Abstract
A new series of flavonoid derivatives have been designed, synthesised and evaluated as acetylcholinesterase inhibitors that could bind simultaneously to the peripheral and catalytic sites of the enzyme. Among them, fifteen derivatives were found to inhibit the enzyme in the micromolar range and isoflavone derivatives possessed more potent inhibitory activity than other flavonoid derivatives. The best compound 9a had its inhibitory activity (IC50 = 0.093μM) in the same range as the reference compound, donepezil (IC50 = 0.025μM). Preliminary structure-activity relationships and a molecular modeling study for 9a have revealed that the isoflavone moiety plays a key role in the interaction of this series of derivatives with AChE by acting as an anchor in its peripheral anionic site.
Introduction
Alzheimer's disease (AD), the most common cause of dementia in the elderly, is a chronic neurodegenerative disorder which is characterized by loss of memory and progressive deficits in different cognitive domains[Citation1]. Although many factors are implicated in the disease, its pathogenesis remains unclear. The so-called “cholinergic hypothesis” postulated that the cognitive decline experienced by AD patients is due to an extensive loss of cholinergic neurons. To date, the enhancement of the central cholinergic function is the only clinically effective approach, mainly by means of reversible acetylcholinesterase (AChE) inhibitors[Citation2]. AChE inhibitors such as tacrine, galantamine, donepezil and rivastigmine are used for the symptomatic treatment of Alzheimer's disease, by preventing the degradation of the released neurotransmitter, thereby enhancing neurotransmission at cholinergic synapses[Citation3,Citation4].
On the other hand, β-amyloid (Aβ) is considered to be critical for inducing the pathology of AD, as its accumulation may result in a cascade of biochemical events leading to neuronal dysfunction[Citation5]. It is reported that AChE can interact with Aβ and promote amyloid fibril formation through the residues located in the peripheral anionic site (PAS) of the enzyme[Citation6]. Considering the non-cholinergic aspects and the role of the peripheral site of the cholinergic enzyme AChE, some dual-binding inhibitors binding simultaneously to both the catalytic and peripheral sites of AChE have been synthesized which will not only inhibit the hydrolysis of AChE toward ACh, but also prevent the aggregation of Aβ induced by AChE[Citation7,Citation8].
Flavonoids, a large group of natural compounds which possess a broad spectrum of biological activities have been proven to possess neuroprotective effects[Citation9]. Some of which exhibit inhibitory effects on Aβ fibril formation[Citation10], and some show reduction effect of H2O2-induced ROS formation[Citation11]. On the other hand, the crystallographic structure of donepezil-TcAChE complex reveals that N-benzyl piperidine moiety of donepezil interacts with the central site of AChE[Citation12]. Following this reasoning, various flavonoids (flavone, isoflavone, flavonone or chalcone) and N-benzyl piperidine were chosen as the two pharmacophoric moieties and linked with oxygen or alkoxyl group. Four series of flavonoid derivatives 4, 5, 9, 11 were synthesized and assayed for their AChE inhibitory activity.
To explore the possible binding conformation of synthesized compounds and protein-ligand interaction mode, the docking study was performed using the flexX program in SYBYL software.
Materials and methods
Materials
Melting points were obtained on a B-540 Buchi melting-point apparatus and uncorrected. 1H NMR and 13C NMR spectra were recorded on a Brucker Advance DMX 400 MHz spectrometer with TMS as the internal standard. Proton Chemical shifts are expressed in parts per million (ppm) and coupling constants in Hz. Infrared spectra were recorded on a JASCO FT/IR-4100 spectrophotometer. Analytical TLC was carried out on Merck 0.2mm precoated silica gel (60 F-254) aluminium sheets, with visualization by irradiation with a UV lamp. Column chromatography was accomplished on Qingdao silica gel 60 (230–400 mesh).
Chemistry
The synthetic routs to the four series target compounds are outlined in Scheme . The flavanone series 4a-d were obtained by reaction of the key intermediate 3a or 3b with either 1-benzylpiperidin-4-ol or (1-benzylpiperidin-4-yl)methanol according to the known method [Citation13]. Intermediate 3a and 3b were prepared as follows: Condensation of 2-hydroxy-4,5-dimethoxy-acetophenone with substituted-benzaldehyde resulted in chalcone 1a or 1b[Citation14], which was treated with NaOAc[Citation15] under reflux to give flavanone 2a or 2b. Then 2a or 2b was deprotected with dilute HCl in methanol to give 3a, b; The flavone series 5a-d were directly prepared by oxidation of 4a-d with iodine in pyridine [Citation16].
Scheme 1. Reagent and conditions: (i) KOH, ethanol/water, rt, 10h (ii) NaOAc, 95% ethanol reflux 20h (iii) 3N HCl, methanol, 65°C, 0.5h (iv) PPh3, DEAD, CH2Cl2, Et3N, rt, 48h (v) I2, pyridine, 90°C, 6h (vi) BzCl, DMF, 60°C, 3h (vii) a DIB-TsOH, methanol, rt, 4h b aq. NaOH, rt, 24h.
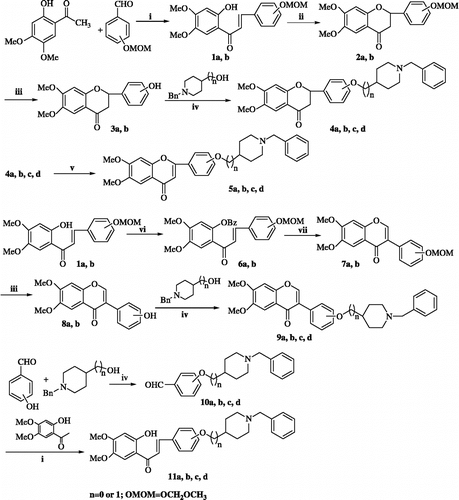
To prepare the isoflavone series 9a-d, the starting material 1a, b was reacted with BzCl to afford 2′-benzoyloxychalone 6a, b[Citation17], which was treated with DIB/TsOH and followed by NaOH/MeOH-H2O got isoflavone 7a, b[Citation18]. 7a, b was deprotected to afford 8a, b, and then coupled with benzylpiperidin alcohol.
