Abstract
Human and murine lanosterol synthases (EC 5.4.99.7) were studied as targets of a series of umbelliferone aminoalkyl derivatives previously tested as inhibitors of oxidosqualene cyclases from other eukaryotes. Tests were carried out on cell cultures of human keratinocytes and mouse 3T3 fibroblasts incubated with radiolabeled acetate, and on homogenates prepared from yeast cells expressing human lanosterol synthase, incubated with radiolabeled oxidosqualene. In cell cultures of both human keratinocytes and mouse 3T3 fibroblasts, the observed inhibition of cholesterol biosynthesis was selective for oxidosqualene cyclase. The most active compounds bear an allylmethylamino chain in position-7 of the coumarin ring. The inhibition was critically dependent on the position and length of the inhibitor side chain, as well as on the type of aminoalkyl group inserted at the end of the same chain. Molecular docking analyses, carried out to clarify details of inhibitors/enzyme interactions, proved useful to explain the observed differences in inhibitory activities.
| Abbreviations | ||
| OS, | = | 2,3-oxidosqualene |
| DOS, | = | 2,3-22,23-dioxidosqualene |
| OSC, | = | oxidosqualene cyclase |
Introduction
Treatment of hypercholesterolemia and related cardiovascular disease was revolutionized years ago by the introduction of statins, a class of powerful inhibitors of HMG-CoA reductase, the rate-limiting enzyme of cholesterol biosynthesis [Citation1]. Over the last years, an impressive mass of research has been devoted to the comprehension of the mechanism by which these inhibitors work and to the characterization of the target enzyme.
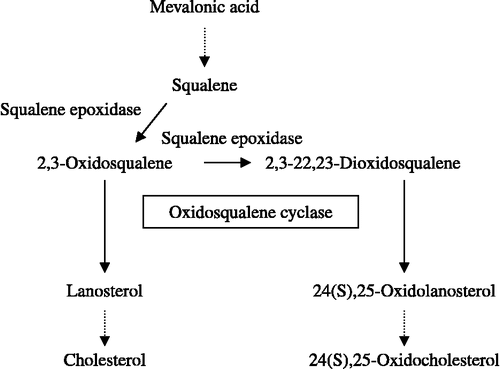
The appearance of statins, however, did not stop the search for other target enzymes and novel cholesterol-lowering drugs. Statins block cholesterol biosynthesis early in its pathway, by inhibiting the formation of farnesylpyrophosphate (FPP), which is the precursor not only of cholesterol but also of dolicols, coenzyme Q and prenylated proteins. Thus, in principle, a prolonged treatment with statins, especially at high doses, might cause undesirable side effects arising from a reduced production of the above FPP-derivatives. Search for target enzymes acting downstream of FPP formation has since been a major task in many pharmaceutical laboratories. Among potential targets, squalene epoxidase and oxidosqualene cyclase (OSC) have been most intensively studied Citation2, Citation3, Citation4. OSC is very attractive target as its partial inhibition affects several aspects of lipid metabolism besides reducing the formation of the steroid nucleus. The monoepoxysqualene that accumulates when OSC is inhibited is effectively converted to 2,3-22,23-diepoxysqualene, which is channelled into the pathway of oxysterol synthesis (). Oxysterols in turn, with other ligands, trigger a process mediated by the liver X receptor (LXR), enhancing the expression of a series of genes that regulate cellular lipid metabolism [Citation5]. In a recent paper, the observed decrease of foam cell formation caused by the treatment with OSC inhibitors is attributed to the enhanced synthesis of epoxycholesterol in macrophages [Citation6]. The design of novel specific inhibitors, potential tools for cholesterol-lowering therapy, can take great advantage of the information deriving from the crystallization of human OSC recently obtained [Citation7].
Figure 1. Schematic representation of the relevant steps in cholesterol biosynthesis.
In the present work we have investigated a series of umbelliferone aminoalkyl derivatives as inhibitors of human and murine OSCs. Some of these compounds are effective inhibitors of OSCs from other organisms Citation8, Citation9. The inhibitory effect has been evaluated both on cell cultures (human keratinocytes and murine fibroblasts) and homogenates prepared from yeast (S. cerevisiae) cells expressing human lanosterol synthase. The human enzyme has been characterized by determining the effect of pH, temperature, and detergent concentration on the enzymatic activity. The inhibitory properties of the umbelliferone aminoalkyl derivatives have been compared with Ro 48-8071, a known powerful inhibitor of human OSC [Citation3]. Molecular docking experiments have been used to study some key interactions that could give insight into the activities of inhibitors.
