Abstract
The malonyl-CoA decarboxylase (MCD) inhibition activity of derivatives of N-alkyl-N-(1,1,1,3,3,3-hexafluoro-2-hydroxypropylphenyl)amide has been analyzed through combinatorial protocolin multiple linear regression (CP-MLR) using different topological descriptors obtained from Dragon software for the energy minimized 3D-structures of these molecules. Among the topological descriptor classes considered in the study, the MCD inhibition activity is correlated with simple topological descriptors (TOPO) and 2D-autocorrelation descriptors (2DAUTO). The complementary information contents having neighborhood symmetry of 2-order, CIC2 from the TOPO class, the Geary autocorrelations-lag 8, weighted by atomic Sanderson electronegativities, GATS8e and the Moran autocorrelations-lag 6, weighted by atomic Sanderson electronegativities, MATS6e both from 2DAUTO class have contributed significantly in the development of a statistical significant model.
Introduction
The enzyme malonyl-CoA decarboxylase (MCD, EC 4.1.1.9; CoA = coenzyme A) catalyzes the conversion of malonyl-CoA to acetyl-CoA and thus regulates malonyl CoA levels. Malonyl-CoA is a potent endogenous inhibitor of carnitine palmitoyl-transferase-I (CPT-I), an enzyme necessary for the mitochondrial metabolism of long-chain fatty acids. CPT-I, the rate-limiting enzyme in fatty acid oxidation, catalyzes the formation of acylcarnitine, which is transported from the cytosol across the mitochondrial membranes by acylcarnitine translocase. Inside the mitochondria, the long-chain fatty acids are transferred back to their CoA compounds by a complementary enzyme, CPT-II, where acyl-CoA enters the β-oxidation pathway, generating acetyl-CoA.
A number of studies indicate that shifting energy metabolism in the heart towards glucose oxidation is an effective approach to decrease the symptoms associated with myocardial ischemia [Citation1,Citation2]. The antianginal drugs such as ranolazine inhibit fatty acid β-oxidation and stimulate glucose oxidation [Citation3]. Trimetazidine has been shown to specifically inhibit the long-chain 3-ketoacetyl CoA thiolase (3-KAT), an essential step in fatty acid oxidation [Citation4]. Inhibiting CPT-I activity through increasing malonyl-CoA levels with MCD inhibitors would result in a novel and perhaps a much safer method, compared to other known small-molecule CPT-I inhibitors, to the prophylaxis and treatment of ischemic heart disease [Citation5].
Recently, Cheng et al. [Citation6] have argued that increasing the malonyl-CoA level in tissues has potential application in heart disease and diabetes therapy. They have screened in-housed library compounds employing either MCD from rat heart or maltose-binding protein (MBP)-fused human MCD protein and reported a number of small molecules as MCD inhibitors. These compounds were derived from the phenylhexafluoroisopropanol moiety, which was found to be essential for the high in vitro MCD inhibitory activity having favorable pharmacokinetic properties with efficacy in regulating energy metabolism in isolated working rat hearts. However, their structure-activity relationship (SAR) study was targeted at the alterations of substituents at different positions of the said moiety and provided no rationale to reduce the trial-and error factors. Hence, in the present communication, a 2D-quantitative SAR (2D-QSAR) study on these analogues (, along with the generalized structure) has been conducted to provide the rationale for drug-design and to explore the possible mechanism of their action at molecular level. Recently a 3D-QSAR study on similar type of inhibitors has been reported [Citation7], using the comparative molecular field analysis (CoMFA) and the comparative molecular similarity indices analysis (CoMSIA). This study could reveal the importance of steric, electrostatic, hydrophobic and H-bond acceptor fields. Additionally the 3D-contour maps, obtained through these fields, were proposed to be used in the design of more potent MCD inhibitors. Both the 2D- and 3D-QSAR studies are equally important if the developed statistical models under them have high predictive power for the new potential congeners. The emerged models from such studies may assist to identify the type of interactions involved between a drug molecule and the receptor sites. The possible mechanism of action of the compounds anticipated by one study may, therefore, be corroborated through the other one. However, a 2D-QSAR study is quite simple to interpret the biological data in terms of different descriptors obtained from the two dimensional structures of the compounds without involving energy minimization procedures. In a congeneric series, where a relative study is being carried out, the 2D- descriptors may play important role in deriving the significant correlations with biological activities of the compounds. Thus the novelty and importance of a 2D-QSAR study is mainly due to its simplicity for the calculations of different descriptors and their interpretation (in physical sense) to explain the inhibition actions of compounds in a congeneric series.