The chalcone series 11a-d could be obtained by coupling of 3- or 4-hydroxybenzaldehyde with benzylpiperidin alcohol, and then condensing with 2-hydroxy-4,5-dimethoxy-acetophenone.
General procedure for the synthesis of 3a, b
2-Hydroxy-4,5-dimethoxy-acetophenone (0.98g, 0.005mol) and 3- or 4- (methoxymethoxy) benzaldehyde (1.25g, 0.0075mol) were added to a solution of 9N KOH (5mL) in EtOH (20mL) under ice cooling, and the mixture was stirred overnight at room temperature. 2N HCl (25mL) was added slowly to the mixture under ice cooling until a yellow solid precipitated, which was filtered and dried to obtain 1a(70%) or 1b(67%). The mixture of 1a or 1b (0.0025mol) and NaOAc (0.03mol) in EtOH (10mL)was refluxed for 20h. The solvent was evaporated in vacuum, and water was added to the residue. The mixture was extracted with EtOAc (3 × 50mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated in vacuum. The residue was purified by a silica-gel column chromatography (PE: EtOAc: CH2Cl2 = 4: 1: 1) to afford 2a (63%) or 2b (69%). To a solution of 2a, b (1mmol) in methanol (8mL), was added dropwise 3N HCl (8 mL) under ice cooling. The mixture was kept at 65°C for 0.5h. After cooling to room temperature, The products were obtained as yellow precipitates. The precipitate was filtered and recrystallized from aqueous ethanol to afford 3a or 3b.
2-(4-Hydroxyphenyl)-6,7-dimethoxychroman-4-one (3a). Yield 86%; m.p. 204-206°C; 1H NMR (400MHz, DMSO) 9.57 (s, 1H), 7.33 (d, 2H, J = 8.4Hz), 7.16 (s, 1H), 6.78 (d, 2H, J = 8Hz), 6.66 (s, 1H), 5.42 (dd, 1H, J = 13.2, 2.8Hz), 3.81 (s, 3H), 3.75 (s, 3H), 3.11 (dd, 1H, J = 16.8, 13.2Hz), 2.62 (dd, 1H, J = 16.8, 2.8Hz); IR (KBr), ν(cm− 1): 3182, 2949, 1619, 1507, 1473, 1356
2-(3-Hydroxyphenyl)-6,7-dimethoxychroman-4-one (3b). Yield 79%; m.p. 189-191°C; 1H NMR (400MHz, DMSO) 9.52 (s, 1H), 7.18 (t, 1H, J = 8.4Hz), 7.16 (s, 1H), 6.91-6.93 (m, 2H), 6.74 (d, 1H, J = 7.6Hz), 6.68 (s, 1H), 5.47 (dd, 1H, J = 12.8, 8Hz), 3.83 (s, 3H), 3.75 (s, 3H), 3.01 (dd, 1H, J = 16.8, 12.8Hz), 2.69 (dd, 1H, J = 16.8, 2.8Hz); IR (KBr), ν(cm− 1): 2999, 2784, 1633, 1610, 1507, 1469
General procedure for the synthesis of 8a, b
Benzoylchloride (9mmol) was added dropwise to a mixture of 1a or 1b (3mmol), K2CO3 (15mmol) in DMF (30mL) under ice cooling, and then the mixture was heated at 60°C for 3h. The reaction mixture was poured into ice-water, and the aqueous mixture was extracted with EtOAc (150mL × 2). The combined organic layer was washed successively with water and brine, evaporated in vacuum to give crude prisms 6a (77%) or 6b (68%). 6a or 6b (3.6mmol) dissolved in CH2Cl2 (19mL) and CH3OH (57mL), and the solution of DIB (7.2mmol) and TsOH (14.4mmol) in CH3OH (38mL) was added dropwise under ice cooling. The mixture was stirred at room temperature for 4h under nitrogen atmosphere, and then the solution of NaOH (22.5mmol) in CH3OH-H2O (5:1, 100mL) was added under ice cooling. The reaction mixture was stirred at room temperature for 24h. The solvent was evaporated in vacuum, and water was added to the residue. The aqueous mixture was extrated with EtOAc (50mL × 3) and evaporated in vacuum. The residue was purified by a silica-gel column chromatography (PE: EtOAc: CH2Cl2 = 1: 1: 1) to obtain 7a(52%) or 7b(46%). To a solution of 7a, b (1mmol) in methanol (8mL), was added dropwise 3N HCl (8 mL) under ice cooling. The mixture was kept at 65°C for 0.5h. After cooling to room temperature, The products were obtained as yellow precipitates. The precipitate was filtered and recrystallized from aqueous ethanol to afford 8a or 8b as white crystals.
3-(4-Hydroxyphenyl)-6,7-dimethoxy-4H-chromen-4-one (8a). Yield 85%; m.p. 193-195°C; 1H NMR (400MHz, DMSO) 9.52 (s, 1H), 8.35 (s, 1H), 7.37-7.41 (m, 3H), 7.19 (s, 1H), 6.78 (d, 2H, J = 8.8Hz), 3.85 (s, 3H), 3.90 (s, 3H). IR (KBr), ν(cm− 1): 3005, 2837, 1630, 1565, 1506, 1459, 1306.
