Abstract
The anti- and pro-oxidant effects of green tea catechins have been implicated in the alterations of cellular functions determining their chemoprotective and therapeutic potentials in toxiCIT000y and diseases. The glutathione S-transferases (GSTs; EC 2.5.1.18) family is a widely distributed phase-II detoxifying enzymes and the GST P1-1 isoenzyme has been shown to catalyze the conjugation of GSH with some alkylating anti-cancer agents, suggesting that over-expression of GST P1-1 would result in tumor cell resistance. Here we report the docking study of four green tea catechins and four alkylating anticancer drugs into the GST P1-1 model, as GSTs were found to be affected by tea catechins. The EGCG ligands exhibit higher docking potential with respect to the anticancer agents, with a ligand-receptor interaction pattern indicating an high conformational stability. Consequently, the competition mechanisms favourable for the green tea catechins could lead to enzyme(s) desensitisation with a reduction of the alkylating drugs metabolism. The results provide a useful theoretical contribution in understanding the biochemical mechanisms implicated in the chemotherapeutic use of green tea catechins in oxidative stress-related diseases.
| Abbreviations | ||
| GSH, | = | L-glutamyl-L-cysteinyl-glycine |
| HGST, | = | human glutathione S-transferase |
| GTP, | = | green tea polyphenols |
| EGCG, | = | epigallocatechin gallate |
| ECG, | = | epicatechin gallate |
| GCG, | = | gallocatechin gallate |
| CG, | = | epicatechin gallate |
| RGS, | = | regulators of G-protein signaling |
Introduction
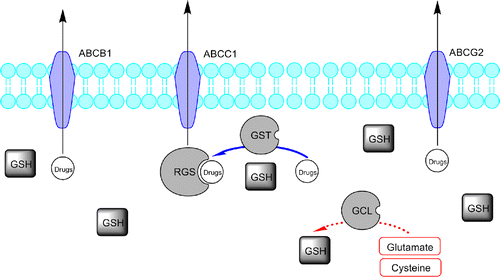
Advances in molecular medicine resulting in new drugs with specific cellular targets are not still sufficient to cover the multidisciplinary aspects related to drug resistance and individual patient variability [Citation1]. In particular, the resistance of human tumor to multiple chemotherapeutic drugs (multidrug resistance, MDR) is known to be a major reason for the failure of cancer therapy [Citation2]. Drug–metabolizing enzymes (DME) play a key role in the activation and deactivation of drugs, including a number of cytotoxics. They are subject to significant inter- and intra-individual variability, displaying both genetic polymorphism and in some cases inducibility. The glutathione S-transferases (GSTs; EC 2.5.1.18), is a family of widely distributed phase-II detoxifying enzymes (DME) involved in the cells protection (against many xenobiotic substances and products of oxidative stress) and detoxification, by catalytic conjugation of toxic electrophiles (including carcinogens and cytotoxic drugs) with the tripeptide glutathione [L-glutamyl-L-cysteinyl-glycine, (GSH)], making them less toxic and more readily excretable from the body [Citation3,Citation4]. Beside detoxification, the GST system possesses an additional function in the development of cytotoxic resistance in cancer cells, playing a negative regulatory role on the mitogen-activated protein (MAP) kinase pathway (through which many cytotoxics induce apoptosis). This effect is mediated by a direct binding to c-Jun-N-terminal kinase I (JNK) that is involved in the stress response and apoptosis by activation of the MAP kinase system [Citation5]. Glutathione is the most abundant intracellular non-protein thiol [Citation6] and its levels are primarily regulated by the enzyme glutamate-cysteine ligase (GCL), which catalyzes the rate-limiting step in overall GSH biosynthesis ().
Figure 1 . Representative scheme of the glutathione pathway and ABC-transporters family role in drug elimination and defence. Glutathione S-transferases (GSTs) conjugate GSH to drugs and drug metabolites facilitating the ATP-dependent elimination of drugs or drug-GS conjugates, involving ABC transporters (like ABCB1, ABCC1 and ABCG2). The drug/drug metabolites and intracellular GSH equilibrium is a major determinant of blood drug level.