Table I. Observed and modeled malonyl coenzyme A decarboxylase inhibition activity of N-alkyl-N-(1,1,1,3,3,3-hexafluoro-2-hydroxypropylphenyl)amide derivatives.
Material and methods
Data set
In the present study, the compounds derived from phenylhexafluoro- isopropanol moiety () have been considered from the literature [Citation6] along with their in vitro malonyl coenzyme A decarboxylase (MCD) inhibition activity. The activity, IC50, was measured as the concentration of an inhibitor to achieve 50% inhibition of the MCD enzyme. For present work, the same is expressed as —logIC50 on a molar basis. DRAGON software [Citation8] has been used for the parameterization of the compounds of this study (). This software offers several hundreds of descriptors from different perspectives relating to empirical, constitutional and topological indices characteristic to the molecules under multi-descriptor class environment. The structures of the compounds under study have been drawn in 2D ChemDraw [Citation9] using the standard procedure. These structures were converted into 3D objects using the default conversion procedure implemented in the CS Chem3D Ultra. The generated 3D-structures of the compounds were subjected to energy minimization in the MOPAC module, using the AM1 procedure for closed shell systems, implemented in the CS Chem3D Ultra. This will ensure a well defined conformer relationship across the compounds of the study. All these energy minimized structures for the varying R1 and R2 groups of respective compounds have been ported to DRAGON software for computing the parameters corresponding to 0D-, 1D-, and 2D-descriptor classes. The descriptor classes considered in the study along with their definitions and scope in addressing the structural features have been presented in . As the total number of descriptors involved in this study is very large, only the names of descriptor classes and the actual descriptor involved in the models have been listed. The combinatorial protocolmultiple linear regression (CP-MLR) computational procedure used for present work in developing QSAR models is briefly described below.
Table II. Descriptor classes used for the analysis of malonyl coenzyme A decarboxylase inhibitiory activity of N-alkyl-N-(1,1,1,3,3,3-hexafluoro-2-hydroxypropyl phenyl)amide derivatives and identified categories in modeling the activity.
Model development
The CP-MLR is a ‘filter’ based variable selection procedure for model development in QSAR studies Citation10–13. The procedure employs a combinatorial strategy with MLR to result in selected subset regressions for the extraction of diverse structure-activity models, each having unique combination of descriptors from the generated data set of the compounds under study. The ‘filters’ set in CP-MLR are intended at (i) having inter-parameter correlation to a predefined cutoff value (filter-1; default acceptable value ≤ 0.3, (ii) optimize the variable entry to a model through t-value of regression coefficients (filter-2; default acceptable value ≥ 2.0, (iii) comparability of models (regression equations) with different number of descriptor in terms of square root of adjusted multiple correlation coefficient (filter-3; r-bar, default acceptable value ≥ 0.74), and (iv) addressing the external consistency of the model with leave-one-out (LOO) cross-validation as default option (filter-4; cross-validated Q2 criteria, default acceptable limits are 0.3 ≤ Q2 ≤ 1.0). All these filters make the variable selection process efficient and lead to unique solution. Further, to find out any chance correlations associated with the models recognized in CP-MLR, each cross-validated model has been subjected to randomization test [Citation14,Citation15] by repeated randomization of the biological responses. The datasets with randomized response vector have been reassessed by multiple regression analysis. The resulting regression equations, if any, with correlation coefficients better than or equal to the one corresponding to unscrambled response data were counted. Every model has been subjected to 100 such simulation runs. This has been used as a measure to express the percent chance correlation of the model under scrutiny. The CP-MLR protocolhas been applied with default filter thresholds to identify the all possible models that could emerge from the descriptors of compounds.
Results and discussion
In multi-descriptor class environment, exploring for best model equation(s) along the descriptor class provides an opportunity to unravel the phenomenon under investigation. In other words, the concepts embedded in the descriptor classes relate the biological actions revealed by the compounds. With this, attempts have been made to develop one-, and two-descriptor models for the malonyl coenzyme A decarboxylase inhibition activity of N-alkyl-N-(1,1,1,3,3,3-hexafluoro-2-hydroxypropylphenyl)amide derivatives. The study has led to forty-one models in the topological (TOPO) class descriptors, one model each in modified Burden eigenvalues (BCUT), and Galvez topological charge indices (GVZ) and twenty-six models in 2D-autocorrelations (2DAUTO) class descriptors (). In these individual classes, the models derived in two descriptors, compared to the models in one- or three-descriptors, are only statistically sound. Thus, the collective descriptors of these classes may further result into the improved models. However, no derived model in collective descriptors could explain the variance in activity beyond 67%. This prompted us to search out for the models in three-descriptors from the collective class, resetting filter-3 at 0.84 without altering the other filters. The extract of all such models obtained from the study has been provided in in the form of identified descriptor's average of regression coefficients along with standard deviation across the models and the total incidence corresponding to all the models. This, while providing the averages of the estimated regression coefficients of all the identified descriptors, shows their variance across the models emerging from the study as well. To be concise, the complete regression equations have been shown for selected models only. The following regression equations represent collective class structure-activity model of the compounds.