Materials and methods
Materials
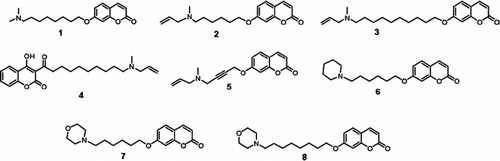
Chemicals and culture media were obtained from Sigma-Aldrich (Milan, Italy) unless otherwise specified. Coumarin derivatives () and the OSC substrate, 2,3-oxidosqualene (OS), were synthesized as previously reported [Citation10]. The 3-acyl-4-hydroxycoumarin 4 is a new compound prepared in three steps: 1) acylation of 4-hydroxycoumarin with 10-undecenoyl chloride [Citation11]; 2) Oxidative demolition of the terminal double bond obtaining the 10-formyl derivative [Citation12]; 3) reductive amination with excess allylmethylamine [Citation12].
Figure 2. Structures of umbelliferone aminoalkyl derivatives.
Labeled [14C]-(3S)2,3-oxidosqualene was prepared as previously reported [Citation13], according to Popjak [Citation14] by incubating pig liver S10 supernatant with R,S[2-14C]mevalonic acid (1 μCi, 55 mCi/mmol, 2.04 GBq/mmol; Amersham Pharmacia Biotech, UK), in the presence of OSC inhibitor U14266-A [Citation15]. [2-14C]acetate (50 mCi/mmol) and R,S[2-14C]mevalonic acid (55 mCi/mmol, 2.04 GBq/mmol) were obtained from Amersham Pharmacia Biotech (U.K).
Restriction enzymes and T4 DNA ligase were from Promega (Milan, Italy).
Mammalian cells and culture conditions
3T3 fibroblasts (ATCC CCL 92 3T3 Swiss albino) and human keratinocytes (NCTC 2544, kindly provided by Prof. Angela Rosa Canuto, Department of Medicina ed Oncologia Sperimentale, University of Turin, Italy) were routinely maintained as monolayers in Dulbecco-modified Eagle medium (DMEM) high (4500 mg/L) or low (1000 mg/L) in glucose respectively, supplemented with 10% adult bovine serum (ABS). Cells were grown at 37°C in a humidified atmosphere (air 95%/ CO2 5%), and the same conditions were used for the [2-14C]acetate incorporation assay.
S. cerevisiae strain and culture conditions
The lanosterol-synthase mutant strain of S. cerevisiae SMY8 (MATα erg7::HIS3 hem1::TRP1 ura3-52-trpl-Δ63 leu2-3.112 his3-Δ200 ade2 Gal+) was kindly provided by Professor S.P.T. Matsuda (Department of Chemistry and Biochemistry and Cell Biology, Rice University Houston, Texas-USA) [Citation16]. The yeast strain was preserved in 40% glycerol at − 80°C.
Cells were grown aerobically at 30°C to early stationary phase in YPD medium (1% yeast extract, 2% peptone, 2% dextrose) supplemented with hemin (0.013 mg/mL) and ergosterol (0.02 mg/mL). Hemin had to be added to the growth medium because the SMY8 strain contains a mutation (hem1::TRP1) affecting the heme biosynthesis. The presence of a heme mutant background is necessary for the viability of lanosterol synthase mutants in aerobic conditions [Citation16].
Cloning and expression of recombinant human OSC in S. cerevisiae SMY8
Clone pOTB7 containing human OSC cDNA was kindly provided by Professor S.P.T. Matsuda. The cDNA was sequenced by the C.R.I.B.I. - BMR Servizio Sequenziamento DNA, Padova (Italy). pOTB7 was digested using XhoI and BamHI and the resultant fragment was subsequently subcloned into pYES2 yeast plasmid vector (Invitrogen, Milan, Italy) under the control of Gal1 promoter to yield the pSOB1.1 plasmid. Lithium acetate was used to transform the S. cerevisiae lanosterol synthase mutant SMY8 with the pSOB1.1 plasmid [Citation17]. SMY8 transformants were selected on synthetic complete medium plates without uracil (SC-UraD) (0.17% yeast nitrogen base, 0.2% amino acids, 0.5% ammonium sulphate, 2% dextrose, 2% agar) supplemented with hemin (0.013 mg/mL) and ergosterol (0.02 mg/mL). Human OSC expression was induced in SC-Ura medium supplemented with hemin (0.013 mg/mL) in the presence of 2% galactose (SC-UraG).
