total variance in the biological activity. The F statistic of the model is significant at 99% level as the calculated F-test value exceed the critical F value (F(3,12) = 5.95) by a large margin. Since the p value less than 0.01, there exist a statistically significant relationship between the variables in the model at 99% confidence level. Furthermore, the intercorrelation coefficient between the descriptors in the model is less than 0.7 as indicated by correlation matrix () and the variance inflation factors of all the descriptors in the model as shown in are less than 5, which indicate the absence of multicollinearity in the models.
Table III. Correlation matrix for series 1.
The molecular descriptors in the model 1 include petitjeanSC, KierFlex and b_1rotR. Among the three descriptors found in the model, petitjeanSC and KierFlex are topological descriptors and b_1rotR is a constitutional descriptor, which indicates that variation in the uPA inhibitory activity of pyridinyl guanidines can be described in terms of structural components of the molecule. The topological descriptor petitjeanSC refers to petitjean shape coefficient, which encodes information regarding the shape of the molecule. The regression coefficient of the descriptor petitjeanSC bears a negative sign, which suggest that shape of the molecule is an important determinant for uPA inhibition exhibited by pyridinyl guanidines. The topological descriptor KierFlex [Citation19] encodes the structural properties that restrict a molecule from being “infinitely flexible”, the model for which is an endless chain of C(sp3) atoms. The value of KierFlex decreases with the presence structural features considered to preventing a molecule from attaining infinite flexibility are fewer atoms, cyclicity, branching, conjugation and presence of atoms with a covalent radii smaller than C (sp3). However, an observation of the substituents in the pyridinyl ring indicates none of the substituents except for phenyl ring seem to exhibit cyclicity, branching or conjugation. So, it appears that descriptor might actually characterize the covalent radii of the atoms in the substituents. Hence, the positive coefficient of the descriptor in the model may be interpreted that the presence of atom with larger covalent radii than carbon such as chlorine, bromine will increase the uPA binding affinity of pyridinyl guanidines. The constitutional descriptor b_1rotR [Citation12] represents fraction of rotatable single bonds in the molecule. The negative coefficient of the descriptor in model suggests that increase in the fraction of rotatable single bonds will decrease the uPA inhibitory potency of pyridinyl guanidines. Furthermore, it is quite evident from that fractional increase in number of rotatable single bonds occurs with methoxy and hydroxyl substitution in the pyridinyl ring hence it may be interpreted that presence of aforementioned substituents is not conducive for uPA inhibitory activity.
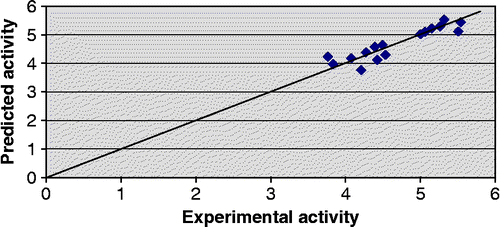
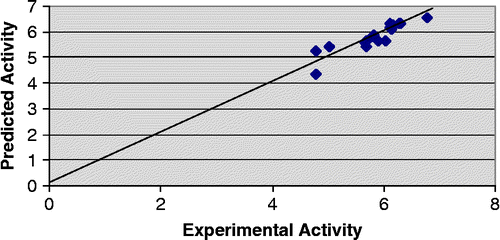
The predictive potential of model 1 as judged by leave-one-out procedure is fairly high (Q2 > 0.8) suggesting that the models will be useful for meaningful predictions. Further support in this regard is obtained from the low values of the cross-validation parameters Spress and Sdep. Predicted activity values for the compounds in the training set alongwith their corresponding experimental activity values are recorded in . The predicted − log IC50 values of the compounds in the test set are in good agreement with the corresponding experimental values as shown in .
Figure 1. Experimental vs predicted activity of model 1.
QSAR models for series 2
The best QSAR models generated for series 2 are given below alongwith regression statistics as well as crossvalidation statistics.
N = 18, R = 0.92,R2 = 0.85, SEE = 0.28, F = 26.86, P>0.000, Q2 = 0.74, SPRESS = 0.37, SDEP = 0.33.
N = 17, R = 0.94, R2 = 0.89, SEE = 0.19, F = 35.22, P>0.000, Q2 = 0.78, SPRESS = 0.27, SDEP = 0.24
Both the models given above manifest a satisfying correlation, as they account for more than 80% of the observed variance in the biological activity. Accuracy in the analysis is shown by low values of standard error of estimate. The F statistics are significant at 99% level and the p value less than 0.000 indicates that the models are not a mere result of chance. The correlation matrix and VIF data recorded in and respectively depicts the orthogonal nature of the descriptors in the selected correlations.
Table IV. Correlation matrix for series 2.
Table V. Variance inflation factor (VIF) value of descriptors in QSAR models.
