Abstract
Synthesis and enzymatic evaluation of new series of N4-substituted piperazine naphthamide derivatives as BACE-1 inhibitors for the treatment of Alzheimer's disease are reported.
Introduction
The most prominent neuropathological finding in Alzheimer's disease (AD) brains is the occurrence of cortical plaques containing beta-amyloid (Aβ) peptide. Genetic evidence points to a causal role of Aβ during development of AD as it is postulated by the amyloid cascade hypothesis [Citation1]. Consequently, blocking the production of Aβ peptides by specific inhibition of key proteases required for Aβ peptide generation is a major focus of research into AD therapy. The identification of secretase inhibitors has triggered a race to develop drug-like inhibitors of these enzymes, which have become major AD targets. Since it has been established that γ-secretase cleaves various substrates, some of which might have important physiological roles (Notch cleavage), γ-secretase inhibitors might have insurmountable mechanism-based toxicity [Citation2]. Taking into account those observations developing β-secretase inhibitors appear to be more encouraging. Several independent approaches led to the identification of β-site amyloid precursor protein cleavage enzyme (BACE-1) as the first enzyme of the amyloid cascade. BACE-1 cleaves the 695 or 751 amino acid amyloid precursor protein to a 99 amino acid long precursor peptide which is later trimmed by γ-secretase to form the pathological 40 or 42 amino acid long β-amyloid peptide. BACE-1 is an integral membrane protein with high homology to other aspartic proteases. Given the lack of adverse effects in gene knock out mice, BACE-1 is considered a prime target for the development of AD therapeutics. BACE-1 possesses a bilobal structure [Citation3]. Typical for eukaryotic aspartic proteases [Citation4], with the catalytical aspartate residues Asp32 and Asp228 located in the substrate-binding cleft between the N- and C-terminal lobes [Citation3]. To exert their pharmacological activity in vivo, BACE-1 inhibitors have to traverse the blood–brain barrier. Most peptidomimetic BACE-1 inhibitors reported so far are large [Citation5], moderately permeable compounds not ideally suited for a central nervous system indication. An overview on the design and development of BACE-1 inhibitors has been published by Cummings et al. [Citation6].
Materials and methods
Starting from already published data on arylpiperazine amide scaffolds as a possible framework for the design of β-secretase inhibitors [Citation7–9], we investigated original and subtitle modifications at different positions on the aryl piperazine scaffold. Molecular modelisation and Protein Data Bank revealed that the aryl piperazine scaffold allows the design of a large diversity of BACE-1 inhibitors through the substitution possibilities offered at various positions as shown in .
Figure 1. General structure of new N4-substituted piperazine naphthamide derivatives as BACE-1 inhibitors.
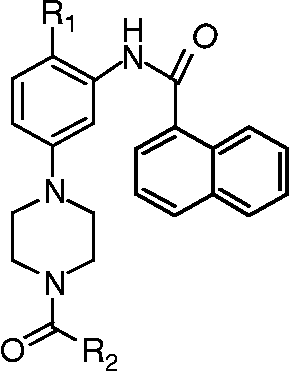
Through variation temperature NMR and modelisation studies, we have recently confirmed that introduction at the N4-position of the piperazine ring allows productive interactions within the BACE-1 active site, interactions which appear to be determinative for high BACE-1 inhibitory activity [Citation9]. This observation led us to focus our synthetic efforts on the synthesis of a library of derivatives in which the N4-position of the piperazine ring was substituted by a wide diversity of R2 substituents. We reported here the synthesis and the inhibitory potency of a new series of N4-substituted piperazine naphthamide.
The required intermediates 4a (R1 = H) and 4b (R1 = Br; Scheme ), have been synthesised according to reported procedure [Citation9].
Scheme 1. Reagents and conditions: (i) (a) N-Boc-piperazine (b) or N-benzylpiperazine, i-PrOH, reflux, 24 h; (ii) Zn powder, 1 M KH2PO4/THF, reflux, 24 h; (iii) 1-naphthoyl chloride, CH2Cl2, TEA, rt, 4 h; (iv) HCO2NH4, 10 wt% Pd/C, CH3OH, reflux, 6 h; (v) TFA, CH2Cl2, rt, 12 h; (vi) R2COCl, CH2Cl2, Et3N; (vii) R2CO2H, DCC, HOBt, DMAP, rt, 2 h.
