Abstract
Monoamine transporters have emerged as important drug targets with a multitude of therapeutic potentials for their inhibitors. With the purpose of designing new chemical entities with enhanced inhibitory potencies against norepinephrine and serotonin transporters, the QSAR study carried out on N-arylmethylpiperidinamine derivatives as known inhibitors of these transporters is presented. The developed model was validated by standard QSAR parameters and through a detailed structural analysis on how it reproduces and explains the differences in the experimentally known activity data. The model showed a good correlative and predictive ability having a squared cross validated correlation co-efficient of 0.716 and 0.700 respectively for SET and NET inhibition. The squared conventional correlation coefficient was found to be 0.731 for SET antagonism and 0.777 for norepinephrine reuptake inhibition. The study confirmed that the serotonin reuptake inhibitory activity exhibited by the series is largely explained by steric factors of substituents emphasizing the role of size and shape of the inhibitors in making effective inhibitor-SET binding interactions whereas substituent lipophiliCIT000y was found to govern inhibitor-NET interaction chemistry. A detailed comparative investigation was made between the two models and the insights gleaned from the study could be usefully employed to design inhibitors with a much more enhanced potency and selectivity.
Introduction
Depression is a severe mental disorder characterized by exaggerated and pervasive feelings of sadness, loss of interest, and decreased energy that affects a considerable part of the population. According to the WHO, it is a worldwide mental health problem affecting an estimated 121 million people that is linked with a significant morbidity and mortality [Citation1,Citation2]. Moreover, depression is a multifaceted disease in terms of symptoms, co-morbidities and health complications, and treatment is complicated by the heterogeneity of the disease population in terms of these variables. The disease has serious implications in terms of quality of life and has serious economic burdens associated with loss of work and health-care costs. The high prevalence of suicide in depressed patients (up to 15%) coupled with complications arising from stress and its effects on the cardiovascular system have suggested that it will be the second leading cause of death by the year 2020.
The monoaminergic hypothesis for depression assumes that depression is caused mainly by a defiCIT000 of two monoamines, serotonin (5-HT) and norepinephrine (NE), in corticolimbic synaptic clefts [Citation3,Citation4] and this hypothesis has been used to explain the efficacy of existing antidepressant therapies. Among these available therapies, selective serotonin reuptake inhibitors (SSRIs) have become the standard treatment for depression. However, there are some limitations associated with the use of SSRIs including a delayed onset of action (2–4 weeks), partial treatment response (60–70%), exCIT000ation during early treatment response, nausea, and sexual dysfunction Citation5–7. It is widely accepted that the older tricyclic antidepressants (TCAs) such as amitriptyline and imipramine remain unsurpassed in their antidepressant efficacy and remain the gold standard for efficacy against which all potentially new antidepressants are compared in clinical trials. One possible reason for the efficacy of the TCAs, particularly in severe depression, has been attributed to their combined actions on both the noradrenergic and serotonergic systems [Citation8]. Indeed, an increasing body of evidence suggests that changes in both these aminergic systems are important for the success of treatment of severe depression [Citation9,Citation10]. By contrast, the selective serotonin reuptake inhibitors, while better tolerated than TCAs as a result of their less severe adverse effects at normal therapeutic doses, appear to be less efficacious than the TCAs in severe depression. Experimental evidence for the greater efficacy of dual action antidepressants has also been provided in the study of the effects of the SSRI sertraline, and the selective noradrenaline reuptake inhibitor reboxetine, alone and in combination, on the locomotor activity of the olfactory bulbectomized rat in a novel, stressful environment. Both drugs were effective in reducing the hyperactivity of the bulbectomized rats, a property exhibited by all therapeutically effective antidepressants [Citation11]. This indicates the need for dual modulation of the two transporters for effective antidepressant activity. Given the importance of monamine transporters in the progression of depression, serotonin and norepinephrine transporter proteins have increasingly become important targets and the hunt for their inhibitors has been intensified and attracted a great attention in drug discovery over the years Citation12–15.