3-(3-Hydroxyphenyl)-6,7-dimethoxy-4H-chromen-4-one (8b). Yield 82%; m.p. 213-215°C; 1H NMR (400MHz, DMSO): 9.47 (s, 1H), 8.43 (s, 1H), 7.44 (s, 1H), 7.19-7.23 (m, 2H), 7.02 (s, 1H), 6.95 (d, 1H, J = 7.6Hz), 6.76 (d, 1H, J = 8Hz), 3.92 (s, 3H), 3.87 (s, 3H); IR (KBr), ν(cm− 1): 3004, 2807, 1626, 1577, 1513, 1481.
General procedure for the synthesis of 4a-d, 9a-d
A mixture of triphenylphosphine (0.36mmol), 3 or 8 (0.3mmol) and benzylpiperidine alcohol (0.3mmol) in dry CH2Cl2 (2 mL) was cooled in an ice bath under nitrogen atmosphere, diethyl azodicarboxylate (DEAD, 0.36mmol) was slowly added, and Et3N (0.36mmol) was added subsequently. The mixture was stirred at room temperature for 48h. The solvent was removed under reduced pressure, and EtOAc (20mL) was added to the residue. The mixture was extracted with 2N HCl (20mL × 3). After the combined aqueous solution was washed with EtOAc (20mL × 2), it was neutralized using 28% NH4OH and extracted with CH2Cl2 (20mL × 3). The combined organic layer was washed successively with water and brine, dried over anhydrous Na2SO4, evaporated in vacuum. The residue was purified by a silica-gel column chromatography (PE: EtOAc: TEA = 100: 25: 1).
2-(4-(1-Benzylpiperidin-4-yloxy)phenyl)-6,7-dimethoxychroman-4-one (4a). Yield 23%; m.p. 166-168°C; Anal. Calc. for C29H31NO5: C, 73.55; H, 6.60; N, 2.96. Found: C, 73.58; H, 6.64; N, 2.92; 1H NMR (400MHz, CDCl3) δ 7.35 (d, 2H, J = 8.4Hz), 7.30-7.32 (m, 5H), 7.24 (m, 1H), 6.92 (d, 2H, J = 8.8Hz), 6.50 (s, 1H), 5.35 (dd, 1H, J = 13.6, 2.8Hz), 4.35 (m, 1H), 3.88 (s, 6H), 3.54 (s, 2H), 3.00 (dd, 1H, J = 16.8, 13.6Hz), 2.76 (dd, 1H, J = 16.8, 3.2Hz), 2.74 (s, 2H), 2.31 (s, 2H), 2.02 (s, 2H), 1.84 (s, 2H); 13C NMR (100MHz, CDCl3) δ 190.8, 158.0, 157.9, 156.2, 144.6, 138.3, 130.7, 129.1, 128.2, 127.7, 127.0, 116.1, 113.1, 106.6, 100.2, 80.0, 76.7, 62.9, 56.2, 56.1, 50.4, 43.8, 30.7; IR (KBr), ν(cm− 1): 3022, 2944, 1676, 1612, 1505, 1453, 1423.
2-(4-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-6,7-dimethoxychroman-4-one (4b). Yield 19%; m.p. 155-157°C; Anal. Calc. for C30H33NO5: C, 73.90; H, 6.82; N, 2.87. Found: C, 73.87; H, 6.81; N, 2.87; 1H NMR (400MHz, CDCl3) δ 7.38 (d, 2H, J = 8.4Hz), 7.32-7.34 (m, 5H), 7.28 (m, 1H), 6.93 (d, 2H, J = 8.8Hz), 6.51 (s, 1H), 5.38 (dd, 1H, J = 13.6, 2.4Hz), 3.90 (s, 6H), 3.82 (d, 2H, J = 6Hz), 3.54 (s, 2H), 3.02 (dd, 1H, J = 16.8, 13.6Hz), 2.95 (m, 2H), 2.76 (dd, 1H, J = 17.6, 8Hz), 2.00 (m, 2H), 1.82 (m, 2H), 1.43 (m, 2H), 1.25 (m, 1H); 13C NMR (100MHz, CDCl3) δ 190.6, 159.2, 157.8, 155.9, 144.3, 137.9, 130.4, 128.9, 127.9, 127.4, 126.7, 114.4, 112.8, 106.3, 100.0, 79.7, 72.4, 63.1, 56.0, 55.9, 53.0, 43.6, 35.5, 28.7; IR (KBr), ν(cm− 1): 3013, 2924, 1676, 1612, 1504, 1467, 1423.
2-(3-(1-Benzylpiperidin-4-yloxy)phenyl)-6,7-dimethoxychroman-4-one (4c). Yield 15%; m.p. 134-136°C; Anal. Calc. for C29H31NO5: C, 73.55; H, 6.60; N, 2.96. Found: C, 73.56; H, 6.59; N, 2.93; 1H NMR (400MHz, CDCl3) δ 7.20-7.29 (m, 7H) 6.96-6.98 (m, 2H), 6.85 (d, 1H), 6.48 (s, 1H), 5.34 (dd, 1H, J = 13.6, 3.2Hz), 4.30 (m, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.50 (s, 2H), 2.93 (dd, 1H, J = 17.2, 13.6Hz), 2.74 (dd, 1H, J = 17.2, 3.2Hz), 2.70 (m, 2H), 2.26 (m, 2H), 1.94 (m, 2H), 1.79 (m, 2H); IR (KBr), ν(cm− 1): 3026, 2935, 1678, 1606, 1501, 1445, 1443.