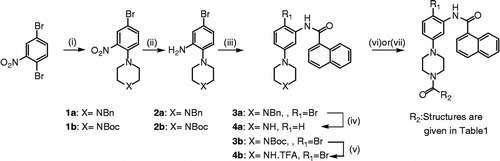
The new analogues have been synthesised, using standard methodologies, which involved the direct condensation of the corresponding acid chlorides (compounds 6a, 7a, 9a, 10a, 6b, 7b, 9b and 10b), or chloroformiate (compounds 11a and 11b), in the presence of triethylamine in methylene chloride at room temperature, and in the case of the carboxylic acids, the new derivatives (compounds 5a, 8a, 5b and 8b) were obtained in good yields using standard coupling reactions using dicyclohexylcarbodiimide (DCC), hydroxybenzotriazole (HOBt) in the presence of dimethylaminopyridine (DMAP) in methylene chloride at room temperature. It should be underlined that the corresponding 4′,4-diallyloxybenzoic acid was obtained through a methodology reported by Eicher et al. [Citation10]
Synthesis
General procedures
Unless otherwise noted, starting materials and reagents were obtained from commercial suppliers and were used without purification. Methylene dichloride (CH2Cl2) was distilled over P2O5 just prior to use. NMR spectra were recorded at 250 MHz for 1H and 62.9 MHz for 13C on a Brüker AC-250 spectrometer. Chemical shifts are expressed as d units (parts per million) downfield from tetramethylsilane. Electrospray mass spectra were obtained on a Waters Micromass ZMD spectrometer by direct injection of the sample solubilised in acetonitrile. Elemental analyses were within ± 0.4% of theorical values for all compounds. Analytical thin layer chromatographies (TLC) and preparative thin layers chromatographies were performed using silica gel plates 0.2 and 1 mm thick, respectively (60F254, Merck, Darmstadt, Germany). Preparative flash column chromatographies were carried out on silica gel (230–240 mesh, G60, Merck).
General procedure for compounds 3a and 3b
To a mixture of 2,5-dibromonitrobenzene (2.23 g, 7.94 mmol) in i-PrOH (40 ml) was added N-benzyl piperazine (2.1 g, 11.9 mmol). The resulting mixture was stirred at 70°C for 24 h. The reaction mixture was cooled to room temperature and then concentrated under reduced pressure. The brown residue was purified by chromatography on silica gel. Elution with increasing proportions of EtOAc–cHex (1:9–1:1) gave compound 1a (2 g, 69%) as an orange oil.
Zinc dust (0.55 g, 8.45 mmol) was added to a stirred solution of 1a (0.64 g, 1.69 mmol) in tetrahydrofuran (THF; 45 ml) followed by KH2PO4 (30 ml, 1 M). The resulting mixture was stirred at 80°C for 24 h. The reaction mixture was cooled to room temperature, filtered and evaporated to a small volume. The mixture was extracted with CH2Cl2, and the organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel, with EtOAc–cHex as eluant (1:9), giving aniline 2a (0.40 g, 68%) as a pale yellow foam.
To a solution of 2a (0.2 g, 1.15 mmol) in methylene chloride (10 ml) with diisopropylethylamine (DIEA) (300 μl, 1.73 mmol) was added 1-naphthoyl chloride (1.5 mmol). The reaction mixture was stirred for 4 h at room temperature. The mixture was extracted with water (15 ml), and the organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel, with EtOAc–cHex eluant (1:3) giving 3a (0.50 mg, 90%) as a yellow solid. Compound 3b was prepared by a method similar to that described for 3a using 1b.