The intense research on small molecule inhibitors of NET and SET has produced a diverse class of chemical scaffolds, which includes benzopyrano[4,3-c]isoxazoles [Citation16,Citation17], piperidinylpropanols [Citation18], benzo[1,4]oxazine [Citation19], tricyclic isoxazoles [Citation20]. shows some inhibitors of these monamine transporters. Although diverse in structure and large in number, most of them are beset with the problem of non-selectivity and weak binding affinity. TSAR, in common with other QSAR tools, is generally employed to enhance and optimize the binding affinity using a series of compounds acting on the same target with the same mechanism of actions. As a quantitative pharmacophore mapping tool, such a methodology is valuable in pinpointing the structural requirements for the observed pharmacotoxicological properties by a given series. Such insights are an aid to design a new entity having an acceptable level of potency and selectivity. In this paper, we report the 2D-QSAR study carried out on the dual NET and SET inhibitors in the anticipation of developing a model that would account for the quantitative differences in bioactivity seen in this series and to capitalize upon the insights to design ligands with pronounced inhibitory potency and selectivity.
Figure 1. Examples of monoamine transporter inhibitors.
Computational details
Dataset for analysis
The in vitro biological activity data reported as Ki for inhibition of serotonin and norepinephrine reuptake by a series of N-arylmethylpiperidinamine derivatives [Citation21] was used for the current study. As biological activities are generally skewed and since QSARs are measures of the free energy of ligand binding, the reported Ki values were converted into the corresponding pKi.
Molecular modeling
The structures of N-arylmethylpiperidinamine derivatives selected for the present QSAR study are shown in . The structures were sketched using ChemDraw ultra 8.0 and were exported to TSAR 3.3 software (Accelrys, www.accelrys.com). Three-dimensional structures of all the molecules were generated. Partial charges were derived using Charge-2 CORINA 3D package implemented in TSAR 3.3 and their geometries were optimized using Cosmic module of TSAR. The calculations were terminated when the energy difference or the energy gradient were smaller than 1e-005 and 1e-010 kcal/mol respectively.
Molecular descriptors were calculated with TSAR 3.3. The descriptors were obtained for the substituents which vary from one molecule to another at a common point on the generic structure. TSAR affords calculation of the following descriptors: molecular surface area and volume, moments of inertia, ellipsoidal volume, Verloop parameters, dipole moments, lipole moments, molecular mass, Wiener index, molecular connectivity indices, molecular shape indices, electrotopological state indices, log P, number of defined atoms (carbon, nitrogen, etc.), rings (aromatic and aliphatic), and groups (methyl, hydroxyl, etc.). Vamp which is a semiempirical molecular orbital package in TSAR 3.3 was used to calculate electrostatic properties like total energy, electronic energy, nuclear repulsion energy, accessible surface area, atomic charge, mean polarizability, heat of formation, HOMO and LUMO eigenvalues, ionization potential, total dipole, polarizability, and dipole components. Structure optimization was performed in vacuo using default parameters with the AM1 Hamiltonian. Pairwise correlation analysis of the calculated descriptors was performed. The model was obtained using descriptors that are strongly correlated with the antidepressant activity.
Statistical analysis
The relationship between the structural parameters (TSAR descriptors) and the biological activities has been quantified by the multiple linear regressions implemented in TSAR 3.3. Values for F-to-enter and F-to-leave were set to 4. The cross-validation analysis was performed using leave-one-out (LOO) method where one compound is removed from the dataset and its activity is predicted using the model derived from the rest of the dataset. The cross-validated r2 and conventional r2 that resulted in lowest error of prediction was taken. Unless otherwise stated, the default values for the other parameters were used. The predictive capabilities of the 2D-QSAR models were determined using test set compounds that were excluded during model development. The optimization, charge derivation, and all other steps of the test sets were the same as that of the training set compounds as described above, and their activities were predicted using the model produced by the training set.
Results and discussion
QSAR study for the SET reuptake inhibition
The 2D-QSAR TSAR study was carried out using arylmethylpiperidinamine derivatives which are reported for their antidepressant activity. Molecules, which lack biological inhibitory activity in numerical form, have been removed from the analysis. Following this, 56 molecules were left for SET reuptake inhibitory study. This was partitioned into a training set of 42 and a test set of 14 compounds for the SET inhibition at random with bias given to both chemical and biological diversity in both the training and test set molecules and so as to form 4:1 training set to test set ratio for a standard QSAR study. Despite the ambiguity of drug–receptor interaction in general, a statistically significant model were obtained from both the TSAR studies.