Mammalian cytosolic glutathione S-transferases (GSTs) exist either as homo- or hetero-dimers with a subunit molecular mass of about 25 kDa and with one active site per monomer [Citation7]. The active site can be divided into two adjacent functional regions, a highly specific G-site binding the tripeptidic substrate and a non-specific H-site for binding non polar electrophilic substrate. Most GSTs exist as soluble enzymes, although a small family of microsomal GSTs has been characterized [Citation8,Citation9] and a mitochondrial GST (GST Kappa) has also been identified [Citation10]. GSTs have been classified into at least five distinct gene classes (alpha, mu, pi, sigma and theta) based on substrate specifiCIT000y and primary structures Citation11–13. Three polymorphisms have been reported to decrease or abolish the GST activity [Citation5,Citation14,Citation15], one of these, the human pi-class GST (hGST P1-1), was found to be over expressed in many tumor types, where it catalyzes the conjugation of GSH with some alkylating anti-cancer agents (such as chlorambucil, cyclophospamide and metabolites and melphalan) [Citation16]; hGST P1-1 is also implicated in the development of tumor resistance towards various anti-cancer drugs Citation17–20. The introduction of the enzymes in colon, stomach, pancreas, uterine cervix, breast and lung tumor cell lines reduces the cytotoxic activity of these drugs, suggesting that over-expression of GSTs (in particular the hGST P1-1 isozyme [Citation21]) would result in tumor cell resistance. Moreover, hGST P1-1 selective inhibitors increase the efficacy of anti-cancer drugs chemotherapeutic in resistant tumor cells [Citation22]. Both results stress the importance of using highly potent hGST P1-1 selective inhibitors, since they could increase the therapeutic outcome of anti-cancer agents Citation23–26.
Green tea polyphenols, mainly catechins like epigallocatechin gallate (EGCG), epicatechin gallate (ECG) and epicatechin (EC), have been implicated, in protection against cancer, diabetes, cardiovascular and neurodegenerative disorders [Citation27]. Following oral ingestion, EGCG are found in the blood and tissues in high concentrations [Citation28]. Catechins are believed to react with biomolecules either directly or after cellular metabolism and thus alter their functions. Catechins are antioxidants and scavengers of reactive oxygen (ROS) and nitrogen species, and are used in the prevention of oxidative stress related disorders [Citation29]. On the contrary, the most abundant green tea catechin EGCG and other catechins have been shown to spontaneously generate H2O2 in vitro, with subsequent cell death [Citation30]. EGCG targets multiple cells signalling pathways and control the cell proliferation and apoptosis, especially in cancer cells [Citation31]. In particular, EGCG has important effects on the expression of GSTs and these effects are cancer-specific [Citation32].
Nowadays, the use of antioxidant help to alleviate the toxic chemotherapic side effects and increase the efficacy of chemotherapy Citation33–35, Citation37, but much debate has arisen about whether and how antioxidant supplementation alters the efficacy of cancer chemotherapy.
Even if numerous cancer-related proteins are affected by green tea polyphenols [Citation38,Citation39], the molecular bases for green tea polyphenol-mediated cancer prevention remain unknown [Citation25,Citation26]. In particular, they significantly interfere with GSTs activity [Citation40] and the induction of these enzymes by either naturally or synthetic agents represents a promising chemoprevention strategy [Citation41,Citation42]. A recent study have shown that a four weeks green catechins administration in healthy subjects leads to differential effects on lymphocytes GST activity, depending on the its baseline activity. In fact, in this study it has been observed that individuals with the highest level of GST baseline exhibited a statistically significant decrease in mean GST activity. This effect is particularly relevant in clinical protocols where the green tea catechins are combined with anti-cancer drugs, as the GST baseline level is already high for the chemotherapy. In this case the catechins principally could reduce the anti-cancer agents metabolism through GSTs inhibition, affecting the pharmacokinetics and pharmacodynamics of the anticancer drugs. For this reason the achieving of further insight into the mechanism of interaction between green tea catechins and GST isoform(s) could give deepen confidence to their chemotherapeutic efficacy.
As an X-ray diffraction model of the complexes between the isoform hGST-P1 with the clinically used anti-cancer drugs are still absent, we have performed computational docking calculations to define the putative interactions of four alkylating anti-cancer drugs (cyclophosphamide, ifosfamide, melphalan and chlorambucil) and four natural green tea polyphenols (( − )-EGCG, ( − )-GCG, ( − )-ECG and ( − )-CG) [Citation30] (). In the binding model obtained, the natural EGCG ligand exhibits high docking potential over the other studied anti-cancer drugs to the hGST-P1-1, with a ligand-receptor interaction pattern indicating an high conformational stability. This competition in the ligand-enzyme interaction favourable for the catechins with respect to the conventional alkylating drugs, could lead to a decreased tumour cell drug resistance.
Figure 2 . Chemical structures of the green tea catechins and of the alkylating anti-cancer drugs docked. The oxygen numbering scheme and the ring nomenclature [Citation43] used throughout the text is indicated only for ( − )-EGCG.