Procedure for compounds 4a
To a stirred suspension of the appropriate N-benzyl compound 3a (0.36 g, 0.72 mmol) and an equal weight of 10% Pd–C in dry methanol (10 ml) was added anhydrous ammonium formate (0.23 g, 3.6 mmol) in a single portion under nitrogen. The resulting mixture was stirred at reflux temperature, and the reaction was monitored by TLC. After completion, the catalyst was removed by filtration through Celite and washed with methylene chloride (10 ml). The solvent was removed under reduced pressure, and the crude product was purified by chromatography on silica gel with methylene dichloride (DCM), 0.5% MeOH as eluent, to yield the desired compound 4a (0.23 g, 98%) as a yellow oil. Rf 0.30 (CH2Cl2/MeOH 10%). 1H NMR (CDCl3, 250 MHz) δ: 9.16 (1H, s), 8.69 (1H, d), 8.47 (1H, dd), 7.98 (1H, d), 7.91 (1H, dd), 7.75 (1H, d), 7.60–7.50 (3H, m), 7.28–7.20 (2H, m), 7.14 (1H, dd), 2.91 (8H, s), 2.80 (1H, l). ESI-MS m/z [M + H]+ 332. Anal. (C21H21N3O) C, H, N.
Procedure for compound 4b
To a stirred solution of the appropriate N-Boc compound 3b (0.51 mg, 1 mmol) in dry CH2Cl2 (10 ml) was added trifluoroacetic acid (TFA; 770 μl, 10 mmol). The reaction mixture was stirred overnight at room temperature. After completion, the solvent and excess TFA were removed under reduced pressure, and the corresponding TFA salt was identified as the desired compound 4b (0.38 g, 93%) as a TFA salt. Rf 0.11 (CH2Cl2/MeOH 10%) 1H NMR (CDCl3, 250 MHz) δ: 9.20 (1H, s), 8.94 (1H, s), 8.50 (1H, t), 8.03 (1H, d), 7.94 (1H, t), 7.76 (1H, d), 7.59 (3H, m), 7.27 (1H, dd), 7.09 (1H, d), 2.87 (8H, l), 1.97 (1H, l). ESI-MS m/z [M + H]+ 411. Anal. (C21H20BrN3O) C, H, N.
General procedure for compounds 6a, 7a, 9a, 10a, 6b, 7b, 9b and 10b
To a solution of 4a (60 mg, 0.18 mmol) in methylene chloride (8 ml) with DIEA (0.27 mmol) was added in 4-trifluoromethylbenzoyl chloride (47 mg, 0.23 mmol). The reaction mixture was stirred for 12 h at room temperature. The mixture was extracted with water (15 ml), and the organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel, with EtOAc–cHex eluant (1:2), to give the desired compound 6a (85 mg, yield 94%) as a white solid. Rf 0.53 (cHex–AcOEt, 1:1). 1H NMR (CDCl3, 250 MHz) δ: 8.98 (s, 1H), 8.62 (d, 1H), 8.37 (t, 1H), 7.93 (d, 1H), 7.86 (t, 1H), 7.68–7.41 (m, 7H), 7.23–7.08 (m, 3H), 3.51 (d, 4H), 2.85 (d, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 169.01, 167.22, 140.31, 138.87, 134.74, 132.16, 131.27, 133.82, 130.10, 128.61, 127.45, 126.72, 125.75, 124.89, 124.31, 120.87, 119.84, 52.44, 48.52. ESI-MS m/z [M + H]+ 504. Anal. (C29H24F3N3O2) C, H, N.
Compound 7a
Yield: 74%. Rf 0.11 (cHex–AcOEt, 2:1). 1H NMR (CDCl3, 250 MHz) δ: 9.11 (s, 1H), 8.70 (d, 1H), 8.45 (t, 1H), 8.00 (d, 1H), 7.92 (t, 1H), 7.73 (d, 1H), 7.57 (m, 3H), 7.37 (d, 2H), 7.30–7.10 (m, 3H), 6.90 (d, 2H), 3.82 (s, 3H), 3.62 (m, 4H), 2.91 (m, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 169.69, 166.39, 160.11, 139.75, 133.91, 133.11, 130.41, 129.28, 128.38, 127.74, 126.55, 125.85, 125.51, 124.39, 124.06, 123.41, 120.080, 118.88, 112.95, 54.53, 51.69, 26.075. ESI-MS m/z [M + H]+ 466. Anal. (C29H27N3O3) C, H, N.
