Abstract
Comparative molecular field analysis (CoMFA) was performed on twenty-three pyrimethamine (pyr) derivatives active against quadruple mutant type (Asn51Ile, Cys59Arg, Ser108Asn, Ile164Leu) dihydrofolate reductase of Plasmodium falcipaarum (PfDHFR). The represented CoMFA models were evaluated based on the various three different probe atoms, Csp3 (+1), Osp3 ( − 1) and H (+1), resulting in the best model with combined three types of probe atoms. The statistical results were ![]() = 0.702, Spress = 0.608,
= 0.702, Spress = 0.608, ![]() = 0.980, s = 0.156, and
= 0.980, s = 0.156, and ![]() = 0.698 which can explain steric contribution of about 50%. In addition, an understanding of particular interaction energy between inhibitor and surrounding residues in the binding pocket was performed by using MP2/6-31G(d,p) quantum chemical calculations. The obtained results clearly demonstrate that Asn108 is the cause of pyr resistance with the highest repulsive interaction energy. Therefore, CoMFA and particular interaction energy analyses can be useful for identifying the structural features of potent pyr derivatives active against quadruple mutant type PfDHFR.
= 0.698 which can explain steric contribution of about 50%. In addition, an understanding of particular interaction energy between inhibitor and surrounding residues in the binding pocket was performed by using MP2/6-31G(d,p) quantum chemical calculations. The obtained results clearly demonstrate that Asn108 is the cause of pyr resistance with the highest repulsive interaction energy. Therefore, CoMFA and particular interaction energy analyses can be useful for identifying the structural features of potent pyr derivatives active against quadruple mutant type PfDHFR.
Introduction
Malaria is one of the most common infectious diseases and an enormous public-health problem which kills approximately 400 million people every year, especially in tropical and subtropical regions [Citation1]. The disease is caused by protozoan parasites of the genus Plasmodium. The most serious form of the disease is caused by Plasmodium falciparum (Pf). Dihydrofolate reductase (DHFR) of Pf is an important target for malaria chemotherapy. In malaria, DHFR is a bifunctional enzyme with Thymidilate synthase (TS), which is known to be essential for DNA synthesis Citation2, Citation3, Citation4. Inhibition of DNA synthesis through inhibition of PfDHFR can be done by preventing dihydrofolate to forming tetrahydrofolate by using an antifolate such as pyrimethamine (pyr) and cycloguanil drugs. Pyr is an effective, sentitive and selective antifolate drug against the wild type PfDHFR but its antimalarial action is slow [Citation5]. Generally, pyr is used in combinations with other antifolates, such as Maloprim (pyr-dapsone) [Citation6] and Fansidar (pyr-sulfadoxine) [Citation7,Citation8].
Unfortunately, resistance to antifolates has been found after using them as antimalarial drugs. It has been reported that antifolate drug resistance is due to point mutations in PfDHFR. The most important one is Ser108 to Asn108 mutation in the active site of PfDHFR. Additional point mutations in Asn51Ile, Cys59Arg and Ile164Leu were associated with higher levels of resistance to the conventional antifolate drugs Citation9, Citation10, Citation11, Citation12, Citation13, Citation14, Citation15.
In developing more potent pyr antimalarial derivatives against pyr-resistant parasites, Kamchonwongpaisan et al. [Citation16] reported a series of pyr derivatives including the structural modification, synthesis, and biological assay of the PfDHFR enzyme. Based on the structure of pyr modifications at position X and Y and R substitutions on 5-phenyl ring of the 2,4 diaminopyrimidine ring contribute to the improvement in its antimalarial activity, as shown in . Both structures and biological activities, in terms of inhibition constant (Ki), of these pyr derivatives are shown in . Following the discovery of these effective pyr derivatives, a 3D-QSAR (Three Dimentional-Quantitative Structure Activity Relationship) study of these compounds was carried out in order to investigate the local physicochemical properties involving in drug and receptor interactions. In this report, comparative molecular field analysis (CoMFA), one of the most useful tools for 3D-QSAR study Citation17, Citation18, Citation19, Citation20, Citation21, by relating the biological activity of a series of molecules with their steric and electrostatic fields was employed. CoMFA was performed on pyr derivatives to determine the structural requirements for improving binding to quadruple mutant type of PfDHFRs (Asn51Ile, Cys59Arg, Ser108Asn, Ile164Leu). In addition, the particular interaction energy between an inhibitor and surrounding amino acids in the binding pocket of quadruple mutant PfDHFR was also determined by quantum chemical calculations. Combination of the tools has led to investigation of inhibitor-enzyme interactions at the molecular level. Due to limitations of molecular systems by its complexity size [Citation22,Citation23], the analysis was therefore performed by limiting the interaction boundary to only 4 Å surrounding the ligand molecule in the active site of quadruple mutant PfDHFR. Comparisons of the particular interaction energy were made between pyr and the potent derivative, compound 6. Both the CoMFA method and quantum chemical calculations provide crucial information not only on the spatial orientation of the ligands and the main amino acid interactions but also on design of new potent inhibitors effective against the mutant PfDHFR enzyme.