The TSAR multiple regression analysis is summarized in . The squared cross-validated correlation coefficient defines the goodness of prediction whereas the non-cross-validated squared conventional correlation coefficient indicates goodness of fit of a QSAR model. The F-test value stands for the degree of statistical confidence. As it is evident from the body of , a squared cross-validated correlation coefficient of 0.716 was obtained using leave-one-out cross-validation procedure. This indicates a very good internal predictive capability of the developed model. The model also exhibited a non-cross validated squared correlation coefficient of 0.731. The external predictive capability of a QSAR model is generally checked using test sets. All the other procedures including geometry optimization, charge computation, calculation of structural descriptors of the 14 test set molecules were done in a manner analogous to the training set molecules. The prediction of the test set molecules presented in shows a satisfactory prediction indicating its usefulness in predicting activities of external molecules. Yet another way to further evaluate the usefulness of the developed model is to test for statistical stability. To this end, standard error of estimate and predictive residual sum of squares may be employed. The low values of standard error of estimate (0.258) and that of PRESS for both training set (2.651) and for test sets (3.728) further add to the statistical significance of the developed models. shows the descriptors included in the final QSAR model and their statistical significance.
Table I. Statistical parameters obtained for the SET antagonistic model.
Table II. Structures of inhibitors used for QSAR analysis with corresponding actual and predicted activities for SET Inhibition.
Table III. Descriptors and their significance.
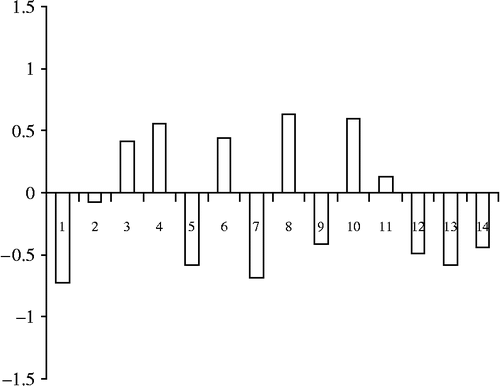
The structures of the monoamine transporter inhibitors chosen and their actual and predicted activity are displayed in . shows plots of actual versus predicted pKi values for the training set molecules. The histogram of residuals of the test set compounds is presented in . It is a plot of residuals against observations, each observation representing the data for a single structure. These two plots are important to graphically observe the predictive capability of QSARs. The shorter the heights of the residuals and the fact that the training set molecules are on or near to the best fit line as shown in further adds to the usefulness of the developed QSAR.
Figure 2. Plots of actual versus predicted Ki values for Training set molecules for SET inhibition.
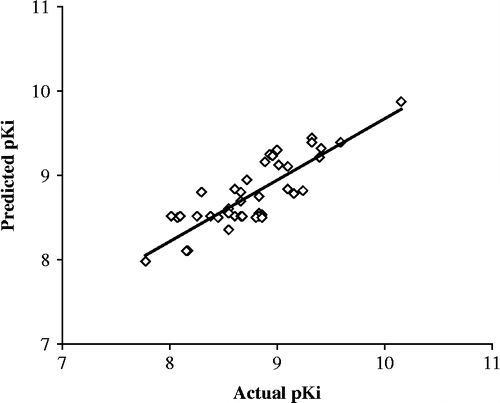
Figure 3. Histograms of residuals of Test set Test molecules for SET inhibition.
The QSAR model with a high statistical significance is represented by Equation (1):
Where B3 is Verloop B3 of substituent 1, B4 is Verloop B4 of substituent 4 and MR is molar refractivity of substituent 3.