In above and all the follow up regression equations, n is the number of compounds, r is the correlation coefficient, Q2 is cross-validated index from leave-one-out (LOO) procedure, s is the standard error of the estimate and F is the F-ratio between the variances of calculated and observed activities. The values given in the parentheses are the standard errors of the regression coefficients. Also, in the randomization study, where 100 simulations per model were carried out, none of the identified models has shown any chance correlation. The six models, including the above-shown two, emerged from the collective class of descriptors belonging to TOPO, BCUT, GVZ and 2DAUTO classes. However the above two models, being more significant compared to remaining four (not given here), shared the descriptors belonging to TOPO and 2DAUTO classes only. Among these descriptors, the bond information contents, BIC2 and the complementary information contents, CIC2, both being neighborhood symmetry of 2-order, belong to TOPO class. The 2DAUTO descriptors, MATSke and GATSke have their origin in autocorrelation of topological structure of Moran and of Geary [Citation16,Citation17], respectively. The computation of these descriptors involves the summation of different autocorrelation functions corresponding to the different fragment lengths and lead to different autocorrelation vectors corresponding to the lengths of the structural fragments [Citation18]. Also a weighting component in terms of a physicochemical property has been embedded in these descriptors. As a result, these descriptors address the topology of the structure or parts thereof in association with a selected physicochemical property. In these descriptors' nomenclature, the penultimate character, a number, indicates the number of consecutively connected edges considered in its computation and is called as the autocorrelation vector of lag k (corresponding to the number of edges in the unit fragment). The very last character of the descriptor's nomenclature indicates the physicochemical property considered in the weighting component— e for atomic Sanderson electronegativity— for its computation. In above two equations, Geary autocorrelation lag 7 and 8 weighted by electronegativity (GATS7e and GATS8e) and Moran autocorrelation lag 6 and 8 weighted by electronegativity (MATS6e and MATS8e) are correlated with the activity. From , it appears that though the descriptors of (Equation 1) have participated in more number of models compared to the descriptors of (Equation 2) but the later one is statistically more significant. This implies that the descriptors of (Equation 2) are more enriched in information content to influence the malonyl coenzyme A decarboxylase inhibition activities of the compounds under study. This equation was, therefore, considered further to draw some meaningful conclusions. The lone compound 19, bearing a CH2CH2NMe2 substituent at R1, does not fit into the trend followed by other congeners of the series. A basic group such as the N,N-dimethylamino group at the terminal position, separated through 4-lags, is undesirable and may significantly decrease the potency [Citation6]. Possibly, an acidic or neutral moiety present at lag-4 is essential for desired interaction of R1 substituents. This compound was, therefore, eliminated from the data-set and the derived correlation is shown in (Equation 3)
All the statistical parameters of this equation are improved over to that of (Equation 2). The r2 now accounts for 83% of variance in observed activity values while F-value remained significant at 99% level [F3,43(0.01) = 4.30]. Also, the value obtained for cross-validated Q2 index is in favor of a robust QSAR model. Above model equation was further subjected to randomization process, where 100 simulations per model were carried out but none of the identified models has shown any chance correlation.
Table III. Descriptors identified for modeling the malonyl coenzyme A decarboxylase inhibitory activity of N-alkyl-N-(1,1,1,3,3,3-hexafluoro-2-hydroxypropylphenyl)amide derivatives along with the average regression coefficients, standard deviation and the total incidence.
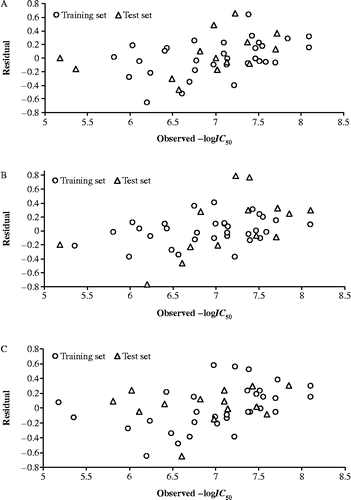
Additionally, the above model equation was also subjected to external validation. For this purpose three test sets, each containing 14 compounds out of the 46 active ones listed in , were considered. Of the three test sets, two were generated in the SYSTAT [Citation19] using the single linkage hierarchical cluster procedure involving the Euclidean distances of the respective descriptors or the activity as the case may be. The selection of the test set from the cluster tree was done in such a way to keep the test compounds at a maximum possible distance from each other. The third test set of the compounds corresponds to the random selection procedure. With this, these test sets represent different cross-sections of compounds. The predictions of the test sets have been done with the models developed using the 46 compounds remaining in the training sets.