![Figure 2 . Chemical structures of the green tea catechins and of the alkylating anti-cancer drugs docked. The oxygen numbering scheme and the ring nomenclature [Citation43] used throughout the text is indicated only for ( − )-EGCG.](/cms/asset/b4890c3e-f870-4cbb-aa1e-149bad4808f8/ienz_a_317895_f0002_b.gif)
Materials and methods
Molecular modelling
Molecular modelling and graphics manipulations were performed using an optimized Mac OSX version NAMD [Citation44], AutoDock [Citation45] and UCSF Chimera software packages [Citation46] on an Apple® MacPro quad-Xeon workstation running Mac OSX 10.4 (Tiger). Model building and geometry optimizations of the studied compounds were accomplished with the Gaussian 03 (6-31g* base set) [Citation47] quantum mechanical calculations package. The output from AutoDock and all modeling studies as well as images were build with PyMOL [Citation48] and Accelrys DSVisualizer [Citation49] and rendered with POVRay [Citation50]. DSVisualizer was used to calculate the hydrogen bonds distances measured between the hydrogen and its assumed binding partner.
Ligand setup
The green tea polyphenols starting conformations were optimized with quantum mechanical calculations by means of Gaussian 03 suite (6-31g* base set) [Citation47]. All ligands atomic charges were assigned using the Gasteiger-Marsili formation, which is the atomic charges type used in calibrating the AutoDock empirical free energy function.
Protein setup
The three-dimensional GST P1-1 starting model was obtained from the Protein Data Bank (PDB code: 1J9H [Citation51]) and it was checked through the OSX version of the AutoDock Tools (ADT, [Citation52]) and UCSF Chimera to guarantee the system conformity with the molecular modelling programs. All the protonation states were set to the normal ionisation forms at pH 7.0 and His residue and both molecular topology and connectivity have been created. The GST P1-1 aminoacidic chain has been terminated with –COO− and –NH3+ groups in their zwitterionic form and the polar hydrogen atoms were added in their calculated positions. The GST-substrate complex was built docking the green tea polyphenols into the equilibrated GST P1-1 structure and the resulting system was then equilibrated with a series of minimizations interspersed by short molecular dynamics simulations. The resulting structure was submitted for the 1 ns molecular dynamics simulation and then it was optimized by means of an AMBER force field as implemented in the NAMD package [Citation44]. The simulation was performed at constant temperature and pressure (NPT ensemble) in a periodic cubic box of TIP3P water molecules [Citation53,Citation54]. The water bond distances and angles were constrained using the SETTLE algorithm [Citation55], while the protein bond lengths were constrained with the LINCS algorithm [Citation56]. The coupling time was set to 1.0 ps and the isothermal compressibility was set to 4.6 × 10− 5 bar− 1. Both protein and solvent were independently coupled to a temperature of 298°K, with a coupling time of 0.1 ps and the pressure was held at 1 bar, with a coupling time of 0.2 ps, using a Berendsen thermostat maintaing temperature and pressure constant. The time step used was 1.0 fs. A total of 6000 snapshots were saved every 0.2 ps. Hydrogen bonds and close contacts were automatically identified using the “contact” module of CCP4 [Citation57] and UCSF Chimera [Citation46], while the other interactions were identified visually.
Docking simulations
Docking of green tea cathechins and alkylating drugs to hGST P1-1 was carried out using AutoDock version 4.0 [Citation45]. The AutoDock suite uses an automated docking approach that allows ligand flexibility, and it is able to locate docking poses in a consistent way with respect to the X-ray crystal structures [Citation58]. Default parameters (including a distance-dependent dielectric “constant”) were used as described in the AutoDock manual and both the protein crystal structure and the ligands were prepared for docking by following the default protocols (except for the changes mentioned below). AutoDock uses an empirical scoring function able to approximate the binding free energies, as it includes a solvation free energy term. The energy-scoring grid was prepared as a 20 × 20 × 20 Å box centered around the S-Hexylglutathione crystallographic molecule, with a 0.375 Å grid resolution. The ligands were limited to move inside this searching space during docking. Atomic solvation parameters were assigned to the protein and the default parameters for the Lamarckian genetic algorithm were used as search protocol, except for the maximum number of energy evaluations, which were changed to 10 million (the population size was raised to 100). For the genetic algorithm, the default parameters were kept for mutation, crossover, and elitism. The docked energy also includes the ligand internal energy or the ligand intramolecular interaction energy. AutoDock also reports a binding free energy excluding the ligand internal energy but including a torsional free energy term, based on the rotatable ligand bonds.