The statistics for this equation are shown in . As the model shows, the SET inhibitory activity increases with an increase in the verloop B4 parameter of substituent 4 and the molar refractivity of substituent 3 while the activity was found to decrease with an increase in the values for the Verloop B2 parameter of substituent 1. The Verloop parameters Citation22–24 are a set of multi-dimensional steric descriptors that define a box that can be used to characterize the shape and volume of the substituent which are very important in explaining the steric influence of substituents in the interaction of organic compounds with macromolecular drug receptors. The Verloop B1-B5 parameters describe the width of the substituent in the direction perpendicular to the length of the substituent. The study suggests that SET reuptake inhibitor activity is strongly correlated with variations in the substituents at three positions of the general skeleton: namely, substitution on the nitrogen atom that bridges the piperidine and the benzyl group; two substitutions on the meta and para position of the benzyl moiety. The QSAR model shows that substitution on para position is strongly correlated with the antidepressant activity as it is evident from the higher Verloop B4 parameter (0.76) of substituents at this position. This apparently explains the difference in the activity of cpds 19, 22, 25, 28 and 17 which differ only in their substitution on the para position. Cpd 19 has got the highest activity for it showed a higher value for the width parameter at this position when compared to cpds 22, 25, 28 and 17. Indeed cpd 17, which has the least Verloop B4 at the para position is found to be the least active as compared to compounds 19, 22, 25 and 28. The higher activity of cpd 40 as compared to compounds 42, 43 and 29 may be explained by the same reasoning: cpd 40 which has higher value for the substitution at the para position is found to be more active. That the molar refractivity is also positively correlated with the antidepressant activity is what explains the better activities of cpds 18, 21, and 24 as compared to 17. Cpd 17 has the lowest MR value for substitution 3 and hence was found to be the least active in comparison with cpds 18, 21 and 24 which differ only by their substitution at this position. The same factor also explains the better activity of cpd 41 as compared to 29. The study suggests that the SET inhibitory activity exhibited by the series taken is largely explained by steric factors and that subsituents with a higher Verloop's B4 parameter on substitution 4 and molar refractivity value on substitution 3 are expected to enhance the antidepressant activity. Considering the fact that the 2D-QSAR model was able to reproduce the experimental facts and that it was validated by the appropriate statistical procedures, it could be useful in designing a more potent inhibitor.
QSAR study for the NET reuptake inhibition
The 2D-QSAR TSAR study was carried out using arylmethylpiperidinamine derivatives reported by Boot et al. Two molecules were found not to possess biological inhibitory activity in numerical form and these were removed from the analysis. Following this, 57 molecules were left for the NET reuptake inhibitory study which subsequently was partitioned into a training set of 41 and a test set of 16 compounds for the NET inhibition at random with bias given to both chemical and biological diversity in both the training and test set molecules. Despite the ambiguity of drug–receptor interaction in general, a statistically significant model were obtained from both the TSAR studies.
The TSAR multiple regression analysis is summarized in . As it is evident from the body of Table, a squared cross-validated correlation coefficient of 0.700 was obtained using leave-one-out cross-validation procedure. This indicates a very good internal predictive capability of the developed model. The model also exhibited a squared non-cross validated correlation coefficient of 0.777. The external predictive capability of the derived QSAR model was checked using test set of 16 molecules. All other procedures including geometry optimization, charge computation, calculation of structural descriptors of the test set molecules were done in a manner analogous to the training set molecules. The prediction of the test set molecules presented in shows a satisfactory prediction indicating its usefulness in predicting activities of external molecules. Yet another way to further evaluate the usefulness of the developed model is to test for statistical stability. To this end, standard error of estimate may be employed. The low values of standard error of estimate (0.389) further add to the statistical significance of the developed models. shows the descriptors included in the final QSAR model and their statistical significance.
Table IV. Statistical parameters obtained for the TSAR model of NET Antagonism.
Table V. Actual and predicted activities and Structural descriptors of inhibitors included in the final QSAR model for NET Inhibition.
Table VI. Descriptors and their significance.
The structural descriptors included in the final QSAR model together with the actual and predicted activity for NET inhibition are displayed in . shows plots of actual versus predicted pKi values for both the training and test molecules. The histograms of residuals of the training set and test set molecules is presented in .
Figure 4. Plots of actual versus predicted PKi values for Training set molecules for NET inhibition.
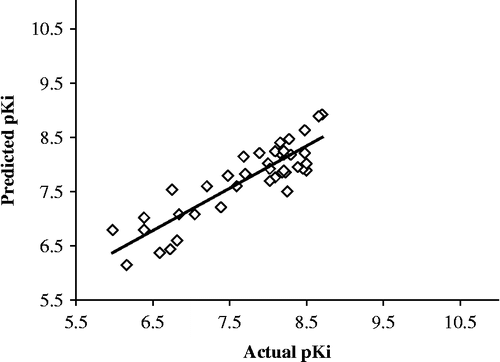
Figure 5. Histograms of residuals of Test set molecules for NET inhibition.
The QSAR model with a high statistical significance is represented by Equation (2):
Where L1 is Verloop L parameter of substituent 1, L3 is Verloop L of Substituent 3, log P2 is log P of substituent 2, log P4 is log P of substituent 4, TL is total lipole of substituent 2.