The activity values of all three inactive compounds (12, 29 and 31) have also been predicted from these sets. The residuals of the predictions and the corresponding predictive R2-values of each test set have been given in . The predictions corresponding to all compounds in three test sets are within the reasonable limits of their actual values. Also, all the models from the training sets have predicted the two out of the three inactive compounds as low active ones. The exception is with compound 31 which is bearing a morpholinyl moiety () as the varying group in this congener and predicted to have activity values, from three test sets, in the range of slight active compounds. The calculated activity value of outlier compound 19, using (Equation 3), has significantly deviated from the observed ones (). Also, the predicted activity of this congener, from three test sets (), is comparatively much higher. This favors our consideration that compound 19 is an obvious outlier. The overall satisfactory performance of all the test sets provided us the confidence about the procedure adopted in the study and the variables selected therein. Additionally, the plot in is given to show the goodness of fit and to identify the systematic variation between observed and calculated—logIC50 values (Equation Equation 3). Similarly, the plot in is given to demonstrate the trend set by the compounds of three training sets, derived through clustering of descriptors, activity values and random selection. The similar trend is followed by the compounds present in corresponding test set. The residuals (observed— calculated activity values) obtained for the aforesaid test sets are found to be evenly distributed about the zero residual line. The predictions of three test sets are, therefore, in parity of what an excellent model can yield.
Table IV. Predicted residual activity of three test-sets (14 compounds in each) derived from the compounds in Table I.
Figure 1. Plot of observed versus calculated –logIC50 values.
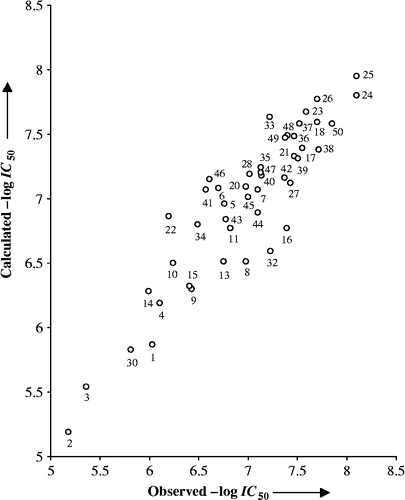
Figure 2. Plot of observed –logIC50 versus residual values of training and test sets developed from (A) Descriptors, (B) Activity values and (C) Random selection of the compounds.
From (Equation 3), it appeared that the complementary information contents having neighborhood symmetry of order-2, CIC2 and the Geary autocorrelations-lag 8, weighted by atomic Sanderson electronegativities, GATS8e have both added negatively to inhibition activity while the Moran autocorrelations-lag 6, weighted by atomic Sanderson electronegativities, MATS6e contributed positively to it. This implies that the smaller positive values of the parameters CIC2 and GATS8e and the larger positive (or smaller negative) value of MATS6e are preferred to improve the activity of a compound. The descriptors, MATS6e and GATS8e have emphasized the importance of electronic enriched centers at the path length-6 and -8, respectively. These lags, in particular, are important for the substituents appeared at R1 and are apparent in the most potent compounds 24, 25, and 50 (–logIC50 > 7.80; ). It appears that there are two important sites of the receptor which may be 7 and/or 8 bonds away from the benzene ring. These sites being opposite in nature (acidic or basic but not neutral) may engage themselves in electronic type of interaction. The functionality, such as carboxyl group in compounds 24 and 50 and tetrazolyl ring in compound 25, appears to be electronically enriched through lag-6, while the hydrogen present at lag-8 is unable to do so. The doubly bonded oxygen (of carboxyl group) or nitrogen (of tetrazolyl ring) atom may, therefore, engage itself with proton-donor (acidic) site while hydrogen bonded oxygen or nitrogen perhaps interact with proton-acceptor (basic) site of the receptor. This implies that a descriptor weighted by electronegativity at lag-6, contributes to enhance the activity and the descriptor weighted similarly at lag-8 reduces the same. The positive and negative regression coefficients associated, respectively to MATS6e and GATS8e, in (Equation 3), have substantiated the same.