2-(3-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-6,7-dimethoxychroman-4-one (4d). Yield 21%; m.p. 143-145°C; Anal. Calc. for C30H33NO5: C, 73.90; H, 6.82; N, 2.87. Found: C, 73.89; H, 6.84; N, 2.85; 1H NMR (400MHz, CDCl3) δ 7.25-7.36 (m, 7H), 7.02-7.04 (m, 2H), 6.89 (d, 1H), 6.55 (s, 1H), 5.41 (dd, 1H, J = 13.6, 3.2Hz), 3.92 (s, 3H), 3.91 (s, 3H), 3.83 (d, 2H, J = 6Hz), 3.53 (s, 2H), 3.00 (dd, 1H, J = 17.2, 13.6Hz), 2.96 (m, 2H), 2.80 (dd, 1H, J = 16.8, 3.2Hz), 1.95 (m, 2H), 1.83 (m, 2H), 1.62 (m, 2H), 1.28 (m, 1H); IR (KBr), ν(cm− 1): 3005, 2928, 1612, 1605, 1510, 1465.
3-(4-(1-Benzylpiperidin-4-yloxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (9a). Yield 23%; m.p. 118-120°C; Anal. Calc. for C29H29NO5: C, 73.87; H, 6.20; N, 2.97. Found: C, 73.80; H, 6.22; N, 2.94. 1H NMR (400MHz, CDCl3) δ 7.94 (s, 1H), 7.63 (s, 1H), 7.48 (d, 2H, J = 8.8Hz), 7.31 (m, 5H), 6.96 (d, 2H, J = 8.8Hz), 6.88 (s, 1H), 4.36-4.40 (m, 1H), 3.99 (s, 6H), 3.56 (s, 2H), 2.77 (m, 2H), 2.32 (m, 2H), 2.01 (m, 2H), 1.84 (m, 2H); 13C NMR (100MHz, CDCl3) δ 175.9, 157.8, 154.6, 152.6, 152.2, 148.0, 130.4, 129.5, 129.4, 128.6, 127.4, 124.7, 124.7, 118.2, 116.3, 105.7, 99.8, 63.3, 60.7, 56.8, 56.7, 50.8, 31.1, 21.4, 14.5; IR (KBr), ν(cm− 1): 3278, 2808, 1735, 1631, 1606, 1509, 1471.
3-(4-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (9b). Yield 18%, m.p. 136-138°C; Anal. Calc. for C30H31NO5: C, 74.21; H, 6.43; N, 2.88. Found: C, 74.19; H, 6.43; N, 2.92; 1H NMR (400MHz, CDCl3) δ 7.93 (s, 1H), 7.63 (s, 1H), 7.47 (d, 2H, J = 8.8Hz), 7.23 (m, 5H), 6.93 (d, 2H, J = 8.8Hz), 6.87 (s, 1H), 3.98 (s, 3H), 3.98 (s, 3H), 3.83 (d, 2H), 3.50 (s, 2H), 3.38 (m, 2H), 2.86 (m, 2H), 1.92 (m, 2H), 1.63 (m, 2H), 1.23 (m, 1H); IR (KBr), ν(cm− 1): 3296, 2802, 1713, 1627, 1603, 1509, 1452.
3-(3-(1-Benzylpiperidin-4-yloxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (9c). Yield 25%, m.p. 127-129°C; Anal. Calc. for C29H29NO5: C, 73.87; H, 6.20; N, 2.97. Found: C, 73.85; H, 6.21; N, 2.94; 1H NMR (400MHz, CDCl3) δ 7.97 (s, 1H), 7.63 (s, 1H), 7.42 (t, 1H, J = 8Hz), 7.26 (m, 6H), 7.19 (d, 1H, J = 8Hz), 7.08 (d, 1H, J = 8Hz), 6.89 (s, 1H), 4.44 (m, 1H), 4.00 (s, 3H), 3.99 (s, 3H), 3.66 (s, 2H), 2.81 (m, 2H), 2.52 (m, 2H), 2.11 (m, 2H), 1.93 (m, 2H); IR (KBr), ν(cm− 1): 2926, 2847, 1729, 1635, 1605, 1505, 1470.
3-(3-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (9d). Yield 15%, m.p. 143-145°C; Anal. Calc. for C30H31NO5: C, 74.21; H, 6.43; N, 2.88. Found: C, 74.22; H, 6.45; N, 2.90; 1H NMR (400MHz, CDCl3) δ 7.97 (s, 1H), 7.64 (s, 1H), 7.33 (t, 1H, J = 8Hz), 7.26 (m, 6H), 7.20 (d, 1H, J = 8Hz), 7.05 (d, 1H, J = 8Hz), 6.88 (s, 1H), 4.02 (s, 3H), 3.98 (s, 3H), 3.88 (d, 2H, J = 6Hz), 3.62 (s, 2H), 3.06 (m, 2H), 2.11 (m, 2H), 1.88 (m, 2H), 1.56 (m, 2H), 1.27 (m, 1H); IR (KBr), ν(cm− 1): 3280, 2810, 1735, 1608, 1505, 1456.
General procedure for the synthesis of 5a-d
A mixture of compound 4 (0.1mmol) and I2 (0.1mmol) in pyridine (2mL) was heated at 90°C for 6h. After cooling to room temperature, the reaction mixture was poured into ice-water, and extracted with EtOAc (30mL × 2). The combined organic layer was washed successively with saturated Na2S2O3 solution and brine, dried over anhydrous Na2SO4, evaporated in vacuum. The residue was purified by a silica-gel column chromatography (PE: acetone: TEA = 99: 30: 1).