AutoDock docking and hybrid QM/MM scoring
To find out the binding regions of the green tea polyphenols derivatives into the GST structure automated docking simulations were implemented with the AutoDock program version 4.0. As stated above, 100 independent docking runs were performed for each docking experiment. The best generated conformations of each ligand were ranked (clustered) into families of similar binding modes, with a root mean square deviation (RMSD) clustering tolerance of 2 Å. In the second step, we have docked the ligands in the binding sites previously found (“refined docking”). The poses obtained from the docking run were scored using the pseudo-bond ab-initio QM/MM approach as implemented in Gaussian03 [Citation47]. For the QM/MM calculations, the GST-ligands resulting from the docking were partitioned into a QM and MM subsystems. The reaction system used a smaller QM subsystem formed by the ligand and aminoacids side chains within 3.5 Å from the centre of the catalytic site, while the rest of the protein (the MM subsystem) was treated using the AMBER force field, together with a low memory convergence algorithm. The boundary problem between the QM and MM subsystems was treated using the pseudo-bond approach. An iterative optimization procedure was applied to the QM/MM system, using a B3LYP/3-21G* QM/MM calculations, leading to an reactants optimized structure. The convergence criterion was set in order to obtain an energy gradient less than 10− 4 using the twin − range cut-off method for non-bonded interactions, with a long- and short-range cut-off of 14 Å and 8 Å, respectively.
Results and discussion
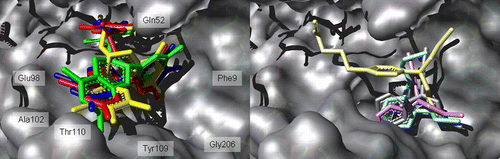
Docking experiments were performed using the crystal structure of hGST P1-1 complexed with the S-Hexylglutathione (PDB entry code: 1J9H), in order to get a better insight into the interaction of the natural EGCG analogs and of the antitumoral alkylating agents with hGST P1-1. An analysis of principles and methods adopted by AutoDock for energy calculations, conformational search and clustering, and for the QM/MM interaction energy ranking is briefly presented in Materials and Methods. The conformations resulting from the automated docking runs of the green tea polyphenols were clustered and most of them (up to 89% of the docking solutions) were found at the interface between the two GST P1-1 subunits, close to the crystallographic S-Hexylglutathione ligand. The most important residues (within 5 Å from the ligands) of this binding site are Asp95, Glu98, Asp99 and Ala102 (α-subunit), Tyr8, Phe9, Arg14, Trp39, Gln52, Pro54, Gln65, Ser66, Asn67, Glu98, Arg101, Lys103 and Tyr104, Tyr109, Thr110, Asn205 and Gly206 (β-subunit). The conformer population generated during the docking simulations into the GST P1-1 active site revealed a convergent binding mode of the EGCG analogs, in contrast to what found for the considered alkylating drugs, where the antitumoral chlorambucil is characterized by a divergent orientation with respect to the other (see ).
Figure 3 . Left: binding into hGST P1-1 of ( − )-EGCG red, ( − )-GCG yellow, ( − )-EGC blue and ( − )-CG green. Right: binding into hGST P1-1of cyclophosphamide gray, ifosfamide light blue, melphalan pink and chlorambucil gold. The protein is represented as a light grey Connolly surface, while the ligands as stick models.
The ( − )-EGCG ligand exhibits the most favourable energy score (see ), with a ligand-receptor interactions pattern indicative of a high conformational stability.
Table I. Heavy atoms (HA), molecular weight (MW) and QM/MM interaction energies (kcal/mol) for the eight studied compounds.
The rather hydrophobic A–C rings (see ) are oriented towards the β–subunit binding pocket, with the gallate group projected up into the α–β subunits interface, bridging the two walls of the binding cleft. The B ring points towards Tyr8(β), which is partially exposed to the solvent and located in a hydrophobic pocket surrounded by Phe8, Val10, Gly12, Arg14 and Pro53. ( − )-EGCG fills the majority of the binding cleft, as evidenced by drawing the water-accessible surface after the docking into the binding site, as depicted by a ribbon structure ().
Figure 4 . Left: 3D structure of hGST P1-1 (solid ribbon) in complex with ( − )-EGCG (top) and ( − )-GCG (bottom). Residues lining the ligand position are represented as a white Connolly surface, while the ligands are represented by a green Connolly surface. Right: binding mode of ( − )-EGCG (top) and ( − )-GCG (bottom) within hGST P1-1. Ligand (CPK) and the residues involved in interactions (orange, labelled) are represented in stick while the hydrogen bonds are shown as green dotted lines.