Figure 1. Template structure of 5-phenyl-2,4-diamonopyrimidine derivatives.
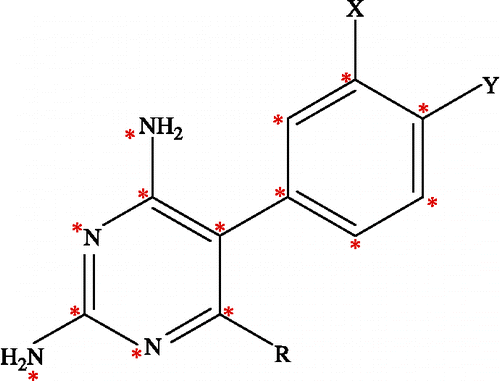
Table I. Data set used for CoMFA analysis with Ki (nM) and pKi values in the quadruple mutant (Asn51Ile, Cys59Arg, Ser108Asn, Ile164Leu) of PfDHFR.
Methods
Biological data
Twenty-three pyr derivatives used for the CoMFA study were selected from Kamchonwongpaisan et al. [Citation16] as shown in . In this table, eighteen compounds served as the training set. In addition, five compounds that were selected from the diversity and ranges of biological activities were kept to evaluate the predictive power of the models as the test set. As the structure of the X and Y substituents of compound 2 are different from others due to the fused X-Y substitution, therefore, this compound was selected into the test set. For each set of biological data, the Ki (nM) was measured in vitro under the same experimental conditions. Consequently, in vitro antimalarial activities were converted into the corresponding pKi ( − log Ki) values. These values were used as dependent variables in the CoMFA study.
Methods of CoMFA study
The CoMFA method is a ligand-based QSAR technique. In this study, the CoMFA method was employed by Sybyl (version 7.0, Tripos Inc.) installed on a Silicon Graphics Octane2 workstation at the National Electronics and Computer Technology Center of Thailand [Citation24]. The structures of pyr derivatives were built using the SKETCH module in Sybyl. The skeleton and conformation of pyr (compound 1) was extracted from the crystal structure of a pyr complex with double mutant PfDHFR with PDB code 1J3J. The other molecules were built taking compound 1 as a template and changing their substituents. Then energy minimizations of all the generated structures were performed using the tripos force field and the Gasteiger Hückel charges were calculated [Citation25].
CoMFA is a 3D-QSAR method that provides steric and electrostatic values from Lennard-Jones and Coulombic potentials equations, respectively. The 3D cubic lattices, with 2 Å grid spacing, were generated around the aligned compounds based on the molecular volume of the structure. The lattices were defined automatically, and were extended by 4.0 Å in all directions. All the generated structures were aligned in a 3D lattice based on atom superposition by aligning 5-phenyl-2,4-diamonopyrimidine as shown with the asterisk in , In this investigation, three different atoms, sp3 carbon atom with +1 charge, sp3 oxygen atom with − 1 charge and H atom with +1 charge, served as probe atoms. The probe atoms were placed at each lattice point and their interactions with the steric and electrostatic fields with each atom in the molecule were calculated with CoMFA standard scaling. The default value of 30 kcal/mol was used as the maximum electrostatic and steric energy cutoff. The CoMFA fields were scaled by CoMFA-STD method in Sybyl. Then, a partial least squares technique (PLS) was employed to derive a CoMFA model expressing the correlation between the steric and electrostatic properties and the biological activities [Citation26,Citation27]. The orthogonal latent variables were extracted by the NIPALS algorithm and subjected to full cross-validation with the Leave-One-Out method (LOO) [Citation28,Citation29]. In order to speed up the analysis and reduce the amount of noise, the minimum sigma value was set to 2.0 kcal/mol. The analyses were carried out with a maximum of six components, and using the number of components (noc) at which the difference in the ![]() value between components was less than 0.05.
value between components was less than 0.05.
Predictive ability
The predictive ability of the model derived from the training set is expressed by the predictive ![]() value. The
value. The ![]() value is defined as
value is defined as
where, SSY stands for the variance of the biological activities around the mean value, and PRESS is the prediction error sum of squares derived from LOO.