2-(4-(1-Benzylpiperidin-4-yloxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (5a). Yield 57%; m.p. 131-133°C; Anal. Calc. for C29H29NO5: C, 73.87; H, 6.20; N, 2.97. Found: C, 73.83; H, 6.24; N, 2.98; 1H NMR (400MHz, CDCl3) δ 7.75 (d, 2H, J = 8.8Hz), 7.48 (s, 1H), 7.20-7.27 (m, 5H), 6.91-6.94 (m, 3H), 6.63 (s, 1H), 4.37 (m, 1H), 3.94 (s, 3H), 3.91 (s, 3H), 3.52 (s, 2H), 2.71 (m, 2H), 2.33 (m, 2H), 1.97 (m, 2H), 1.82 (m, 2H); IR (KBr), ν(cm− 1): 3006, 2806, 1642, 1606, 1507, 1457.
2-(4-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (5b). Yield 51%; m.p. 155-157°C; Anal. Calc. for C30H31NO5: C, 74.21; H, 6.43; N, 2.88. Found: C, 74.18; H, 6.45; N, 2.89; 1H NMR (400MHz, CDCl3) δ 7.84 (d, 2H, J = 9.2Hz), 7.57 (s, 1H), 7.28-7.35 (m, 5H), 6,99-7.01 (m, 3H), 6.72 (s, 1H), 4.03 (s, 3H), 4.00 (s, 3H), 3.88 (d, 2H, J = 6Hz), 3.56 (s, 2H), 2.97 (m, 2H), 2.02 (m, 2H), 1.84 (m, 2H), 1.46 (m, 2H), 1.25 (m, 1H); IR (KBr), ν(cm− 1): 3004, 2803, 1632, 1604, 1510, 1470.
2-(3-(1-Benzylpiperidin-4-yloxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (5c). Yield 42%; m.p. 120-122°C; Anal. Calc. for C29H29NO5: C, 73.87; H, 6.20; N, 2.97. Found: C, 73.89; H, 6.20; N, 2.96; 1H NMR (400MHz, CDCl3) δ 7.56 (s, 1H), 7.47 (d, 1H, J = 8Hz), 7.31-7.43 (m, 7H), 7.04 (dd, 1H, J = 8, 1.6Hz), 7.01 (s, 1H), 6.77 (s, 1H), 4.49 (m, 1H), 4.03 (s, 3H), 3.99 (s, 3H), 3.70 (s, 2H), 2.86 (m, 2H), 2.55 (m, 2H), 2.16 (m, 2H), 1.95 (m, 2H); IR (KBr), ν(cm− 1): 3005, 2846, 1636, 1602, 1503, 1468.
2-(3-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-6,7-dimethoxy-4H-chromen-4-one (5d). Yield 38%; m.p. 118-120°C; Anal. Calc. for C30H31NO5: C, 74.21; H, 6.43; N, 2.88. Found: C, 74.23; H, 6.45; N, 2.88; 1H NMR (400MHz, CDCl3) δ 7.57 (s, 1H), 7.47 (d, 1H, J = 7.6Hz), 7.29-7.43 (m, 7H), 7.02-7.05 (m, 2H), 6.79 (s, 1H), 4.04 (s, 3H), 4.00 (s, 3H), 3.88 (d, 2H, J = 6Hz), 3.61 (s, 2H), 3.05 (m, 2H), 2.11 (m, 2H), 1.88 (m, 2H), 1.56 (m, 2H), 1.27 (m, 1H); IR (KBr), ν(cm− 1):3009, 2856, 1630, 1604, 1508, 1472.
General procedure for the synthesis of 10a-d
Triphenylphosphine (1.2mmol) was dissolved in a mixture of 4- or 3-hydroxybenzaldehyde (1.2mmol) and benzylpiperidine alcohol (1.0mmol) in dry CH2Cl2 (3mL) under nitrogen atmosphere. After the mixture was cooled in an ice bath, diethyl azodicarboxylate (DEAD, 1.2mmol) was slowly added, and then Et3N (1.2mmol) was added. The solution was stirred at room temperature for 48h. The solvent was removed under reduced pressure, and EtOAc (30mL × 3)was added to the residue. The mixture was extracted with 2N HCl (30mL × 3). After the combined aqueous solution was washed with EtOAc (20mL × 2), it was neutralized using 28%NH4OH and extracted with CH2Cl2 (20mL × 3). The combined organic layer was washed successively with water and brine, dried over anhydrous Na2SO4, evaporated in vacuum. The residue was purified by a Al2O3 column chromatography (PE: CH2Cl2 = 2: 1).
4-(1-Benzylpiperidin-4-yloxy)benzaldehyde (10a). Yield 49%; 1H NMR (400MHz, CDCl3) δ 9.874 (s, 1H), 7.82 (d, 2H, J = 8.8Hz), 7.26-7.35 (m, 5H), 6.99 (d, 2H, J = 8.8Hz), 4.47 (m, 1H), 3.56 (s, 2H), 2.76 (m, 2H), 2.35 (m, 2H), 2.03 (m, 2H), 1.88 (m, 2H); IR (CDCl3), ν(cm− 1): 3028, 2806, 1691, 1600, 1575, 1507, 1453.
4-((1-Benzylpiperidin-4-yl)methoxy)benzaldehyde (10b). Yield 32%; 1H NMR (400MHz, CDCl3) δ 9.88 (s, 1H), 7.82 (d, 2H, J = 8.8Hz), 7.26-7.35 (m, 5H), 7.00 (d, 2H, J = 8.4Hz), 3.85 (d, 2H, J = 6.4Hz), 3.53 (s, 2H), 2.95 (m, 2H), 2.05 (m, 2H), 1.81 (m, 2H), 1.46 (m, 2H), 1.25 (m, 1H); IR (CDCl3), ν(cm− 1): 3026, 2809, 1698, 1640, 1596, 1505, 1455.