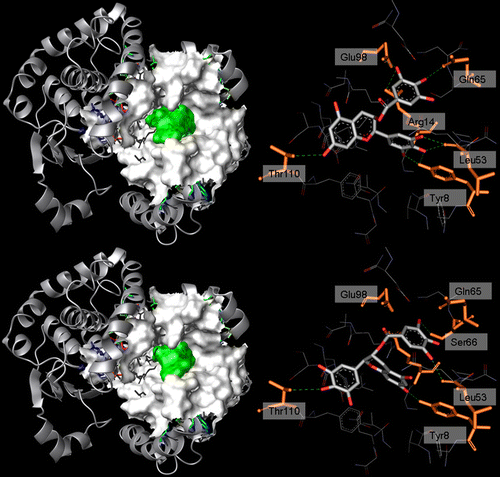
In addition, ( − )-EGCG is characterized by one carbonyl oxygen and eight polar hydrogen's available for H-bonding and among these three actively participate to the formation of an extensive hydrogen bonds network (). The gallate ring interacts with Arg14, Gln65 and Glu98 through O(6) and O(7), while the only interaction of the A–C ring is between Oγ1Thr110(β) and O(2). The B ring is located near Arg14, Leu53 and Tyr8 (H-site), and is involved in five H-bonds, O(3)…OLeu53, O(4)…OLeu53, O(4)…HOTyr8, O(5)…HOTyr8 and O(6)…NArg14. In particular the O(5)…HOTyr8 interaction has a key role for the hGST activity, as indicated by site-directed mutagenesis studies [Citation59,Citation60] showing that its catalytic role is due to the formation of an hydrogen bond between the HOTyr8 and GSH stabilizing the thiolate anion form of the substrate. In hGST the direct hydrogen bond between Tyr8 and GSH is replaced by an indirect one, mediated through a water molecule [Citation61]. The same binding behaviour is common to all the EGCG analogs except ( − )-GCG, the catechin presenting the worse interaction energy. In GCG the A–C ring flips to exchange its position with the B ring, leading to an interaction between the gallate ring and Leu53 and Ser66 through O(6) and O(8). The A–C ring O(2) oxygen is now in contact with HOTyr8(β) and the O(4) B ring oxygen hydrogen bonds Oγ1Thr110(β), replacing the A–C ring O(2) oxygen atom, while the gallate ring points towards Tyr8(β).
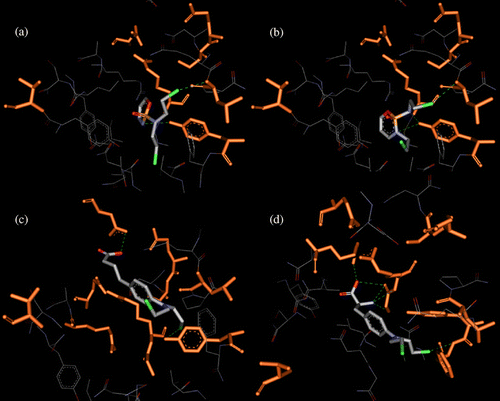
The binding mode of the alkylating anti-cancer agents shows the conservation of the Tyr8 H-bond, confirming the importance of this residue for GST enzymatic activity. Cyclophosphamide and ifosfamide are completely buried within the H-Site and present the worst energy scores compared with the other studied compounds (). Their phosphamidic nitrogen atoms bind to Tyr8, giving rise to one and two H-bonds, respectively. The H-bond network is completed by an additional contact for both compounds between chlorine and NLeu53 (see ). On the other hand, chlorambucil and melphalan are located close to the catechin positions, with the carboxylic group projected up into the α–β subunits interface and the chloro-ethane groups directed towards the H-Site. Consequently, chlorambucil makes two strong H-bonds, between Cl…HOTyr8 and between the carboxylic oxygen and Oδ2Asp99. Melphalan forms a network of H-bonds: two between the carboxylic oxygen and NH1Arg101-OGly13, two between the aliphatic nitrogen and NArg14-NGly13 and the last between the chlorine and NPhe9 and it is the only ligand that do not contact Tyr8.
Figure 5 . Binding within hGST P1-1 of the considered drugs: cyclophosphamide (a), ifosfamide (b), chlorambucil (c) and melphalan (d). Ligand (CPK) and interacting key residues (orange) are represented as stick models, hydrogen bonds are shown as green dotted lines.