Since the biological activity of a compound is by virtue of the mere presence of its constituent atoms and bonds, therefore, depending upon the number and type of atoms and the arrangement of different bonds, a molecule may conceal unique information content in the form of electron probability fields distributed in space. This information content of different symmetry orders is of considerable importance as its interpretation by the receptor site determines the efficacy of the drug molecule. The descriptor CIC2, being the function of molecular structure is, therefore, embodies the complementary information content of the 2-order symmetry and emerged as one of the promising parameter for the inhibition of MCD. Its negative but significant contribution to inhibition activity has required for those substituents in parent structure which could yield the lower values of CIC2.
Thus the criteria mentioned above may be used to explore new potential compounds of the series.
Acknowledgements
We express sincere thanks to our Institutions for providing necessary facilities to complete this work.
Related Research Data
References
- D Hearse. Metabolic approaches to ischemic heart disease and its management. New York: Science Press Ltd; (1998).
- JA Kennedy, SA Unger, and JD Horowitz. (1996). Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol 52:273–280.
- JG McCormack, RL Barr, AA Wolff, and GD Lopaschuk. (1996). Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 93:135–142. (b) McCormack JG, Stanley WC, Wolff AA. Ranolazine: A novel metabolic modulator for the treatment of angina. Genet Pharmacol1998;30:639-645. (c) Pepine CJ, Wolff AA. A controlled trial with a novel anti-ischemic agent, ranolazine in chronic stable angina pectoris that is responsive to conventional antianginal agents. Ranolazine study group. Am J Cardiol 1999;84:46-50
- PF Kantor, A Lucien, R Kozak, and GD Lopaschuk. (2000). The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res 86:580–588.
- JR Dyck, J-F Cheng, W Stanley, R Barr, MP Chandler, S Brown, D Wallace, T Arrhenius, C Harmon, G Yang, A Nadzan, and GD Lopaschuk. (2004). Malonyl-CoA decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res 94:e78–e84.
- J-F Cheng, M Chen, D Wallace, S Tith, M Haramura, B Liu, CC Mak, T Arrhenius, S Reily, S Brown, V Thorn, C Harmon, R Barr, JRB Dyck, GD Lopaschuk, and AM Nadzan. (2006). Synthesis and structure-activity relationship of small-molecule malonyl coenzyme A decarboxylase inhibitors. J Med Chem 49:1517–1525.
- MR Patel, and TT Talele. (2007). 3D-QSAR studies on malonyl coenzyme A decarboxylase inhibitors. Bioorg Med Chem 15:4470–4481.
- Dragon software (version 1.11-2001) by R Todeschini ,V Consonni. Milano, Italy.
- Chemdraw ultra 6.0 and Chem3D ultra. Cambridge Soft Corporation, Cambridge, USA.
- YS Prabhakar. (2003). A combinatorial approach to the variable selection in multiple linear regression: Analysis of Selwood. data set – A case study. QSAR Comb Sci 22:583–595.
- MK Gupta, R Sagar, AK Shaw, and YS Prabhakar. (2005). CP-MLR directed QSAR studies on the antimycobacterial activity of functionalized alkenols-topological descriptors in modeling the activity. Bioorg Med Chem 13:343–351.
- YS Prabhakar, RK Rawal, MK Gupta, VR Solomon, and SB Katti. (2005). Topological descriptors in modeling the HIV inhibitory activity of 2-aryl-3-pyridyl-thiazolidin-4-ones. Comb Chem High Thro Screen 8:431–437.
- MK Gupta, and YS Prabhakar. (2006). Topological descriptors in modeling the antimalarial activity of 4-(3',5'-disubstituted aniline)quinolines. J Chem Inf Model 46:93–102.
- S-S So, and M Karplus. (1997). Three-dimensional quantitative structure-activity relationship from molecular similarity matrices and genetic neural networks 1. Method and validation. J Med Chem 40:4347–4359.
- YS Prabhakar, VR Solomon, RK Rawal, MK Gupta, and SB Katti. (2004). CP-MLR/PLS directed structure-activity modeling of the HIV-1 RT inhibitory activity of 2,3-diaryl-1,3-thiazolidin-4-ones. QSAR Comb Sci 23:234–244.
- PAP Moran. (1950). Notes on continuous stochastic phenomena. Biometrika 37:17–23.
- RC Geary. (1954). The contiguity ratio and statistical mapping. Incorp Statist 5:115–145.
- P Broto, G Moreau, and C Vandycke. (1984). 2D autocorrelations. Eur J Med Chem 19:66–70.
- SYSTAT, Version 7.0; SPSS Inc., 444 North Michigan Avenue, Chicago, IL, 60611.