3-(1-Benzylpiperidin-4-yloxy)benzaldehyde (10c). Yield 41%; 1H NMR (400MHz, CDCl3) δ 9.96 (s, 1H), 7.43 (d, 2H, J = 4.8Hz), 7.38 (s, 1H), 7.26-7.34 (m, 5H), 7.17-7.17 (m, 1H), 4.42 (m, 1H), 3.56 (s, 2H), 2.75 (m, 2H), 2.35 (m, 2H), 2.02 (m, 2H), 1.86 (m, 2H). IR (CDCl3), ν(cm− 1): 3028, 2930, 2810, 1698, 1640, 1595, 1452.
3-((1-Benzylpiperidin-4-yl)methoxy)benzaldehyde (10d). Yield 27%; 1H NMR (400MHz, CDCl3) δ 9.96 (s, 1H), 7.42 (d, 2H, J = 4.8Hz), 7.39 (s, 1H), 7.26-7.37 (m, 5H), 7.18 (m, 1H), 4.41 (m, 1H), 3.59 (s, 2H), 3.54 (s, 2H), 2.73 (m, 2H), 2.33 (m, 2H), 2.01 (m, 2H), 1.86 (m, 2H); IR (CDCl3), ν(cm− 1): 3028, 2809, 1697, 1594, 1483, 1452.
General procedure for the synthesis of 11a-d
2-Hydroxy-4,5-dimethoxy-acetophenone (0.5mmol) and compound 10 (0.5mmol) was added to a solution of 9N KOH (0.5mL) in EtOH (2.2mL) under ice cooling, and the mixture was heated at 50°C for 5h. After cooling to room temperature, 2N HCl (2.5mL) was added slowly to the mixture under ice cooling The aqueous mixture was extracted with CH2Cl2 (5mL × 3), and the combined organic layer was washed successively with water and brine, dried over anhydrous Na2SO4, and evaporated in vacuum. The residue was purified by a silica-gel column chromatography (PE: EtOAc: TEA = 100: 50: 1.5).
(E)-3-(4-(1-Benzylpiperidin-4-yloxy)phenyl)-1-(2-hydroxy-4,5-dimethoxyphenyl)prop-2-en-1-one(11a). Yield 21%; m.p. 170-172°C; Anal. Calc. for C29H31NO5: C, 73.55; H, 6.60; N, 2.96. Found: C, 73.59; H, 6.58; N, 2.97; 1H NMR (400MHz, CDCl3) δ 13.51 (s, 1H), 7.85 (d, 1H, J = 15.6Hz), 7.59 (d, 2H, J = 8.4Hz), 7.37 (d, 1H, J = 15.6Hz), 7.26-7.34 (m, 6H), 6.93 (d, 2H, J = 8.4Hz), 6.51 (s, 1H), 4.39-4.43 (m, 1H), 3.92 (s, 3H), 3.93 (s, 3H), 3.55 (s, 2H), 2.76 (m, 2H), 2.34 (m, 2H), 2.02 (m, 2H), 1.86 (m, 2H); 13C NMR (100MHz, CDCl3) δ 191.8, 162.0, 160.2, 157.2, 144.7, 142.2, 138.6, 130.8, 129.4, 128.6, 127.7, 127.4, 118.0, 116.4, 112.4, 111.3, 101.1; IR (KBr), ν(cm− 1): 3022, 2943, 1633, 1602, 1564, 1509, 1443
(E)-3-(4-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-1-(2-hydroxy-4,5-dimethoxyphenyl)prop-2-en-1-one (11b). Yield 13%; m.p. 157-159°C; Anal. Calc. for C30H33NO5: C, 73.90; H, 6.82; N, 2.87. Found: C, 73.86; H, 6.83; N, 2.82; 1H NMR (400MHz, CDCl3) δ 13.41 (s, 1H), 7.85 (d, 1H, J = 15.6Hz), 7.59 (d, 2H, J = 8.4Hz), 7.36 (d, 1H, J = 15.6Hz), 7.26-7.33 (m, 6H), 6.91 (d, 2H, J = 8.4Hz), 6.51 (s, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 3.85 (d, 2H, J = 6.4Hz), 3.53 (s, 2H), 2.95 (m, 2H), 2.04 (m, 2H), 1.81 (m, 2H), 1.46 (m, 2H), 1.25 (m, 1H); IR (KBr), ν(cm− 1): 3025, 2929, 1638, 1610, 1559, 1506, 1449.
(E)-3-(3-(1-Benzylpiperidin-4-yloxy)phenyl)-1-(2-hydroxy-4,5-dimethoxyphenyl)prop-2-en-1-one (11c). Yield 23%; m.p. 139-141°C; Anal. Calc. for C29H31NO5: C, 73.55; H, 6.60; N, 2.96. Found: C, 73.56; H, 6.57; N, 2.93; 1H NMR (400MHz, CDCl3) δ 7.82 (d, 1H, J = 15.6Hz), 7.45 (d, 1H, J = 15.6Hz), 7.31-7.35 (m, 5H), 7.23-7.28 (m, 3H), 7.15 (s, 1H), 6.95 (dd, 1H, J = 8, 2.4Hz), 6.51 (s, 1H), 4.37 (m, 1H), 4.02 (s, 3H), 3.98 (s, 3H), 3.69 (s, 2H), 2.85 (m, 2H), 2.54 (m, 2H), 2.15 (m, 2H), 1.95 (m, 2H). IR (KBr), ν(cm− 1): 3024, 2926, 1638, 1605, 1570, 1509, 1452.