Conclusions
In conclusion, this molecular docking study has been carried out to deepen characterize the possible interactions between EGCGs and alkylating agents on the GST catalytic site. In particular, the interaction mechanisms of the main green tea catechins and of four alkylating drugs with GST P1-1 has been investigated, which may be responsible for the greater systemic concentration of several conventional anti-cancer drugs. The docking experiments have shown that catechins and the examined drugs compete for the same catalytic binding site, close to the substrate GSH, using a comparable binding mode. This latter involve hydrophilic and steric interactions and, in particular, a direct and strong contact with Tyr8 (except melphalan), that provides an indirect evidence of the competition between alkylating drugs and catechins. This hypothesis is also supported by the values of the calculated QM/MM interaction energies, showing that all the green tea catechins bind the hGST P1-1 with higher affinity with respect to the alkylating agents. This could lead to a enzyme desensibilization, decreasing the anti-cancer drugs metabolism resulting in a greater systemic concentration of alkylating agents.
The results presented in this study are useful in view of the potential use of the green tea polyphenols both as anticancer as well as chemopreventive agents. In particular, we have determined the potentially bioactive conformations of EGCGs and anticancer drugs which may constitute the basis for generating GSTs binding models. On the other hand, we have confirmed the importance of the interaction with Tyr8 for the GST enzymatic activity. Finally, the observed differences in potency between the EGCGs and anticancer drugs have been explained through their different binding site occupancy.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- YA Elsayad, and EA Sausville. (2001). Selected novel anticancer treatments targeting cell signaling proteins. Oncologist 6:517–537.
- MM Gottesman, and I Pastan. (1993). Biochemistry of multidrug resistance mediated by the multidrug transporter. Ann Rev Biochem 62:385–427.
- RN Armstrong. (1997). Structure, catalytic machanism, and evolution of the glutathione transferase. Chem Res Toxicol 10:2–18.
- DL Eaton, and TK Bammler. (1999). Concise review of the glutathione S-transferase and their significance to toxicology. Toxicol Sci 49:156–164.
- DM Townsend, and KD Tew. (2003). The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 22:7369–7375.
- ML O'Brien, and KD Tew. (1996). Glutathione and related enzymes in multidrug resistance. Eur J Cancer 32A:967–978.
- MCJ Wilce, and MW Parker. (1994). Structure and function of glutathione S-transferases. Biochim Biophys Acta 1–18.
- C Anderson, E Mosialou, R Weinander, and R Morgenstern. (1994). Enzymology of microsomal glutathione S-transferase. Adv Pharmacol 27:19–35.
- PJ Jakobsson, JA Mancini, and AW Ford-Hutchinson. (1996). Identification and characterization of a novel human microsomal glutathione S-transferase with leukotriene C4 synthase activity and significant sequence identity to 5-lipoxygenase-activating protein and leukotriene C4 synthase. J Biol Chem 271:22203–22210.
- SE Pemble, AF Wardle, and JB Taylor. (1996). Glutathione S-transferase class Kappa: Characterization by the cloning of rat mitochondrial GST and identification of a human homologue. Biochem J 319:749–754.
- B Mannervik, PA lin, C Guthenberg, H Jensson, MK Tahir, M Warholm, and H Jornvall. (1985). Identification of three classes of cytosolic glutathione transferase common to several mammalian species: Correlation between structural data and enzymatic properties. Proc Natl Acad Sci USA 82:7202–7206.
- DJ Meyer, B Coles, SE Pemble, KS Gilmore, GM Fraser, and B Ketterer. (1991). Theta, a new class of glutathione transferases purified from rat and man. Biochem J 274:409–414.
- DJ Meyer, and M Thomas. (1995). Characterization of rat spleen prostaglandin H D-isomerase as a sigma-class GSH transferase. Biochem J 311:739–742.
- B Mannervik. (1985). The isoenzymes of glutathione transferase. Adv Enzymol 57:357–417.
- MA Watson, RK Stewart, GB Smith, and et al (1998). Human glutathione S-transferase P1 polymorphisms: Relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis 19:275–280.
- M Michael, and MM Doherty. (2005). Tumoral drug metabolism: Overview and its implications for cancer therapy. J Clin Oncol 23 (1):205–229.
- B Coles, and B Ketterer. (1990). The role of glutathione transferases in chemical carcinogenesis. CRC Crit Rev Biochem Mol Biol 25:47–70.
- JD Hayes, and DJ Pulford. (1995). The glutathione S-transferase supergene familyregulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. CRC Crit Rev Biochem Mol Biol 30:445–600.
- S Tsuchida, and K Sato. (1992). Glutathione transferases and cancer. CRC Crit Rev Biochem Mol Biol 27:337–384.
- DJ Waxman. (1990). Glutathione-S-transferases: Role in alkylating agent resistance and possible target for modulation chemotherapy-a review. Cancer Res 50:6449–6454.