Compound 9a
Yield: 98%. Rf 0.27 (cHex–AcOEt, 1:1). 1H NMR (CDCl3, 250 MHz) δ: 8.62 (d, 1H), 8.38 (t, 1H), 7.92 (d, 1H), 7.84 (t, 1H), 7.67 (d, 1H), 7.49 (m, 3H), 7.23–7.08 (m, 3H), 6.45 (q, 1H), 6.22 (d, 1H), 5.63 (d, 1H), 3.54 (d, 4H), 2.82 (s, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 167.22, 165.51, 140.49, 134.72, 133.96, 131.27, 130.10, 128.61, 127.38, 127.14, 126.70, 125.21, 124.92, 124.27, 120.91, 119.72, 52.53, 52.51, 46.29, 42.00. ESI-MS m/z [M + H]+ 386. Anal. (C24H23N3O2) C, H, N.
Compound 10a
Yield: 94%. Rf 0.89 (CH2Cl2–MeOH, 9:1). 1H NMR (CDCl3, 250 MHz) δ: 9.06 (s, 1H), 8.62 (d, 1H), 8.39 (t, 1H), 7.92 (d, 1H), 7.85 (t, 1H), 7.67 (d, 1H), 7.50 (m, 3H), 7.20–7.02 (m, 3H), 3.36 (m, 4H), 2.77 (t, 4H), 2.12–1.97 (m, 10H), 1.60 (m, 5H). 13C NMR (CDCl3, 62.9 MHz) δ: 167.23, 154.21, 133.9, 131.20, 130.16, 128.53, 127.35, 126.65, 125.32, 124.87, 124.18, 120.83, 79.95, 52.27, 41.68. ESI-MS m/z [M + H]+ 510. Anal. (C32H35N3O3) C, H, N.
Compound 6b
Yield: 87%. Rf 0.13 (cHex–AcOEt, 3:1). 1H NMR (CDCl3, 250 MHz) δ: 8.94 (s, 1H), 8.83 (d, 1H), 8.32 (t, 1H), 7.91 (d, 1H), 7.83 (t, 1H), 7.59 (m, 3H), 7.45 (m, 5H), 7.15 (dd, 1H), 6.95 (d, 1H), 3.69 (m, 2H), 3.29 (m, 2H), 2.78 (m, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 168.99, 167.15, 139.25, 138.75, 135.01, 134.16, 133.95, 131.58, 130.04, 128.66, 127.52, 127.47, 126.81, 124.85, 122.43, 119.81, 52.29, 48.03, 42.60. ESI-MS m/z [M + H]+ 583. Anal. (C29H23BrF3N3O2) C, H, N.
Compound 7b
Yield: 94%. Rf 0.22 (cHex–AcOEt, 2:1). 1H NMR (CDCl3, 250 MHz) δ: 9.00 (s, 1H), 8.84 (s, 1H), 8.35 (t, 1H), 7.92 (d, 1H), 7.84 (t, 1H), 7.63 (d, 1H), 7.48 (m, 3H), 7.28 (d, 2H), 7.17 (dd, 1H), 6.97 (d, 1H), 6.71 (d, 2H), 3.73 (s, 3H), 3.53 (m, 4Hl), 2.78 (s, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 170.53, 167.16, 160.99, 139.49, 135.06, 134.20, 133.95, 131.55, 130.07, 128.63, 127.52, 127.04, 126.79, 125.13, 124.86, 122.45, 119.47, 113.81, 55.39, 52.43, 29.72, 26.92. ESI-MS m/z [M + H]+ 545. Anal. (C29H26BrN3O3) C, H, N.
Compound 9b
Yield: 57%. Rf 0.19 (cHex–AcOEt, 1:1). 1H NMR (CDCl3, 250 MHz) δ: 8.99 (s, 1H), 8.85 (s, 1H), 8.37 (t, 1H), 7.92 (d, 1H), 7.84 (t, 1H), 7.64 (d, 1H), 7.48 (m, 2H), 7.16 (dd, 1H), 6.94 (d, 1H), 6.44 (q, 1H), 6.22 (d, 1H), 5.62 (d, 1H), 3.48 (d, 4H), 2.77 (s, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 167.15, 165.50, 139.38, 135.05, 134.17, 133.95, 131.57, 130.04, 128.62, 127.51, 127.05, 124.97, 122.44, 119.73, 52.44, 52.16, 46.18, 43.45. ESI-MS m/z [M + H]+ 465. Anal. (C24H22BrN3O2) C, H, N.