The uncertainty of the prediction is defined as
where k is the number of variables in the model and n is the number of compounds used in the study [Citation30].
Particular interaction energy
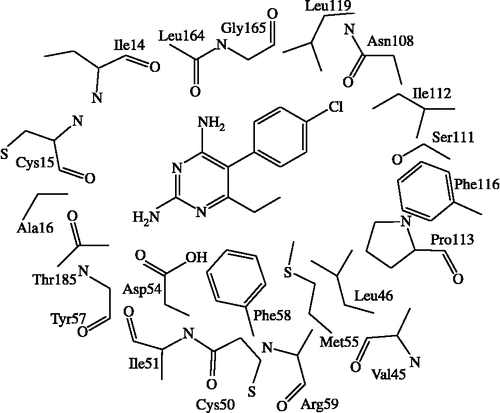
In order to investigate specific interaction of different potency of pyr derivatives in quadruple mutant DHFR, particular interaction was determined by quantum chemical calculations. According to comparison between double mutant complex with pyr (1J3J) and quadruple mutant complex with WR99210 (1J3K) structures superimposition, it was found that the binding sites are quite similar with RMS = 0.435 Å as shown in . Due to there being no pyr/quadruple mutant DHFR available, therefore, in this study, we proposed pyr and quadruple mutant complex, based on atom superposition. Considering the graphical backbone superimpostion, it can be implied that WR99210 and pyr oriented in the same binding position, therefore, pyr can be adapted into the quadruple mutant type PfDHFR to find the estimated particular interaction energy. The selected inhibitors were compounds 1 (pyr drug) and 6 according to their similar structures, but different Ki values. Compound 1 represented a resistance to quadruple mutant PfDHFR while compound 6, Cl substituent at X, gave a good Ki for this enzyme. The model systems contained compounds 1 or 6 and surrounding residues in the binding pocket with at least one atom interacting with any atoms of inhibitor within the interatomic distance of 4 Å that covered van der Waal interactions. The 22 selected residues were Ile14, Cys15, Ala16, Val45, Leu46, Trp48, Cys50, Ile51, Asp54, Met55, Tyr57, Phe58, Arg59, Asn108, Ser111, Ile112, Pro113, Phe116, Leu119, Leu164, Gly165 and Thr185. The four mutations, Asn51Ile, Cys59Arg, Ser108Asn and Ile164Leu, were also included in the system setup. The 2D scheme of the adopted model system of the inhibitor bound to the mutant PfDHFR binding site is shown in . In addition, the eleven inserted residues, 47, 49, 52, 53, 56, 109, 110, 114, 115, 117 and 118, were added into the system to complete the connection between the amino acids in the cutting chains. The N- and C-terminals were capped with a methyl amino group (-NHCH3) and an acetyl group (CH3CO-), respectively, which were retained from the backbone geometries of the nearby residues. Thus, the hydrogen atoms were added to the starting system using Sybyl7.0. Partial optimizations were performed by using the semiempirical PM3 method, implemented in the Gaussian 03 program [Citation31], based on the ‘heavy atoms fixing’ approximation. Therefore, only H atoms of amino acids and all atoms of the inhibitor were optimized. Finally, MP2 calculations with 6-31G(d,p) basis set were applied to investigate the particular interaction energy between inhibitor and each residue surrounding the binding site as shown in the interaction energy formula:
where A and B are the number of basis sets of ligands and amino acids, respectively,
![]() is the energy of the ligand-amino acid complex with the basis set of A plus B.
is the energy of the ligand-amino acid complex with the basis set of A plus B. ![]() and
and ![]() are the energies of ligand and amino acid with its number of basis sets. Moreover, the basis set superposition error based on counterpoise scheme (BSSE-CP) of Boys-Bernardi [Citation32] was also computed to define the interaction energy with BSSE as shown in Equation 6:
are the energies of ligand and amino acid with its number of basis sets. Moreover, the basis set superposition error based on counterpoise scheme (BSSE-CP) of Boys-Bernardi [Citation32] was also computed to define the interaction energy with BSSE as shown in Equation 6:
where
![]() and
and ![]() are the energies of ligand and amino acid, respectively, with the number of basis sets of A plus B [Citation23].
are the energies of ligand and amino acid, respectively, with the number of basis sets of A plus B [Citation23].