- JA Montali, JB Wheatley, and DE Schmidt. (1995). Comparison of glutathione S-transferase levels in predicting the efficacy of a novel alkylating agent. Cellular Pharmacol 2:241–247.
- AS Morgan, PJ Ciaccio, KD Tew, and LM Kauvar. (1996). Isozyme-specific glutathione S-transferase inhibitors potentiate drug sensitivity in cultured human tumor cell lines. Cancer Chemother Pharmacol 37:363–370.
- Y Ohno, K Wakai, K Genka, K Ohmine, T Kawamura, A Tamakoshi, R Aoki, M Senda, Y Hayashi, K Nagao, and et al (1995). Tea consumption and lung cancer risk: A case-control study in Okinawa, Japan. Jpn J Cancer Res 86:1027–1034.
- L Zhong, MS Goldberg, YT Gao, JA Hanley, ME Parent, and F Jin. (2001). A population-based case-control study of lung cancer and green tea consumption among women living in Shanghai, China. Epidemiology 12:695–700.
- BT Ji, WH Chow, AW Hsing, JK McLaughlin, Q Dai, YT Gao, WJ Blot, and JF FraumeniJr. (1997). Green tea consumption and the risk of pancreatic and colorectal cancers. Int J Cancer 70:255–258.
- M Inoue, K Tajima, M Mizutani, H Iwata, T Iwase, S Miura, K Hirose, N Hamajima, and S Tominaga. (2001). Regular consumption of green tea and the risk of breast cancer recurrence: Follow-up study from the hospital-based epidemiologic research program at Aichi Cancer Center (HERPACC), Japan. Cancer Lett 167:175–182.
- JV Higdon, and B Frei. (2003). Tea catechins and polyphenols: Health effects, metabolism and antioxidant functions. Crit Rev Food Sci Nutr 43:89–143. b) Mustata GT, Rosca M, Biemel KM, Reihl O, Smith MA, Viswanathan A, Strauch C, Du Y, Tang J, Kern TS, Lederer MO, Brownlee M, Weiss MF, Monnier VM. Paradoxical effects of green tea (Camellia Sinensis) and antioxidant vitamins in diabetic rats. Diabetes 2005;54:517–526. c) Rice-Evans CA, Miller NJ, Paganga G. Structure antioxidant activity relationships of flavonoids and phenolics acids. Free Radic Biol Med 1996;20:933–956
- MJ Lee, P Maliakal, L Chen, X Meng, FY Bondoc, S Prabhu, G Lambert, S Mohr, and CS Yang. (2002). Pharmacokinetics of tea catechins after ingestion of green tea and ( − )-epigallocatechin-3-gallate by humans: Formation of different metabolites and individual variability. Cancer Epidemiol Biomark Prev 11:1025–1032.
- IA Siddiqui, F Afaq, VM Adhami, N Ahmad, and H Mukhtar. (2004). Antioxidants of the beverage tea in promotion of human health. Antioxid Redox Signal 6:571–582. b) Whiteside MA, Heimburger DC, Johanning GL. Micronutrients and cancer therapy. Nutr Rev 2004;62:142-147
- PC Chai, LH Long, and B Halliwell. (2003). Contribution of hydrogen peroxide to the cytotoxiCIT000y of green tea and red wines. Biochem Biophys Res Commun 304:650–654.
- N Khan, F Afaq, M Saleem, N Ahmad, and H Mukhtar. (2006). Targeting multiple signaling pathways by green tea polyphenol EGCG. Cancer Res 66:2500–2505.
- ZP Chen, JB Schell, CT Ho, and KY Chen. (1998). Green tea epigallocatechin gallate shows a pronounced growth inhibitory effect on cancerous cells but not on their normal counterparts. Cancer Lett 129:173–179. b) Fujiki H. Two stages of cancer prevention with green tea. J Cancer Res Clin Oncol 1999;125:589–597
- D Gupta, CG Lis, TC Birdsall, and JF Grutsch. (2005). The use of dietary supplements in a community hospital comprehensive cancer centre: Implications for conventional cancer care. Support Care Cancer 13:912–919.
- A Molassiotis, G Ozden, N Platin, and et al (2006). Complementary and alternative medicine use in patients with head and neck cancer in Europe. Eur J Cancer Care (Engl) 15:19–24.
- A Molassiotis, P Fernandez-Ortega, D Pud, and et al (2005). Complementary and alternative medicine use in colorectal cancer patients in seven European countries. Complement Ther Med 13:251–257.