Compound 10b
Yield: 97%. Rf 0.38 (cHex–AcOEt, 5:1). 1H NMR (CDCl3, 250 MHz) δ: 9.03 (s, 1H), 8.86 (s, 1H), 8.38 (t, 1H), 7.94 (d, 1H), 7.87 (t, 1H), 7.65 (d, 1H), 7.53 (m, 3H), 7.19 (s, 1H), 6.97 (d, 1H), 3.36 (d, 4H), 2.73 (s, 4H), 2.10–2.02 (m, 10H), 1.57 (m, 5H). 13C NMR (CDCl3, 62.9 MHz) δ: 167.19, 154.15, 139.90, 135.07, 134.22, 133.96, 131.51, 130.12, 128.58, 127.50, 126.76, 125.21, 124.83, 122.35, 119.48, 80.07, 52.21, 41.67, 40.30, 36.17, 35.70, 30.85. ESI-MS m/z [M + H]+ 589. Anal. (C32H34BrN3O3) C, H, N.
General procedure for compounds 5a, 8a, 5b and 8b
Compound 4a (60 mg, 0.18 mmol), 2-chlorobenzoic acid (45 mg, 0.29 mmol), DCC (59 mg, 0.29 mmol), HOBt (39 mg, 0.29 mmol) and DMAP (35 mg, 0.29 mmo) were stirred in dry methylene chloride (15 ml) at room temperature for 10 h. After this time, the insoluble residue was filtered off and washed with 5 ml of methylene chloride. The filtrate was concentrated, and the resulting crude residue was purified by column chromatography with toluene–EtOAc (5:1) as eluent, to yield compound 5a as a white solid (92 mg, 98%; as a white powder). Rf 0.29 (toluene–AcOEt, 4:1). 1H NMR (CDCl3, 250 MHz) δ: 8.98 (s, 1H), 8.63 (d, 1H), 8.36 (t, 1H), 7.92 (d, 1H), 7.84 (t, 1H), 7.66 (d, 1H), 7.47 (m, 3H), 7.33–7.03 (m, 7H), 3.75 (d, 2H), 3.17 (m, 2H), 2.90 (d, 2H), 2.75 (dd, 2H). 13C NMR (CDCl3, 62.9 MHz) δ: 167.24, 167.00, 140.56, 135.38, 134.73, 133.81, 131.28, 130.44, 129.75, 128.61, 127.81, 127.41, 126.715, 126.38, 125.21, 124.93, 124.29, 120.90, 119.78, 52.43, 47.24, 42.18. ESI-MS m/z [M + H]+ 470. Anal. (C28H24ClN3O2) C, H, N.
Compound 8a
Yield: 90%. Rf 0.16 (cHex–AcOEt, 2:1). 1H NMR (CDCl3, 250 MHz) δ: 9.02 (s, 1H), 8.62 (d, 1H), 8.37 (t, 1H),7.93 (d, 1H), 7.86 (t, 1H), 7.67 (d, 1H), 7.50 (m, 3H), 7.18 (m, 3H), 6.87 (m, 3H), 5.97 (m, 2H), 5.29 (m, 4H), 4.54 (t, 4H), 3.52 (d, 4H), 2.83 (s, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 170.20, 167.06, 156.88, 149.85, 148.08, 140.46, 134.58, 133.69, 132.73, 131.08, 129.96, 128.43, 127.24, 126.55, 126.17, 124.75, 124.13, 120.74, 119.60, 120.511, 113.32, 112.95, 69.63, 52.36, 48.63. ESI-MS m/z [M + H]+ 548. Anal. (C34H33N3O4) C, H, N.