Figure 2. The backbone superimposition between the X-ray PfDHFR/WR99210 complex structures of quadruple mutant (PDB entry 1J3K shown in green color) and PfDHFR/pyr double mutant (PDB entry 1J3J shown in orange color), indicating the similarity binding position of both of the inhibitors.
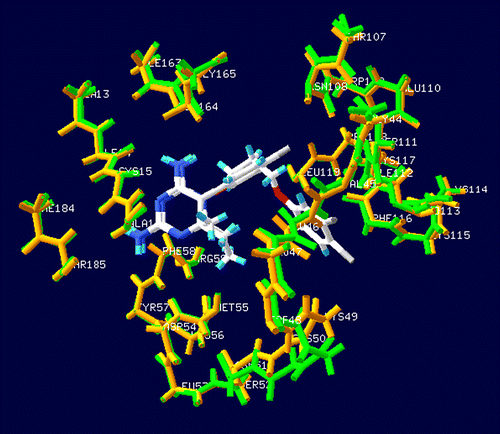
Figure 3. The 2D scheme of the adopted model system of inhibitor bound to the quadruple mutant type (Asn51Ile, Cys59Arg, Ser108Asn, Ile164Leu) of PfDHFR binding site.
Results and discussion
Statistical analysis
The relationship between structural properties of twenty-three pyr derivatives and their biological activities of quadruple mutant type PfDHFR is presented by using the CoMFA model. There are three models that varied the type of probe atoms Csp3 (+1), Osp3 ( − 1) and H (+1) and the statistical results are shown in . All models can be used to well predict the pKi values for training compounds, with a residual not greater than 0.4; these results are summarized in . Evaluation of the model prediction is assessed by test set compounds which all showed acceptable pKi prediction values, except compound 22; this test set compound has a different structure of R substitution ((CH2)2O(CH2)3OPh) from the others, so it shows high residual pKi between actual and predicted values.
Table II. PLS statistical results of CoMFA models for quadruple mutant PfDHFR.
Table III. Actual (Act) and predicted (Pred) pKi values and the residuals (Δ) of the training set and test set molecules for the mutant PfDHFR models.
By considering the statistical results in , model I-III with ![]() values higher than 0.6 (0.724, 0.669 and 0.690, respectively) can be accepted and the conventional r2 or no-validated r2 (
values higher than 0.6 (0.724, 0.669 and 0.690, respectively) can be accepted and the conventional r2 or no-validated r2 (![]() ) values are found to be 0.963, 0.980 and 0.983, respectively. These mean that the three tested probes (Csp3, Osp3 and H) give qualitatively very similar models. The results suggest that all three types of probes form equally important in the enzyme-ligand interactions. Next, the combination of three probe atoms was used, resulting in model IV with
) values are found to be 0.963, 0.980 and 0.983, respectively. These mean that the three tested probes (Csp3, Osp3 and H) give qualitatively very similar models. The results suggest that all three types of probes form equally important in the enzyme-ligand interactions. Next, the combination of three probe atoms was used, resulting in model IV with ![]() = 0.702 and
= 0.702 and ![]() = 0.980. Moreover, the prediction for test set obtained from model IV shows the highest predictive ability (
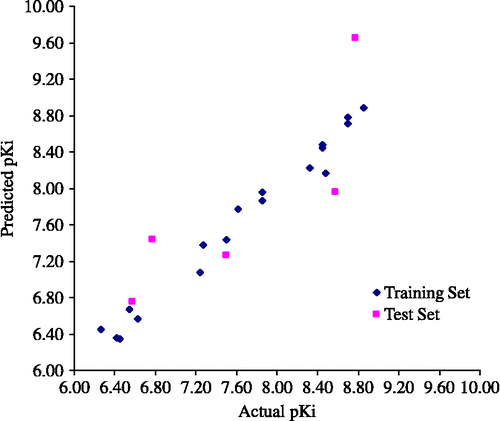
= 0.980. Moreover, the prediction for test set obtained from model IV shows the highest predictive ability (![]() = 0.698). Therefore, the combining three probe atoms in model IV is superior and more general model. Especially, the statistical error (s) of the represented model is 0.156 which is reasonably acceptable for biological activity predictions of the training set and the test set. The graphical plot between actual and predicted pKi of the training set and the test set is shown in . The CoMFA field contributions of the steric and electrostatic interactions contributed approximately 50%. The results indicate that both steric and electrostatic contributions are equivalent that affected the biological activity of mutant PfDHFR.