(E)-3-(3-((1-Benzylpiperidin-4-yl)methoxy)phenyl)-1-(2-hydroxy-4,5-dimethoxyphenyl)prop-2-en-1-one (11d). Yield 18%; m.p. 163-165°C; Anal. Calc. for C30H33NO5: C, 73.90; H, 6.82; N, 2.87. Found: C, 73.91; H, 6.84; N, 2.83; 1H NMR (400MHz, CDCl3) δ 7.83 (d, 1H, J = 15.6Hz), 7.46 (d, 1H, J = 15.6Hz), 7.32-7.35 (m, 5H), 7.22-7.27 (m, 3H), 7.15 (s, 1H), 6.94 (dd, 1H, J = 8, 2.4Hz), 6.51 (s, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 3.85 (d, 2H, J = 6Hz), 3.53 (s, 2H), 2.97 (m, 2H), 2.04 (m, 2H), 1.85 (m, 2H), 1.48 (m, 2H), 1.27 (m, 1H); IR (KBr), ν(cm− 1): 3022, 2758, 1637, 1577, 1511, 1445.
Enzyme inhibition assays
Butylcholinesterase (BuChE) is an enzyme with high structural homology to AChE. Since AChE is mainly located in the central nervous system and BuChE is more abundant in the peripheral system, the selective AChE inhibitors activate mostly the central cholinergic transmission and lack the side effects related to nonselective cholinesterase inhibitors[Citation19]. So, the BuChE inhibitory activity was also tested. AChE and BuChE inhibition activities were measured by the spectrophotometric Ellman's method[Citation20,Citation21] using rat cortex homogenate and rat serum as the resource of AChE and BuChE, respectively. The brain homogenate was preincubated for 5 min with tetraisopropyl pyrophosphoramido (isoOMPA) (0.04 mmol/L), a selective inhibitor of BuChE. For assay of AChE or BuChE activity, a reaction mixture of 200μL containing acetylthiocholine iodide (0.3 mmol/L) 60μL or butyrylthiocholine iodide (0.4 mmol/L) 60μL, sodium phosphate buffer (0.1 mmol/L, pH 7.4) 100μL, homogenate or serum 20μL, different concentration of the tested compounds 20μL was incubated at 37°C for 15 min. The reaction was terminated by adding 50μL 3% sodium lauryl sulphate, then 50μL 0.2% 5,5′-dithio-bis (2-nitrobenzoic acid) to produce the yellow anion of 5-thio-2-nitro-benzoic acid. The rate of color production was measured spectrophotometrically at 450 nm.
Molecular modeling
Molecular modeling studies were performed using a SYBYL software implemented in a Silicon Graphics workstation[Citation22]. The X-ray crystal structure of Torpedo californica AChE complexed with Donepezil (PDB file identificator 1EVE) was retrieved from the Protein Data Bank (PDB). All bound waters and donepezil were removed from the complexes, and hydrogen atoms were added subsequently. The structure of the ligand was prepared in MOL2 format using the sketcher module and Gasteiger-Huckel charges were assigned to the ligand atoms. The minimization was run until converged to a maximum derivative of 0.001 kcal mol− 1 Å− 1. The docking and subsequent scoring were performed using the default parameters of the FlexX program in the SYBYL.
Results and discussion
The newly synthesized compounds were assayed for their AChE inhibitory activity. To allow comparison of the results, donepezil was used as the reference compound. The results are presented in .
Table I. AChE and BuChE inhibition data (IC50, μM)* of prepared compounds.
From these data, some preliminary structure-activity relationships could be derived as follows:
The nature of flavonoid moiety affected in a great extent the AChE inhibitory activity. Isoflavone series generally demonstrated more potent inhibitory activity against AChE than flavone, flavanone and chalcone series. For example, 9a and 9b showed sub-micromolar inhibition of AChE (IC50 = 0.093 and 0.72μM, respectively), followed by the flavone series (IC50 values ranging from 4.05 to 13.7μM). Almost all of the flavanone series and chalcones series displayed weak inhibitory activity.
From the IC50 values of the tested compounds, it appeared that variation of the linker chain between the flavonoids and benzylpiperidine moieties influenced the inhibitory potency. Generally, the compounds with an oxygen atom as linker are more potent than the compounds with OCH2 as linker, which maybe implicated that the molecules with oxygen atom as linker are of more appropriate length and will bind well to the peripheral and catalytic sites of AChE.
With regard to tested intermediates 10a-d, it is worth mentioning that one of these compounds 10b showed far less AChE inhibitory activity compared to flavonoid derivatives, and others are inactive. This result suggests that flavonoid moiety plays a key role in the interaction of flavonoid derivatives with AChE by acting as anchor in its pipepheral anionic site.
Most of the compounds demonstrated high selectivity for AChE over BChE. Especially, the most potent AChE inhibitors 9a is 235-fold more active inhibiting AChE than BChE, being slightly more selective than donepezil.
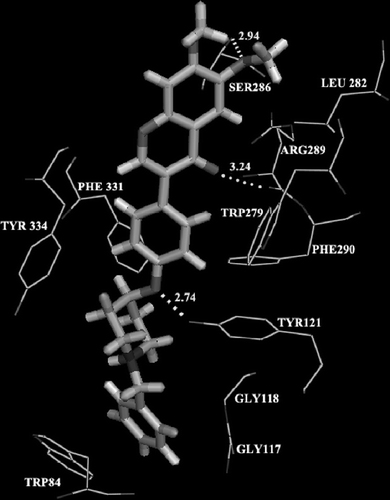
Additionally, according to the biological results, 9a showed the best inhibitory activity and it was chosen for molecular modeling (). The docking result demonstrated that it extended well along the gorge of the enzyme. The isoflavone moiety interacted in the peripheral pocket with the residue Leu282, Phe290, Tyr334 and Trp279 through hydrophobic interaction. More impotantly, The isoflavone moiety form two hydrogen-bonds contact with Ser286 (average O···N distance of 2.94 Å) and with Arg 289 (average O···O distance of 3.24 Å). The ethereal oxygen atom is hydrogen-bonded with the OH group of Tyr121 with the close distance of 2.74 Å. At the active site, the N-benzyl piperidine moiety interacted with Trp84, Phe331, Gly117 and Gly118 through hydrophobic interaction. Binding free energy was calculated (ΔG = − 22.28 kcal/mol). The simultaneous interaction of this compound in the central pocket, gorge, and peripheral pocket of TcAChE suggests the reason for the high inhibitory potency towards AChE.