Compound 5b
Yield: 98%. Rf 0.25 (toluene–AcOEt, 4:1). 1H NMR (CDCl3, 250 MHz) δ: 8.96 (s, 1H), 8.83 (s, 1H), 8.33 (t, 1H), 7.92 (d, 1H), 7.84 (t, 1H), 7.62 (d, 1H), 7.48 (m, 3H), 7.31–6.98 (m, 5H), 6.96 (d, 1H), 3.50 (d, 4H), 2.77 (d, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 169.47, 167.15, 139.30, 136.15, 135.03, 134.18, 133.96, 133.48, 131.57, 130.04, 128.90, 128.66, 127.53, 127.08, 126.81, 124.86, 122.54, 119.80, 52.35, 47.92, 42.80, 29.72, 26.92. ESI-MS m/z [M + H]+ 549. Anal. (C28H23BrClN3O2) C, H, N.
Compound 8b
Yield: 95%. Rf 0.27 (cHex–AcOEt, 2:1). 1H NMR (CDCl3, 250 MHz) δ: 8.99 (s, 1H), 8.85 (d, 1H), 8.34 (t, 1H), 7.93 (d, 1H), 7.85 (t, 1H), 7.64 (d, 1H), 7.49 (m, 3H), 7.16 (dd, 1H), 6.97 (d, 1H), 6.86 (d, 2H), 6.77 (d, 1H), 5.96 (d, 2H), 5.36–5.16 (m, 4H), 4.53 (t, 4H), 3.52 (d, 4H), 2.78 (s, 4H). 13C NMR (CDCl3, 62.9 MHz) δ: 170.39, 167.08, 150.05, 148.23, 139.43, 135.03, 133.95, 133.00, 132.84, 131.55, 130.05, 128.63, 127.52, 127.06, 126.80, 124.96, 122.43, 120.65, 119.70, 118.03, 113.44, 113.04, 69.77, 52.43, 33.88, 30.16, 29.91, 24.96. ESI-MS m/z [M + H]+ 627. Anal. (C34H32BrN3O4) C, H, N.
General procedure for compounds 11a and 11b
To a solution of 4a (94 mg, 0.28 mmol) in methylene chloride (8 ml) with triethylenamine (TEA) (0.28 mmol) was added succinic anhydride (28 mg, 0.28 mmol). The reaction mixture was stirred for 24 h at room temperature. The reaction mixture was adjusted to pH 2 (with 1 N HCl), and the organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel, with CH2Cl2–MeOH eluant (9:1), to give the desired compound 11a (75 mg, yield 62%) as a white solid. Rf 0.41 (CH2Cl2–MeOH, 9:1). 1H NMR (DMSO-d6, 250 MHz) δ: 9.73 (s, 1H), 8.43 (d, 1H), 8.26 (d, 1H), 8.19 (m, 2H), 7.91 (d, 1H), 7.72 (t, 3H), 7.29 (m, 3H), 3.63 (s, 4H), 2.98 (m, 4H), 2.59 (m, 4H). 13C NMR (DMSO-d6, 62.9 MHz) δ: 175.28, 170.69, 167.92, 144.74, 135.46, 134.24, 133.83, 131.35, 130.63, 129.33, 127.99, 127.39, 126.093, 125.44, 123.45, 121.81, 55.86, 52.44, 46.02, 42.48, 30.15, 28.48. ESI-MS m/z [M + H]+ 432. Anal. (C25H25N3O4) C, H, N.
Compound 11b
Yield: 49%. Rf 0.47 (CH2Cl2–MeOH, 9:1). 1H NMR (DMSO-d6, 250 MHz) δ: 8.97 (s, 1H), 8.84 (s, 1H), 8.35 (t, 1H), 7.92 (d, 1H), 7.84 (t, 1H), 7.64 (d, 1H), 7.48 (m, 3H), 7.17 (d, 1H), 6.94 (d, 1H), 3.53 (m, 2H), 3.39 (m, 2H), 2.76 (m, 4H), 2.54 (d, 4H). 13C NMR (DMSO-d6, 62.9 MHz) δ: 177.2, 170.34, 167.20, 139.37, 135.02, 133.95, 131.59, 130.05, 128.66, 127.53, 127.10, 126.81, 125.09, 122.56, 119.77, 52.12, 45.80, 42.35, 29.42, 29.38, 27.89. ESI-MS m/z [M + H]+ 511. Anal. (C25H24BrN3O4) C, H, N.