= 0.698). Therefore, the combining three probe atoms in model IV is superior and more general model. Especially, the statistical error (s) of the represented model is 0.156 which is reasonably acceptable for biological activity predictions of the training set and the test set. The graphical plot between actual and predicted pKi of the training set and the test set is shown in . The CoMFA field contributions of the steric and electrostatic interactions contributed approximately 50%. The results indicate that both steric and electrostatic contributions are equivalent that affected the biological activity of mutant PfDHFR.
Figure 4. Plot of the predicted and actual pKi values of the training set and the test set molecules with CoMFA H model IV.
CoMFA contour analysis
The CoMFA analysis with hundreds or thousands terms, is usually represented as the scalar product of the associated coefficient and the standard deviation of all values in the corresponding column of the data table (STDEV*COEFF) contour plots. Moreover, the contour maps can be showed by merging with the binding pocket of a drug target. In this study, the CoMFA contour maps are merged with 7 Å of binding pocket of crystal structure of quadruple mutant type PfDHFR which is available in the Protein Data Bank with PDB code 1J3K. The template compound 12 is displayed as the inhibitor in the CoMFA contour maps.
shows the steric contour maps of CoMFA model IV. The steric contour map indicates areas in which molecular steric bulk might have a favourable (green) or unfavourable (yellow) effect on the activity of an analogue. A sterically favoured green region is found near X substituent of the aromatic ring. This is further supported by comparing X substituent with Cl and H when these compounds have the same Y and R substituents. In addition, the distribution of steric contours appears around the phenyl side chain of R substituent; this evidence would explain why compound 12, used as the template, is a better quadruple mutant PfDHFR inhibitor than the pyr drug (compound 1). Furthermore, this region is closed to Phe116 of the binding pocket. An unfavourable steric contour region is found at Y substituent on the aromatic ring which can explain the fact that compounds 1, 7 and 10 show lower pKi when compared with unsubstituted structures of compounds 5, 8 and 11, respectively. Therefore, a hydrogen atom is suitable substituent of Y position because the unfavourable steric area of Y is closed to an important mutation position, Asn108, of quadruple mutant PfDHFR. The obtained unfavourable steric contour coincides with the previous publications [Citation3,Citation5,Citation9,Citation10] that reported a steric clash between the Cl substituent of pyr and the side chain of Asn108.
Figure 5. CoMFA (stdev.*coeff.) sterically favored areas are represented by green regions. Sterically unfavored areas are represented by yellow regions (level of steric contour contribution = 80%) and compound 12 is represented by ball and stick.
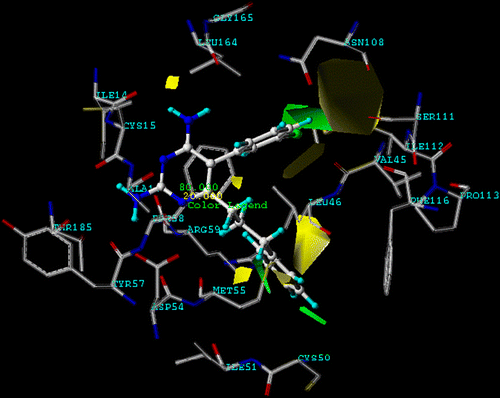
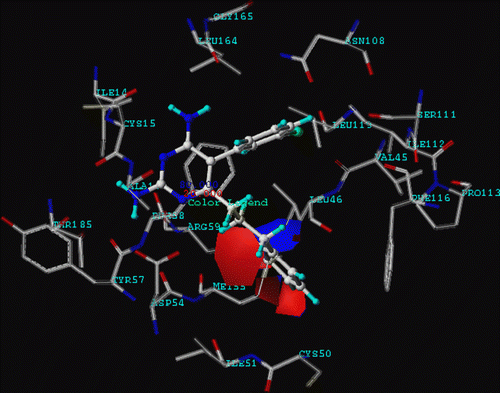
depicts electrostatic contour maps of CoMFA. The electrostatic contour map reveals that blue contours refer to positive charge favouring areas and red contours indicate negative charge favouring areas. The red area is found in the middle of the phenyl ring of R substitution which means high electron density in this area. Furthermore, a large blue contour also surrounds the phenyl ring of R substitution. It can be suggested that high positive charges or low electron density in this area is preferable. Therefore, donating substituents of the phenyl ring will increase the activity of the inhibitors, for example, Cl, F, OCH3, etc.
Figure 6. CoMFA (stdev.*coeff.) negative charge favored area is represented by the red region. Positive charge favored area is represented by the blue region (level of electrostatic contour contribution = 80%) and compound 12 is represented by ball and stick.