- A Molassiotis, JA Scott, N Kearney, and et al (2006). Complementary and alternative medicine use in breast cancer patients in Europe. Support Care Cancer 14:260–267.
- KI Block, AC Koch, MN Mead, PK Tothy, RA Newman, and C Gyllenhaal. (2007). Impact of antioxidant supplementation on chemotherapeutic efficacy: A systematic review of the evidence from randomized controller trials. Cancer Treat Rev 33:407–418.
- CS Yang. (1999). Tea and health. Nutrition 15:946–949.
- N Ahmad, and H Mukhtar. (1999). Green tea polyphenols and cancer: Biologic mechanisms and practical implications. Nutr Rev 57:78–83.
- HH Sherry Chow, IA Hakim, DR Vining, JA Crowell, ME Tome, J Ranger-Moore, CA Cordova, DM Mikhael, MM Briehl, and DS Alberts. (2007). Modulation of human glutathione S-transferase by polyphenon E intervention. Cancer Epidem Biomark 16 (8):1662–1666.
- B Pool-Zobel, S Veeriah, and FD Bohmer. (2005). Modulation of xenobiotic metabolising enzymes by anticarcinogens – Focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colerectal carcinogenesis. Mutat Res 591:74–92.
- P Talalay. (2000). Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors 12:5–11.
- L Bravo. (1998). Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr Rev 56:317–333.
- a) NAMD was developed by the Theoretical Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign. b) JC Phillips, R Braun, W Wang, J Gumbart, E Tajkhorshid, E Villa, C Chipot, RD Skeel, L Kale, K Schulten. Scalable molecular dynamics with NAMD. J Comp Chem 2005; 26:1781–1802.
- GM Morris, DS Goodsell, RS Halliday, R Huey, WE Hart, RK Belew, and AJ Olson. (1998). Automated docking using a lamarkian genetic algorithm and an empirical binding free energy function. J Comp Chem 19:1639–1662.
- EF Pettersen, TD Goddard, CC Huang, GS Couch, DM Greenblatt, EC Meng, and TE Ferrin. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J Comp Chem 25:1605–1612.
- MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, MA Robb, JR Cheeseman, JA Montgomery, T Vreven, KN Kudin, JC Burant, et al. Gaussian 03. Revision A.1. Pittsburgh, PA: Gaussian; (2003).
- WL DeLano. The PyMOL molecular graphics system 2002. San Carlos, CA: DeLano Scientific; (2002). Retrieved from (http://www.pymol.org).
- Retrieved from http://www.accelrys.com.
- Persistence of Vision Pty. Ltd. Persistence of Vision Raytracer (Version 3.6). Retrieved from http://www.povray.org/download/ 2004.
- U Hegazy, K Tars, U Hellman, B Mannervik, PDB code 1J9H.
- MF Sanner. (1999). Python: A programming language for software integration and development. J Mol Graphics Mod 17:57–61.
- HJC Berendsen, JPM Postma, WF van Gunsteren, J Hermans, and B Pullman. Intermolecular forces. Dordrecht: Reidel; (1981).
- WL Jorgensen, J Chandrasekhar, J Madura, RW Impey, and ML Klein. (1983). Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–933.
- S Miyamoto, and PA Kollman. (1992). SETTLE – an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem 13:952–962.
- B Hess, H Bekker, HJC Berendsen, and JGEM Fraaije. (1997). LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472.
- , and Collaborative Computational Project, Number 4. (1994). The CCP4 suite: Programs for protein crystallography. Acta Cryst D50:760–763.
- O Dym, I Xenarios, H Ke, and J Colicelli. (2002). Molecular docking of competitive phosphodiesterase inhibitors. Mol Pharmacol 61:20–25. b) Rao MS, Olson AJ. Modelling of factor Xa-inhibitor complexes: A computational flexible docking approach. Proteins 1999;34:173–183
- RH Kolm, GE Sroga, and B Mannervik. (1992). Participation of the phenolic hydroxyl group of Tyr-8 in the catalytic mechanism of human glutathione transferase P1-1. Biochem J 285:537–540.
- KH Kong, M Nishida, H Inoue, and K Takahashi. (1992). Tyrosine-7 is an essential residue for the catalytic activity of human class PI glutathione S-transferase: Chemical modification and site-directed mutagenesis studies. Biochem Biophys Res Commun 182:1122–1129.
- J Oakley, M Lo Bello, A Battistoni, G Ricci, J Rossjohn, HO Villar, and MW Parker. (1997). The structures of human glutathione transferase P1-1 in complex with glutathione and various inhibitors at high resolution. J Mol Biol 274:84–100.