Computational studies
The development of substrate-based inhibitors has been significantly aided by X-ray crystallographic analysis of BACE-1 complexes with a standard peptide-based inhibitor known as OM-2 [Citation3], since the structure of this complex has allowed the identification of critical ligand–active site interactions. BACE-1 active site was found more open than that of other aspartic proteases and exhibited conformational flexibility. Since peptide-based or hydroxyethylene inhibitors were relatively large and “undrug-like”, other X-ray crystallographic studies of BACE-1 complexes with other inhibitors (non-peptidic) have been performed. The most detailed account of non-peptidic BACE inhibitors comes from Vertex Pharmaceuticals Inc. (Cambridge, MA, USA) [Citation11], which disclosed several hundreds of compounds along with their associated BACE inhibition Ki values. Among the more potent classes reported, were the biaryl naphthalenes represented by compound 4a, in which the N4-position of the piperazine ring is free. Vertex proposed the first 3D pharmacophore map of BACE [Citation11]. Hydrogen interactions within the active site, with other key residues as well as other hydrophobic interactions with BACE sub-sites were determined. Unfortunately crystallographic data to substantiate this 3D model was not provided. Simulated molecular dynamics analysis reveals that a direct contact through amide hydrogen bonding interactions with the catalytic aspartic acids Asp32 and Asp228 [Citation3], could favoured BACE-1 inhibition. The compound 4a was placed in the most favourable position in the BACE-1 active site, allowing both hydrogen bonding within the active site as well as hydrophobic interactions with enzyme sub-sites. From this study, it appears that N4-position of the piperazine ring could be suitable to accommodate various substituents capable of additional interactions with enzyme sub-sites
In this perspective, we have synthesised a new series of compounds (16 analogues) which included at the N4-position different substituents, which differ from each other by different structural parameters: steric hindrance, polarity in terms of hydrophobic or hydrogen bondingand also by the organic function (amides, amines or carbamates) which links the substituents to the N4 nitrogen atom.
Computational studies (experimental procedure)
Molecular dynamics and molecular mechanics (InsightII, Discover module, Gromacs) is used in order to obtain the most realistic structures from known X-ray structures deposited at the Protein Data Bank. Starting from a model generated from the 1FKN structure [Citation3], a virtual docking of known inhibitors into the active site of the BACE-1 allows the selection of the best descriptor among those previously generated. To reach this goal, a small number of compounds with known Kd were used (training set 15–20 molecules), and screened using different docking software. Using a simulated annealing procedure (InsightII, Affinity module), and with quicker programs DOCK, and AUTODOCK it is possible to simulate a flexible ligand bound to a rigid receptor, but also a certain amount of flexibility of the protein. Following this methodology, we have selected some substituents, synthesise the corresponding compounds and then assayed as possible BACE-1 inhibitors.
The molecular structure of BACE-1 was obtained from Protein Data bank (pdb code: 1 FKM). Docking calculations were performed using as protonated either Asp32 or Asp228 inner oxygens for each compound.
Results and discussion
The new compounds were evaluated as BACE-1 inhibitors using BACE-1 fluorescence resonance energy transfer (FRET) Assay. These experiments have been preformed using BACE-1 (β-secretase) FRET Kit assay, from PanVera Corporation (Madison, WI, USA), according to the described protocol and using a multiwell spectrofluorometer instrument capable of 530–545 nm excitation and 570–590 nm emission wavelengths (Wallac Victor [Citation2] 1420, Perkin-Elmer, Turku, Finland). The kinetic assays are performed in duplicate for each inhibitor, using BACE-1 inhibitor (H-Lys-Thr-Glu-Glu-Ile-Ser-Glu-Val-Asn-(3S,4S)-Stat-Val-Ala-Glu-Phe-OH. IC50 = 30 nM; Calbiochem, Beeston, UK) as reference. The biological results are summarised in .