Particular interaction energy
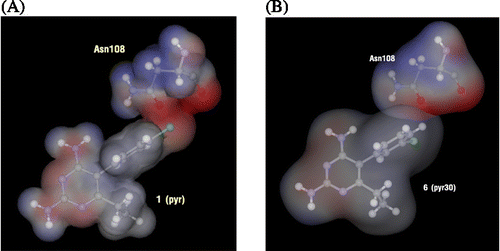
In order to find the particular interaction energy between compound 1 or 6 and the amino acids surrounding the pocket of quadruple mutant type PfDHFR, the MP2 method with basis set 6-31G(d,p) level of calculations was performed. In addition, the BSSE-CP was also calculated to correct the interaction energy. The obtained interaction energies are given in . Asp54 has the strongest interaction energy to compounds 1 and 6 of − 10.139 and − 10.250 kcal/mol, respectively. This amino acid formed H-bond interaction with the inhibitors. In addition, the H-bond interactions are also found with Ile14 and Leu164 for both compounds 1 and 6. All H-bond distances are displayed in . Three mutant amino acids, Ile51, Arg59 and Leu164, show no significantly different interaction energies between compound 1 and 6 (). While Asn108 shows more repulsive interaction to compound 1 by approximately 5 kcal/mol because compound 1 has the Cl substituent at Y position which can occur due to the steric clash with the side chain of Asn108, as shown clearly by electrostatic potential surfaces in . In pervious reviews Citation33, Citation34, Citation35, there were many reports that proposed the cause of pyr resistance in quadruple mutant type PfDHFR came from the steric clash with Asn108 mutation. Our particular interaction energy studies can verify this evidence with the obtained repulsive energy.
Table IV. Particular interaction energy (kcal/mol) of 1 (pyr) and 6 with individual residues, calculated at MP2/6-31G(d,p) and MP2/6-31G(d,p) levels with BSSE-CP.
Figure 7. H-bond distances between inhibitor and residues in the binding pocket; (A) compound 1 and (B) compound 6 (in Å).
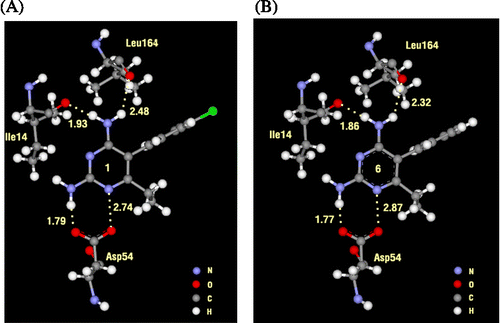
Figure 8. The electrostatic potential is shown on the solvent accessible surface as red for negative and blue for positive values for Asn108 interacted with (A) compound 1 and (B) compound 6.
Conclusion
The CoMFA analysis is a very powerful method for ligand-based drug design. This method has been successful in designing new potent inhibitors for many drugs. Therefore, in this study, the CoMFA method was selected to build a linear equation of the quantitative structure activity relationship of the pyr derivatives that are active against quadruple mutant type PfDHFR. The combined Csp3 (+1), Osp3 ( − 1) and H (+1) probe atoms model was selected to represent the CoMFA moelcular fields for accounting the different types of interactions between mutant PfDHFR binding site and pyr derivatives. The steric contour maps of this model suggest that X and R substitutions favoured a bulky group which is opposite to Y substitution. Electrostatic maps are displayed surrounding the phenyl ring of the R substituent of the template compound which means that an electron donating substituent on the phenyl ring will increase the biological activity. Therefore, the characteristics of new design inhibitors are the bulky group on the X substitution, the hydrogen atom on the Y substitution and the long chain on the R substitution. Moreover, we also performed MP2/6-31G(d,p) quantum chemical calculations with BSSE-CP energy correction to investigate the particular interaction energy of compounds 1 (pyr) and 6 (Cl substituent on X). The obtained results clearly show that Asn108 is the cause of pyr resistance with the highest repulsive interaction energy and negative electrostatic potential. Accordingly, this calculation is consistent with the unfavoured steric region of CoMFA contour maps. The CoMFA and particular interaction energy analyses will be useful for identifying the structural features of potent pyr derivatives active against quadruple mutant type PfDHFR which is an important target of malaria chemotherapy.