Enzyme assays
OSC activity was assayed in cell-free homogenates obtained from yeast cell cultures grown to late exponential phase in SC-UraG at 30°C. Yeast cells expressing human OSC were centrifuged at 3000 x g for 10 min and their walls lysed with lyticase (1 mg/g wet cells in 1.2 M sorbitol, 0.02 M KH2PO4, pH 7.4) for 60 min at 30°C at a final concentration of 0.15 g of cell wet weight/mL. Spheroplasts were sedimented at 3000 × g for 10 min, washed twice with the same buffer and homogenized with a Potter device in 100 mM sodium phosphate buffer pH 7.2 containing PMSF (1 mM) at a final concentration of 0.8 g of cell wet weight/mL. Homogenates could indifferently be used fresh or after storage at − 80°C for several months. Proteins were quantified in the homogenate with the Protein Assay Kit SIGMA, based on the method of Lowry modified by Peterson [Citation18] using bovine serum albumin as a standard. Standard conditions for the OSC assay were as follows. The homogenate was incubated in the presence of labeled [14C]-(3S)-2,3-oxidosqualene (1000 cpm) diluted with unlabeled (R,S)2,3-oxidosqualene to a final concentration of 25 μM. Cold and labeled substrates and inhibitors (when added) were first dissolved in CHCl3, then aliquots were pipetted into test tubes containing Tween-80 (0.5 mg/mL, final concentration) and polidocanol (0.5 mg/mL, final concentration) and the solvent was evaporated under a stream of nitrogen. Residues were redissolved in 100 mM sodium phosphate buffer pH 8 (100 μL) containing glycerol (0.1 g/mL, final concentration), then the amount of homogenate required to obtain a 20% substrate transformation was added and the mixture, made up to a final volume of 200 μL with 100 mM sodium phosphate buffer pH 8, was incubated for 60 min at 45°C. The enzymatic reaction was stopped by adding methanolic KOH (1 mL, 10% w/v) and heating at 80°C for 30 min in a water bath. Extraction of the resulting mixtures was carried out with petroleum ether (2 mL) and extracts were evaporated under a stream of nitrogen; finally, residues were dissolved in a small volume of CH2Cl2 and spotted on TLC plates (Alufolien Kieselgel 60F254, Merck, Darmstadt, Germany), using n-hexane/ethyl acetate (85:15, v/v) as a developing solvent. Percent conversion of the labeled substrate to lanosterol was estimated by integration from radioactivity scans with a System 200 Imaging Scanner (Hewlett-Packard, Palo Alto, CA, USA). Different incubation conditions (different temperature, pH and polidocanol concentrations) were also tested (as reported in the Results).
IC50 values (inhibitor concentrations that reduced the enzymatic conversion by 50%) were calculated by non-linear regression analysis of the residual activity vs the log of inhibitor concentration using statistical software from Genstat (NAG, Oxford, U.K.). Values listed in the Tables are the means of two separate experiments, each one carried out in duplicate.
For the Km determinations, different final substrate concentrations (ranging from 1 to 100 μM) were used and incubation times chosen so as to result in transformation percentages not exceeding 10% (a limit imposed in order to safely confine the results to the range in which a linear relationship obtains between product formation and reaction time). To calculate Km values, reaction rates obtained at different substrate concentrations were fitted by a non-linear regression method to the Michaelis-Menten equation [Citation19].
Incorporation of [2-14C] acetate into non-saponifiable lipids of human keratinocytes and mouse 3T3 fibroblasts
Cells were seeded in 24-well plastic clusters. Each well received 7 × 104 cells suspended in 950 μL DMEM (Dulbecco-modified Eagle medium) containing 10% (v/v) lipid-depleted serum [Citation20]. After 24 h incubation, the cells were pre-incubated for 3 h in the presence of inhibitor (dissolved in absolute ethanol). The final concentration of ethanol in the culture medium did not exceed 0.4% (v/v); the same amount of ethanol was added to control cultures. At any rate, no significant effect of 0.4% ethanol on cell growth was observed during the experiments. Labeled acetate (1 μCi/well) was then added. After 3 h incubation, the medium was removed by aspiration with a pipette and cells were treated with 0.1 N NaOH (500 μL) for 30 min. Phosphate buffer-saline (PBS) (500 μL) was added to each cell lysate, which subsequently was saponified by treatment with methanolic KOH (1 mL, 10% w/v) for 30 min at 80°C.