The statistics for this equation are shown in . As the model shows, the NET inhibitory activity is directly correlated with the Verloop L parameter of substituent 1, and with the log P of substituents 2 and 4 while it was found to decrease with Verloop L of substituent 3 and total lipole of substituent 2. The length parameter, Verloop L, is defined as the maximum length of the substituent along the axis of the bond between the first atom of the substituent and the parent molecule. LipophiliCIT000y is a measure of the ability of molecules to move between fat and water. It is often used to indicate how easily a molecule may be transported across membranes. As lipophiliCIT000y is difficult to measure directly, most of the time the water/octanol partition coefficient (log P) is used as an estimate to it. The lipole of a molecule is a measure of the lipophilic distribution. It is calculated from the summed atomic log P values, as dipole is calculated from the summed partial charges of a molecule.
The study suggests that NET reuptake inhibitory activity is strongly correlated with variations in the substituents at four positions of the general skeleton: namely, substitution on the nitrogen atom that bridges the piperidine and the benzyl group; three substitutions on the ortho, meta and para position of the benzyl moiety. The QSAR model for the NET inhibition shows that substitution on ortho position is directy correlated with the antidepressant activity. This may explain the better activity of cpd 3 as compared to cpd 2 where the former has higher value for the Verloop's L parameter which is found to directly related to the antidepressant activity. The study also suggests that substituents on the meta position of the benzyl moiety with higher Verloop's length parameter are associated with low NET inhibitory activity. This is exemplified by the lower activity of cpd 41 as compared to cpds 40, 42 and 43. Cpd 41 has a trifluoromethyl whereas the rest molecules have H in their meta position instead and thereby having a lower value for the length parameter and hence higher inhibitory value. The QSAR reveals that substituents with higher length are expected to improve activity at the ortho position and to decrease activity if placed at the meta position. The higher activity of cpd 39t as compared to cpd 37t which differ on para substitution emphasizes the fact that higher log P values are required for higher activity. The result shows that for SET inhibitory activity is largely explained by steric factors particularly on position one, three and four of the general structure. The NET inhibitory activity, on the other hand, is found to be predominantly explained by the lipophiliCIT000y of substituents and to a lesser extent by the steric factors of substituent one.
Conclusions
Monoamine transporters have emerged as important drug targets with a multitude of therapeutic potentials for their inhibitors. The QSAR analysis using 58 N-arylmethylpiperidinamines derivatives was successfully carried out to build a statistically significant model possessing good correlative and predictive capability for the inhibition of both NET and SET. The 2D-QSAR modes were validated by standard statistical means and how they reproduce and explains the differences in the experimentally known activity data. The detailed structural investigation revealed that the antidepressant activity exhibited by inhibiting the SET is predominantly explained by the steric factors of the substituent while the lipophiliCIT000y of the substituents was found to govern the inhibitor-NET interaction chemistry. The comparative investigation provided structural insights on how modulation of the steric bulk and lipophiliCIT000y of the substituents could be usefully made to optimize the antidepressant activity. The study provided useful clues about the structural requirement for effective inhibitor-monoamine transporter binding chemistry and hence for the improvement of the observed biological activity. This analysis could be of help in the rational design of potential drug candidates with an enhanced inhibitory potency.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
Related Research Data
References
- C Holden. (2000). Global survey examines impact of depression. Science 288 (5463):39–40.
- F Kerrigan. (1998). Antidepressant patents: 1995–1997. Exp Opin Ther Pat 8 (4):439–460.
- I Hindmarch. (2001). Expanding the horizons of depression: Beyond the monamine hypothesis. Psychopharmacol Clin Exp 16:203–218.
- FT Crews, and CB Smith. (1978). Presynaptic alpha-receptor subsensitivity after long-term antidepressant treatment. Science 202 (4365):322–324.
- C Spyraki, and HC Fibiger. (1980). Functional evidence for subsensitivity of noradrenergic alpha -2-receptors after chronic desipramine treatment. Life Sci 27 (20):1863–1867.
- JJ Rutter, C Gundlah, and SB Auerbach. (1995). Systemic uptake inhibition decreases serotonin release via somatodendritic autoreceptor activation. Synapse 20 (3):225–233.
- M Briley, and C Moret. (1993). Neurobiological mechanisms involved in antidepressant therapies. Clin Neuropharmacol 16 (5):387–400.
- WZ Potter, MV Rudorfer, and H Manji. (1991). The pharmacologic treatment of depression. N Eng J Med 325 (9):633–642.
- HM Van Praag, GM Asnis, and RS Kahn. (1990). Monoamines and abnormal behaviour: A multi-aminergic perspective. Brit J Psychiatry 157 (5):723–734.