Acknowledgements
This work was supported by the Thai Graduate Institute of Science and Technology (TGIST) Funds (TG-22-11-845D) under the National Science and Technology Development Agency, Thailand. S.H. is grateful to the Thailand Research Fund (RSA5080005). The Postgraduate Education and Research Program in Petroleum, Petrochemical Technology and Advanced Materials, Commission on Higher Education (CHE), the Kasetsart University Research and Development Institute (KURDI) and the Graduate School Kasetsart University Scholarship are gratefully acknowledged for financial support. We would like to thank the Large Scale Research Laboratory of the National Electronics and Computer Technology (NECTEC), National Center for Genetic Engineering and Biotechnology (BIOTEC), LCAC and the computing center of KU for research facilities. This work has partially been supported by the National Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Center of Excellence Network. Medicines for Malaria Venture (MMV), Wellcome Trust, and UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR) for YY and SK. SK is an international scholar of Howard Hughes Medical institute (HHMI, USA).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- RW Snow, CA Guerra, AM Noor, HY Myint, and SI Hay. (2005). Estimating clinical episodes of malaria. Nature 434:214–217.
- P Prapunwattana, W Sirawaraporn, Y Yuthavong, and DV Santi. (1996). Chemical synthesis of the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene. Mol Biochem Parasitol 83:93–106.
- J Yuvaniyama, P Chitnumsub, S Kamchonwongpaisan, J Vanichtanankul, W Sirawaraporn, P Taylor, MD Walkinshaw, and Y Yuthavong. (2003). Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol 10:357–365.
- T Dasgupta, and KS Anderson. (2008). Probing the role of parasite-specific, distant structural regions on communication and catalysis in the bifunctional thymidylate synthase- dihydrofolate reductase from Plasmodium falciparum. Biochemistry 47 (5):1336–1345.
- RT Delfino, OA Santos-Filho, and JD Figueroa-Villar. (2002). Type2 antifolates in the chemotherapy of falciparum malaria. J Braz Chem Soc 13:727–741.
- NT Marbiah, E Petersen, K David, E Magbity, J Lines, and DJ Bradley. (1998). A controlled trial of lambda-cyhalothrin-impregnated bed nets and/or dapsone/pyrimethamine for malaria control in Sierra Leone. Am J Trop Med Hyg 58 (1):1–6.
- A Sowunmi, BA Fateye, AA Adedeji, GO Gbotosho, TC Happi, AE Bamgboye, OM Bolaji, and AMJ Oduola. (2006). Predictors of the failure of treatment with pyrimethamine-sulfadoxine in children with uncomplicated falciparum malaria. Acta Trop 98:6–14.
- AP Alker, V Mwapasa, and R Meshnick. (2004). Rapid real-time PCR genotyping of mutations associated with sulfadoxine-pyrimethamine resistance in Plasmodium falciparum. Antimicrob Agents Chemother 48 (8):2924–2929.
- DC Warhurst. (2002). Resistance to antifolate in Plasmodium falciparum, the causative agent of tropical malaria. Sci Prog 85:89–111.
- W Sirawaraporn. (1998). Dihydrofolate reductase and antifolate resistance in malaria. Drug Resist Updat 1:397–406.
- RT Delfino, OA Santos-Filho, and JD Figueroa-Villar. (2002). Molecular modeling of wild-type and antifolate resistant mutant Plasmodium falciparum DHFR. Biophys Chem 98:287–300.
- DS Peterson, WK Milhous, and TE Wellems. (1990). Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proc Natl Acad Sci USA 87:3018–3022.
- W Sirawaraporn, T Sathitkul, R Sirawaraporn, Y Yuthavong, and DV Santi. (1997). Antifolate-resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA 94:1124–1129.
- IM Hastings, and MJ Donnelly. (2005). The impact of antimalarial drug resistance mutations on parasite fitness, and its implications for the evolution of resistance. Drug Resist Updat 8:43–50.
- D Walliker, P Hunt, and H Babiker. (2005). Fitness of drug-resistant malaria parasites. Acta Trop 94:251–259.
- S Kamchonwongpaisan, R Quarrell, N Charoensetakul, R Ponsinet, T Vilaivan, J Vanichtanankul, B Tarnchompoo, W Sirawaraporn, G Lowe, and Y Yuthavong. (2004). Inhibitors of multiple mutants of Plasmodium falciparum dihydrofolate reductase and their antimalarial activities. J Med Chem 47:673–680.
- H Kubinyi, H Kubinyi. QSAR: Hansch Analysis and Related Approaches VCH: Weinheim. 1993.
- C Hansch, and A Leo. Exploring QSAR. Fundamentals and Applications in Chemistry and Biology. American Chemical Society, Washington, DC, (1995) .
- RD CramerIII, DE Patterson, and JD Bunce. (1988). Comparative molecular field analysis CoMFA: 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 110:5959–5967.