Non-saponifiable lipids were extracted twice, with petroleum ether (1.5 and 1 mL, respectively), and combined extracts separated on TLC plates (Alufolien Kieselgel 60F254, Merck, Darmstadt, Germany) using n-hexane/ethyl acetate (85:15 v/v) as a developing solvent and authentic standards of cholesterol, lanosterol, 2,3-22,23-dioxidosqualene, 2,3-oxidosqualene and squalene. Radioactivity in separated bands was measured using a System 200 Imaging Scanner (Hewlett-Packard, Palo Alto, CA, USA).
Molecular modelling
Protein preparation
The crystal structure of OSC complexed with Ro 48-8071 [Citation7] was obtained from the Protein Data Bank (entry 1w6j) [Citation21]. In the crystal structure, the first five residues of the sequence (Met1, Thr2, Glu3, Gly4 and Thr5) were missing. Since these residues are distant from the inhibitor binding site, no addition has been made. Residues with missing atoms (Glu42, Arg43, Ala44) were completed using standard geometries. The orientation of the residue chains was established using the MOE Rotamer Explorer utility (MOE, 2005.06; Chemical Computing Group Inc: Montreal, Quebec Canada, 2005). All of these residues were distant from the cavity where Ro 48-8071 binds and, thus, did not influence the results of the docking procedure. Water molecules were ignored in the calculations. The model was completed adding hydrogen in their standard geometries.
Docking strategy
The 3D coordinates of the four ligands 1, 2, 3, and 4 were obtained using the MOE Builder Application. Then the ligands were aligned with the crystallographic pose of Ro 48-8071 taken as rigid template using the MOE Flexible Alignment application [Citation22] that flexibly aligned each ligand maximizing its overlap over a set of pre-determined features shared with Ro 48-8071 (similarity in shape, aromatic atoms overlap, hydrogen bond donors and acceptors overlap, atoms of similar partial charge overlap, hydrophobic areas overlap and, finally, the strain energy of each molecule). For each ligand the conformation with the best score was selected and manually set in the binding site, and the resulting complex was minimized following a procedure described elsewhere [Citation23]. Energy calculations were performed using the MMFF94x [Citation24] force field implemented in MOE. Electrostatic interactions were treated with a distant-dependent dielectric constant 4ε, as has been suggested by Christensen et al. [Citation25]. To avoid artefact, the crystal structure was minimized to obtain a model to compare with the docking results.
Results
Effects of umbelliferone aminoalkyl derivatives on sterol biosynthesis in cell cultures
The effects of a series of umbelliferone aminoalkyl derivatives () on sterol biosynthesis was studied in mouse 3T3 fibroblasts, a cell line we previously used with other OSC inhibitors [Citation26,Citation27], and human keratinocytes incubated with [2-14C] acetate by determining the amounts of radioactive label taken up by the biosynthetic intermediates.
After extracting from incubated cells the non-saponifiable lipid fractions, using TLC we separated and identified by comparison with authentic reference compounds the following biosynthetic products: cholesterol, lanosterol, 2,3-22,23-dioxidosqualene (DOS), 2,3-oxidosqualene (OS) and squalene (listed in order of decreasing polarity). The amounts of radioactive label incorporated into cholesterol and its biosynthetic intermediates after incubation of cell cultures in the presence of different coumarin-type inhibitors are shown in Tables and . For comparison, results obtained with the potent OSC inhibitor Ro 48-8071 [Citation3] were also reported. The amounts of radioactive label found in cholesterol and its biosynthetic intermediates are expressed as percent of total counts found in non-saponifiable lipids.
Table I. Inhibitory effects of umbelliferone aminoalkyl derivatives on sterol biosynthesis in 3T3 fibroblasts incubated with radiolabeled acetate.
Table II. Inhibitory effects of umbelliferone aminoalkyl derivatives on sterol biosynthesis in human keratinocytes incubated with radiolabeled acetate.
In the absence of inhibitors, both cell types incorporated more than 80% of the label into cholesterol, the remaining 20% being distributed among lanosterol, DOS, OS and squalene.