In addition to statistical significance, the generated models also exhibit good predictive capacity as shown by cross-validation statistics given alongwith the models. Both the models show predicted variance greater than 70% since their Q2 values are greater than 0.7. It is also noteworthy that the PRESS statistics (SPRESS) of the both the models are comparable to their respective standard error of estimate (SEE), suggesting that the models will be useful for prediction. Predicted − log IC50 values of the compounds in the training set together with the experimental − log IC50 values are recorded in . Plot of experimental versus predicted − log IC50 values of the compounds in the training set for model 2 and 3 is illustrated in and respectively.
Table VI. Predicted activity values of selected models.
Figure 2. Experimental vs predicted activity of model 2.
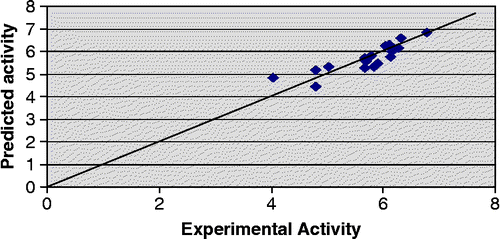
Figure 3. Experimental vs predicted activity of model 3.
Model 2 shows linear positive dependence of uPA inhibitory activity on topological descriptor chi1v_C, steric descriptor PMI − Z and linear negative dependence of activity on potential energy descriptor E_Sol. The topological descriptor, chi1v_C [Citation12] encodes information regarding degree of branching, cyclization, heteroatom content, and heteroatom position in the molecule and is calculated as follows:
Where ![]() and
and ![]() are the vertex connectivity indices of carbon atoms i and j, respectively, and the summation extends to all bonded pairs of non hydrogen carbon atoms in the group or molecule. For second and third rows of atoms, Kier [Citation19] gave a unified definition of δv, as expressed by Equation 5. In this equation,
are the vertex connectivity indices of carbon atoms i and j, respectively, and the summation extends to all bonded pairs of non hydrogen carbon atoms in the group or molecule. For second and third rows of atoms, Kier [Citation19] gave a unified definition of δv, as expressed by Equation 5. In this equation, ![]() is the number of valence electrons of atom i, hi is the number of hydrogen atoms attached to it, and Zi is its atomic number.
is the number of valence electrons of atom i, hi is the number of hydrogen atoms attached to it, and Zi is its atomic number.
The value of the ![]() increase with branching and with increase in the number of heteroatoms in the molecule. Thus, the positive coefficient of the descriptor in model 3 implies that increase in molecular branching and presence of heteroatoms in the molecule will decrease the uPA inhibitory potency of pyridinyl guanidines.
increase with branching and with increase in the number of heteroatoms in the molecule. Thus, the positive coefficient of the descriptor in model 3 implies that increase in molecular branching and presence of heteroatoms in the molecule will decrease the uPA inhibitory potency of pyridinyl guanidines.
The steric descriptor PMI − Z refers to the principal moment of inertia along Z-axis of the molecule. The positive coefficient of the descriptor suggests that orientation of molecular distribution along Z-axis of the molecule is conducive for the uPA inhibitory potency of pyridinyl guanidines. Moreover, the appearance of the descriptor also highlights the likelihood of shape specific steric interactions between molecules and the receptor. The potential energy descriptor E_Sol bears a negative coefficient in model 3, which indicates that increase in the solvation energy of the molecule will decrease the uPA binding affinity of pyridinyl guanidines.
Model 3 comprises of the following descriptors; lipophilicity descriptor logP(o/w), potential energy descriptor E_Sol and constitutional descriptor b_double. The descriptor logP(o/w) refers to log of the octanol/water partition coefficient of the molecule and is considered as a measure of lipophilicity of a molecule. The positive coefficient of the descriptor in model 3 suggests that increase in the overall lipophilicity of the molecule will in turn increase the uPA inhibitory activity of pyridinyl guanidines. The negative coefficient associated with potential energy descriptor E_Sol reaffirms the conclusions drawn from model 2. The third descriptor b_double in model 3 represents number of double bonds in the molecule. From the positive coefficient of this term it appears that increase in the number of double bonds in the molecule have a beneficial effect on the uPA inhibitory activity shown by pyridinyl guanidines. As it can be seen form table the descriptor values are larger when an intermittent double bond is present in the side chain at carbon-3 of pyridine ring. The positive effect of intermittent double bond may be attributed to the fact that incorporation of the double bond makes the molecule assume a trans configuration and orients the substituents attached to the double bond to the opposite direction of the pyridinyl ring which in turn might help in the interaction of the molecule with the enzyme.
Compound number 9 was found to be a statistical outlier (Z score > − 2.72551) during the development of model 2 while compounds 9 and 6 were found to be successive outliers in model 3. The reason for outlying behavior of compound 9 may be attributed to the steric hindrance owing to presence of a phenyl substitution vicinal to guanadino moiety in the pyridine ring. Furthermore, the phenyl ring is directly linked to the pyridinyl ring form a biphenyl like structure, which may not permit proper orientation of the guanidino group in the active site of the enzyme. However, the reason for outlying behavior of compound 6 is not immediately apparent and merits further studies.