- JL Cummings. (1993). The neuroanatomy of depression. J Clin Psychiatry 54 (11):14–20.
- JP Kelly, AS Wrynn, and BE Leonard. (1997). The olfactory bulbectomized rat as a model of depression: An update. Pharmacol Ther 74 (3):299–316.
- F Artigas. (1995). Selective Serotonin Noradrenaline. Reuptake Inhibitors. CNS Drugs 4 (2):79–89.
- PV Tran, FP Bymaster, RK McNamara, and WZ Potter. (2003). Dual Monoamine modulation for improved treatment of major depression. J Clin Psychopharmacol 23 (1):78–86.
- KJ Carrie, JE Brian, BN Anne, and ES Harlan. (2006). Analgesic effects of serotonergic, noradrenergic or dual reuptake inhibitors in the carrageenan test in rats: Evidence for synergism between serotonergic and noradrenergic reuptake inhibition. Neuropharmacology 51 (8):1172–1180.
- FP Bymaster, RK McNamara, and PV Tran. (2003). New approaches to developing antidepressants by enhancing monoaminergic neurotransmission. Expert Opin Investig Drugs 12 (4):531–543.
- JA Ignacio, A Jesus, MA Jose, ID Ana, I Laura, B Ilse, and AM Anton. (2006). Synthesis of 3a,4-dihydro-3H-[1]benzopyrano[4,3-c]isoxazoles, displaying combined 5-HTUptake inhibiting and 2-adrenoceptor antagonistic activities: A novel series of potential antidepressants. Bioorg Med Chem 14 (13):4361–4372.
- JA Ignacio, A Jesus, MA Jose, MA Rosa, MC Jose, AD Lucas, J Fernandez, S Martınez, N Carmen, J Pastor, HB Margot, L Heylen, and AA Megens. (2003). Synthesis of 3α, 4-dihydro-3H-[1]benzopyrano[4,3-c]isoxazoles, displaying combined 5-HTUptake inhibiting and 2-adrenoceptor antagonistic activities: A novel series of potential antidepressants. Bioorg Med Chem Lett 13 (16):2719–2725.
- T Kumiko, JK Todd, AH Nicholas, PR Vincent, GS Patrick, JK Daniel, LN David, W Bradley, JA Laura, S Janice, GT Penny, and TW David. (2003). Advances toward new antidepressants beyond SSRIs: 1-Aryloxy-3-piperidinylpropan-2-ols with Dual 5-HT1A receptor antagonism/SSRI activities. Part 3. Bioorg Med Chem Lett 13 (11):1903–1905.
- Z Dahui, LH Boyd, S Uresh, HA Terrance, AH Geoffrey, S Rosemary, ES Lee, LS Deborah, MS Kelly, and EM Richard. (2006). Studies toward the discovery of the next generation of antidepressants. Part 5: 3,4-Dihydro-2H-benzo[1,4]oxazine derivatives with dual 5-HT1A receptor and serotonin transporter affinity. Bioorg Med Chem Lett 16 (5):1338–1341.
- JI Andres, J Alcazar, JM Alonso, RM Alvarez, MH Bakker, and I Biesmans. (2005). Discovery of a new series of centrally active tricyclic isoxazoles combining serotonin (5-HT) reuptake inhibition with (2-adrenoceptor blocking activity. J Med Chem 48 (6):2054–2071.
- JR Boot, SL Boulet, BP Clark, MJL Cases-Thomas, KD Delhaye, J Fairhurst, J Findlay, PT Gallagher, J Gilmore, JR Harris, JJ Masters, SN Mitchell, M Naik, RG Simmonds, SM Smith, SJ Richards, GH Timms, MA Whatton, CN Wolfe, and VA Wood. (2006). N-Alkyl-N-arylmethylpiperidin-4-amines: Novel dual inhibitors of serotonin and norepinephrine reuptake. Bioorg Med Chem Lett 16 (10):2714–2718.
- A Verloop, and HW Tipker. Development and application of new steric substituent parameters in drug design. In: EJ Ariens. editor Drug design. New York: Academic Press; (1976). p 165–207.
- A Verloop, and J Tipker. (1976). Use of linear free energy related and other parameters in the study of fungicidal selectivity. Pesticide Sci 7 (4):379–390.
- A Verloop, and J Tipker. A comparative study of new parameters in drug design. In: JA Keverling. editor. Biological activity and chemical structure. Amsterdam: Elsevier; (1977). p 63–81.