- H Kubinyi, G Flokers, and YC Martin. (1998). 3D QSAR in drug design: Ligand-protein interactions and molecular similarlity. Perspect Drug Discovery Des 3–398.
- SS Sao, and M Karplus. (2001). Evaluation of designed ligands by a multiple screening method: Application to glycogen phosphorylase inhibitors constructed with a variety of approaches. J Comput Aided Mol. Des 15:613–647.
- S Saen-oon, M Kuno, and S Hannongbua. (2005). Binding energy analysis for wild-type and Y181C Mutant HIV-1 RT/8-Cl TIBO complex structures: Quantum chemical calculations based on the ONIOM method. PROTEINS: Structure, Function, and Bioinformatics 61:859–869.
- M Kuno, R Hongkrengkai, and S Hannongbua. (2006). ONIOM-BSSE scheme for H–¶ system and applications on HIV-1 reverse transcriptase. Chem Phys Lett 424:172–177.
- SYBYL 7.0., Tripos Associates Inc., 1699 South Hanley Road, Suite 303, St. Louis, Missouri 63144, USA.
- A Streitwieser. Molecular Orbital Theory for Organic Chemists. New York, Wiley, (1961) .
- M Clark, and RD CramerIII. (1993). The probability of chance correlation using partial least squares (PLS). Quant Struct-Act Relat 12 (2):137–145.
- BL Bush, and RB NachbarJr. (1993). Sample-distance partial least squares: PLS optimized for many variables, with application to CoMFA. J Comput Aided Mol Des 7:587–619.
- SJ Cho, and A Tropsha. (1995). Cross-validated R2-guided region selection for comparative molecular field analysis: A simple method to achieve consistent results. J Med Chem 38:1060–1066.
- A Golbraikh, and A Tropsha. (2002). Beware of q2!. J Mol Graph Mod 20:269–276.
- S Hannongbua, K Nivesanond, L Lawtrakul, P Pungpo, and P Wolschann. (2001). 3D-quantitative structure-activity relationships of HEPT derivatives as HIV-1 reverse transcriptase inhibitors, based on ab initio calculations. J Chem Inf Comput Sci 41:848–855.
- MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, MA Robb, JR Cheeseman, JA MontgomeryJr., T Vreven, KN Kudin, JC Burant, JM Millam, SS Iyengar, J Tomasi, V Barone, B Mennucci, M Cossi, G Scalmani, N Rega, GA Petersson, H Nakatsuji, M Hada, M Ehara, K Toyota, R Fukuda, J Hasegawa, M Ishida, T Nakajima, Y Honda, O Kitao, H Nakai, M Klene, X Li, JE Knox, HP Hratchian, JB Cross, C Adamo, J Jaramillo, R Gomperts, RE Stratmann, O Yazyev, AJ Austin, R Cammi, C Pomelli, JW Ochterski, PY Ayala, K Morokuma, GA Voth, P Salvador, JJ Dannenberg, VG Zakrzewski, S Dapprich, AD Daniels, MC Strain, O Farkas, DK Malick, AD Rabuck, K Raghavachari, JB Foresman, JV Ortiz, Q Cui, AG Baboul, S Clifford, J Cioslowski, BB Stefanov, G Liu, A Liashenko, P Piskorz, I Komaromi, RL Martin, DJ Fox, T Keith, MA Al-Laham, CY Peng, A Nanayakkara, M Challacombe, PMW Gill, B Johnson, W Chen, MW Wong, C Gonzalez, and JA Pople. . Gaussian, Inc, Pittsburgh, (2003) Gaussian 03, revision B.05.
- SF Boys, and F Bernardi. (1970). The calculations of small molecular interaction by the difference of separate total energies some procedures with reduced error. Mol Phys 19:553–566.
- Y Yuthavong. (2002). Basis for antifolate action and resistance in malaria. Microb Inf 4:175–182.
- G Rastelli, W Sirawaraporn, P Sompornpisut, T Vilaivan, S Kamchonwongpaisan, R Quarrell, G Lowe, Y Thebtaranonth, and Y Yuthavong. (2000). Interaction of pyrimethamine, cycloguanil, WR99210 and their Analogues with Plasmodium falciparum Dihydrofolate reductase: Structural basis of antifolate resistance. Bioorg Med Chem 8:1117–1128.
- Y Yuthavong, J Yuvaniyama, P Chitnumsub, J Vanichtanankul, S Chusacultanachai, B Tarnchompoo, T Vilaivan, and S Kamchonwongpaisan. (2005). Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: Structural basis for antifolate resistance and development of effective inhibitors. Parasitology 130:249–259.