Conclusion
Finally to summarize, two series of pyridinyl guanidines reported by Barber et al. have been analyzed applying a QSAR approach. For the 16 compounds evaluated for uPA inhibitory activity in series 1, a statistically satisfactory QSAR model is developed with topological descriptors Petitjean shape coefficient, Kierflex and constitutional descriptor b_1rotR. Structural information encoded by the aforementioned descriptors suggests that uPA binding affinity of the compounds studied is dependent upon molecular shape, presence of halogen atoms and molecular flexibility. Furthermore, the predictive ability of the model was established by cross-validation procedure following a leave-one-out scheme.
Two QSAR models of good statistical quality and predictive capacity were generated for describing upA inhibitory activity of compounds in series 2. The interpretation of the models suggest that uPA inhibitory activity of pyridinyl guanidines in series 2 increases with lipophilicity of molecules while decreases with molecular branching and increase in the heteroatom content of the molecule. The models also highlight the possibility of shape specific steric interactions between the molecules and the enzyme.
Acknowledgements
One of the authors, C. Karthikeyan wishes to thank CSIR, India for providing a senior research fellowship. The authors wish to thank Tata Elxsi for providing MOE software for the study. Grateful acknowledgements to Prof. P. B. Sharma, Vice Chancellor, Rajiv Gandhi Technical University, Bhopal and Director, SGSITS, Indore for the experimental facilities provided. The authors also wish to thank Prof. L.B. Kier and Prof. M. Petitjean for providing reprints of their research works. This research work is funded by grant from All India Council of Technical Education, New Delhi, India.
Related Research Data
References
- K Dano, PA Andreasen, J Grondahl-Hansen, P Kristensen, LS Nielsen, and L Skriver. (1985). Plasminogen activators, tissue degradation and cancer. Adv Cancer Res 44:139–266.
- SA Rabbani, and RHM Xing. (1998). Role of urokinase and its receptor in invasion and metastasis of hormone-dependent malignancies. Int J Oncol 12:911–920.
- JP Irigoyen, P Munoz-Canoves, L Montero, M Koziczak, and Y Nagamine. (1999). The plasminogen activator system: Biology and regulation. Cell Mol Life Sci 56:104–132.
- N Sidenius, and F Blasi. (2003). The urokinase plasminogen activator system in cancer. Cancer Metast Rev 22:205–222.
- V Ellis, C Pyke, J Eriksen, H Solberg, and K Dano. (1992). The urokinase receptor: Involvement in cell surface proteolysis and cancer invasion. Ann N Y Acad Sci 667:13–31.
- HA Chapman. (1997). Plasminogen activators, integrins, and the coordinant regulation of cell adhesion and migration. Curr Opin Cell Biol 9:714–724.
- A Giovanni, and PF David. (2005). Protease inhibitors in the clinic. Med Chem 1:71–104.
- A Abderrahim, K Stephanie, T Gilles, S Louis-Georges, B Pnina, G David, and AR Shafaat. (1994). Urokinase overproduction results in increased skeletal metastasis by prostate cancer cells in vivo. Cancer Res 54 (9):2372–2377.
- GB Christopher, PD Roger, and AH Valerie. (2002). Selective urokinase-type plasminogen activator (uPA) inhibitors. Part 1: 2-Pyridinylguanidines. Bioorg Med Chem Lett 12:181–184.
- GB Christopher, and PD Roger. (2002). Selective urokinase-type plasminogen activator (uPA) inhibitors. Part 2: (3-Substituted-5-halo-2-pyridinyl) guanidines. Bioorg Med Chem Lett 12:185–187.
- Y Heechung, H Jack, HK Ki, and J Greer. (1990). Selective inhibition of urokinase by substituted phenylguanidines: Quantitative structure–activity relationship analyses. J Med Chem 33 (11):2956–2961.
- AB Bhoomendra, VG Veerappa, and KG Andanappa. (2005). 3D-QSAR CoMFA studies on trypsin-like serine protease inhibitors: A comparative selectivity analysis. Bioorg Med Chem 13:2773–2782.
- BA Bhongade, and AK Gadad. (2004). 3D-QSAR CoMFA/CoMSIA studies on urokinase plasminogen activator (uPA) inhibitors: A strategic design in novel anticancer agents. Bioorg Med Chem 12:2797–2805.
- MOE is a molecular modeling package developed by Chemical Computing Group Inc., Canada, 2002.
- SYSTAT 10.2 is a statistical package developed by SYSTAT software Inc, USA, 2003.
- VALSTAT is a statistical program developed by Dr. Arun Kumar Gupta, SGSITS, Indore, 2004.
- A Lin. J Chem Comput Group, http://www.chemcomp.com/ Journal_of_CCG/Features/descr.htm.
- DH Cho, SK Lee, BT Kim, and KT No. (2001). Quantitative structure-activity relationship (QSAR) study of new fluorovinyloxyacetamides. Bull Korean Chem Soc 22:388–394.
- LB Kier, and LH Hall. (1977). Structure–activity studies on hallucinogenic amphetamines using molecular connectivity. J Med Chem 20 (12):1631–1636.