Figure 1. Interaction between AChE and 9a.
In conclusion, a series of flavonoid derivatives were synthesized and assayed for their acetylcholinesterase and butyrylcholinesterase inhibition activity. Among them, fifteen derivatives were found to inhibit the enzyme in the micromolar range and the optimum inhibitor 9a showed potent inhibitory activity with an IC50 value of 0.09 μM. Preliminary structure-activity relationships and docking results envisaged that it is a dual binding site inhibitor. Isoflavone moiety acts as an efficient ligand for the peripheral site and contributes greatly to the potential activity for this subset of compounds.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (30572239). We also thank Experimental Center of Pharmaceutial Sciences Zhejiang University.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- SO Bachurin. (2003). Medicinal chemistry approaches for the treatment and prevention of Alzheimer's disease. Med Res Rev 23 (1):48–88.
- P Tariot, and H Federoff. (2003). Current treatment for Alzheimer disease and future prospects. Alzheimer Dis Assoc Disord 17:105–113.
- E Giacobini. (2004). Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol Res 50 (4):433–440.
- J Evans, G Wilcock, and J Birks. (2004). Evidence-based pharmacotherapy of Alzheimer's disease. Int J Neuropsychopharmacol 7 (3):351–369.
- J Hardy, and DJ Selkoe. (2002). The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 297 (5580):353–356.
- GV De Ferrari, MA Canales, I Shin, LM Weiner, I Silman, and NC Inestrosa. (2001). A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 40 (35):10447–10457.
- D Alonso, I Dorronsoro, L Rubio, P Munoz, E Garcia-Palomero, M Del Monte, A Bidon-Chanal, M Orozco, FJ Luque, A Castro, M Medina, and A Martinez. (2005). Donepezil-tacrine hybrid related derivatives as new dual binding site inhibitors of AChE. Bioorg Med Chem 13 (24):6588–6597.
- L Piazzi, F Belluti, A Bisi, S Gobbi, S Rizzo, M Bartolini, V Andrisano, M Recanatini, and A Rampa. (2007). Cholinesterase inhibitors: SAR and enzyme inhibitory activity of 3-[omega- (benzylmethylamino)alkoxy] xanthen-9-ones. Bioorg Med Chem 15 (1):575–585.
- CN Wang, CW Chi, YL Lin, CF Chen, and YJ Shiao. (2001). The neuroprotective effects of phytoestrogens on amyloid beta protein-induced toxicity are mediated by abrogating the activation of caspase cascade in rat cortical neurons. J Biol Chem 276:5287–5295.
- H Kim, BS Park, KG Lee, CY Choi, SS Jang, YH Kim, and SE Lee. (2005). Effects of naturally occurring compounds on fibril formation and oxidative stress of β-amyloid. J Agric Food Chem 53 (22):8537–8541.
- J Zhu, R Choi, G Chu, A Cheung, Q Gao, J Li, ZY Jiang, T Dong, and K Tsim. (2007). Flavonoids possess neuroprotective effects on cultured pheochromocytoma PC12 cells: A comparison of different flavonoids in activating estrogenic effect and in preventing β-amyloid-induced cell death. J Agric Food Chem 55 (6):2438–2445.
- G Kryger, I Silman, and JL Sussman. (1998). Three-dimensional structure of a complex of E2020 with acetylcholinesterase from Torpedo Californica. J Physiol (Paris) 92:191–194.
- O Mitsunobu. (1981). The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1:1–28.
- Shaw J, Lee AR, Huang WH. Methods of synthesizing flavonoids and chalcones, US 2004242907.
- L Xiao, WF Tan, and YL Li. (1998). First total synthesis of ( ± )-kenusanone B. Synth Commun 28 (15):2861–2869.
- S Mavel, B Dikic, and S Palakas. (2006). Synthesis and biological evaluation of a series of flavone derivatives as potential radioligands for imaging the multidrug resistance-associated protein 1 (ABCC1/MRP1). Bioorg Med Chem 14 (5):1599–1607.
- T Masao, W Hironari, and K Yasuhiko. (2004). Regioselective synthesis of 6-alkyl- and 6-prenylpolyhydroxyisoflavones and 6-alkylcoumaronochromone derivatives. Chem Pharm Bull 52 (11):1285–1289.
- K Yasuhiko, M Masashi, and T Takanori. (2002). Synthesis of isoflavones from 2′-hydroxy- chalcones using poly[4-(diacetoxy)iodo]styrene or related hypervalent iodine reagent. Synthesis 17:2490–2496.
- MS Cousins, DL Carriero, and JD Salamone. (1997). Tremulous jaw movements induced by the acetylcholinesterase inhibitor tacrine: Effects of antiparkinsonian drugs. Eur J Pharmacol 322:137–145.
- GL Ellman, KD Courtney, V AndresJr., and RM Featherstone. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95.
- DH Cheng, and XC Tang. (1998). Comparative studies of huperzine A, E2020, and tacrine on behavior and cholinesterase activities. Pharmacol Biochem Behav 60 (2):377–386.
- SYBYL 6.9 molecular modeling program., Tripos Associates.