Table I. BACE-1 inhibition activity of N4-substituted piperazine naphthamide derivatives.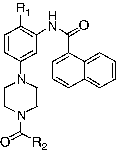
The data reported in show that some compounds are potent BACE-1 inhibitors or their inhibitory activity is noticeably influenced by the substituents on the N4 nitrogen atom of the piperidine ring, confirming that this region is crucial for efficient BACE-1 inhibition. As it could be seen, polar ω-carboxylic acid substituents (compounds 11a and 11b) or bulky substituents like adamantyl carbamate (compounds 10a and 10b) are detrimental for BACE-1 inhibitory activity. In the opposite substitution at the N4 position by halogenated phenyl groups (compounds 5a, 6a, 5b and 6b) or by methoxy groups (compounds 7a and 7b) are the most potent inhibitors. It could be observed that the position of these halogenated substituents on the phenyl ring does affect drastically the inhibitory activity. The presence of a bromine substituent on the phenyl ring instead of hydrogen does not affect the inhibitory activity in the whole series of analogues. One explanation for these observed differences in inhibitory potency could be supported by the differences in lipophilicity inside this new series of analogues. The presence of a substituted phenyl ring (compounds 5a, 6a, 5b and 6b) increased their corresponding log P values ranging between 4 and 5, suggesting that these substituted phenyl moieties seem particularly suitable for simultaneously fit the S1 and S′I pocket, while favourably positioning the protonated nitrogen atom closed to the catalytic diad Asp32 and Asp228. Compounds having lower log P values (compounds 9a, 11a and 11b) or having higher log P values are less potent BACE-1 inhibitors. In conclusion, introduction of suitable substituents at the N4 position of N-substituted piperazine naphthamide allowed to modulate their corresponding BACE-1 inhibitory activity, mainly hydrophobic aromatic substituents.
Acknowledgements
We are grateful to Professor K. Dudley (Université de la Méditerranée, IBDML) for the preparation of the manuscript.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- M Citron, T Oltersdorf, C Haass, L McConlogue, AY Hung, P Seubert, C Vigo-Pelfrey, I Lieberburg, and DJ Selkoe. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature 360 (6405):672–674.
- B De Strooper. (2003). Aph-1, pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 38 (1):9–12.
- L Hong, G Koelsch, X Lin, S Wu, S Terzyan, AK Ghosh, XC Zhang, and J Tang. (2000). Structure of the protease domain of memapsin 2 (b-secretase) complexed with inhibitor. Science (Washington, DC) 290 (5489):150–153.
- BM Dunn. (2002). Structure and mechanism of the pepsin-like family of aspartic peptidases. Chem Rev 102 (12):4431–4458.
- T Guo, and DW Hobbs. (2006). Development of BACE1 inhibitors for Alzheimer's disease. Curr Med Chem 13 (15):1811–1829.
- JN Cumming, U Iserloh, and ME Kennedy. (2004). Design and development of BACE-1 inhibitors. Curr Opin Drug Discov Devel 7 (4):536–556.
- Bhisetti GR, Saunders JO, Murcko MA, Lepre CA, Britt SD, Come JH, Deninger DD, Wang T (Vertex Pharmaceuticals Incorporated, USA). assignee. Preparation of b-carbolines and other inhibitors of BACE-1 aspartic proteinase useful against Alzheimer's and other BACE-mediated diseases. Application: WO patent WO 02/88101. 20020429.
- V John, JP Beck, MJ Bienkowski, S Sinha, and RL Heinrikson. (2003). Human b-secretase (BACE) and BACE inhibitors. J Med Chem 46 (22):4625–4630.
- C Garino, T Tomita, N Pietrancosta, Y Laras, R Rosas, G Herbette, B Maigret, G Quelever, T Iwatsubo, and JL Kraus. (2006). Naphthyl and coumarinyl biarylpiperazine derivatives as highly potent human beta-secretase inhibitors. Design, synthesis, and enzymatic BACE-1 and cell assays. J Med Chem 49 (14):4275–4285.
- T Eicher, M Ott, and A Speicher. (1996). Bryophyte constituents. 7. New synthesis of (+)-rosmarinic acid and related compounds. Synthesis (Stuttgart) (6):755.
- SJ Stachel, CA Coburn, TG Steele, KG Jones, EF Loutzenhiser, AR Gregro, HA Rajapakse, MT Lai, MC Crouthamel, M Xu, and et al (2004). Structure-based design of potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1). J Med Chem 47 (26):6447–6450.