Both cell types responded similarly to the incubation with 7-[10′-(allylmethylamino)-decyloxy]-chromen-2-one (3), an umbelliferone aminoalkyl derivative with a flexible aliphatic chain and a terminal methylallyl tertiary amino group. At a concentration of 1 μM it reduced by about 80% the incorporation of radioactivity in cholesterol and increased from eight to tenfold the labeling of intermediates OS and DOS. The analogue 7-[6′-(allylmethylamino)-hexyloxy]-chromen-2-one (2), bearing a shorter chain, caused a similar effect in 3T3 fibroblasts but was less active on keratinocytes, where the label reduction in cholesterol was only 40% and the label increase in OS and DOS ranged from five to sevenfold. The 7-[8′-(dimethylamino-N-octyloxy)]chromen-2-one (1) bearing a terminal dimethyl amino group was as active as compound 3 in keratinocytes, but in 3T3 fibroblasts the effect on sterol biosynthesis was evident only at 10 μM. 4-Hydroxy-3-[allylmethylamino-decinoyl]-chromen-2-one (4), bearing the same aliphatic chain of compound 3 in the position 3 of the lacton ring, was completely inactive at 10 μM in both cell types. The cumarin derivatives with a rigid acetylenic arm (5) and those with terminal morpholino (7 and 8) or pyrrolidino (6) group on a flexible hexyl or octyl chain, were inactive at 1 μM and weakly active at 10 μM on both 3T3 fibroblasts and keratinocytes. Comparison of Ro 48-8071 with the umbelliferone aminoalkyl derivatives showed that Ro 48-8071 is highly more active than the two most effective compound of the series, 1 and 3.
Characterization and inhibition of human oxidosqualene cyclase expressed in yeast
The human OSC gene was subcloned into the yeast expression vector pYES2 and overexpressed in the lanosterol synthase defective S. cerevisiae strain SMY8. Sequencing of the original cDNA and the whole gene in pYES2 vector showed the same mutation, Leu642Val, present in the reported sequence [Citation28,Citation29]. This conservative mutation is described in the literature as a single-nucleotide polymorphism (SNPs) and seems to be very common [Citation30]. The transformation of OSC defective yeast strain SMY8 with the human OSC gene abolished its sterol auxotrophy: the transformed cells grew in the absence of added ergosterol and produced labeled C27 sterols after incubation with [2-14C]acetate for 4 h at 30°C. In untransformed control cells grown in the same conditions in the presence of [2-14C]acetate, no label was found in C27 sterols, and label accumulated in the OSC substrates 2,3-oxidosqualene and 2,3-22,23-dioxidosqualene. A cell-free homogenate of SMY8 cells expressing the human OSC enzyme was used to evaluate the enzyme activity, expressed as nanomoles of lanosterol formed after the incubation with labeled [14C]2,3-oxidosqualene. All activity assays were performed in the presence of polidocanol previously shown to be the most effective detergent for enhancing the activity of a mammalian OSC [Citation31]. reports the activities obtained at different temperatures, pH and detergent concentrations. Optimal assay conditions were as follows: 45°C, 100 mM sodium phosphate buffer, pH 8, in the presence of 0.05% polidocanol and 10% glycerol. These assay conditions were used to determine the Km and the IC50 values of the inhibitors. The value of the apparent Km obtained in the present work, calculated by a non-linear regression method, was 69.03 ± 22.4 μM, a value slightly higher, yet comparable to Km determined in a previous work with other mammalian OSCs, solubilised from microsomes and tested under the same conditions of detergent, buffer, pH and temperature [Citation26].
Table III. Effect of temperature, pH and detergent concentration on the activity of human OSC expressed in yeast.
The IC50 values for umbelliferone aminoalkyl derivatives 1, 2, 3 and 4 are reported in .
Table IV. Effect of umbelliferone aminoalkyl derivatives on the activity of human OSC expressed in yeast.
The inhibition results essentially confirmed the data obtained with cell cultures: the umbelliferone 7-aminoalkyl derivatives were found to be active inhibitors of human OSC, while the compound 4, bearing the aminoalkyl chain on the position 3 of the lactone ring was definitely less active. The reference compound Ro 48-8071 proved to be more active than the umbelliferone derivatives.
Molecular modelling
The crystal structure of inhibitor Ro 48-8071, taken as reference compound, complexed with human OSC sheds light on the key interactions between the ligand and the enzyme. Four points in particular have to be considered: a) the charged hydrogen bond involving the basic nitrogen atom of the inhibitor and the Asp455 carboxylate group stabilized through cation-π interactions with Phe444, Trp387 and Trp581; b) the stacking of the fluorophenyl group between the side chains of Phe696 and His232; c) the hydrogen bond between the carbonyl group of the reference compound and a water molecule, which in turn interacts with Ile338; d) the occupation of the binding site owing to the inhibitor's terminal phenyl group interacting with Trp192, Trp230 and Phe521, whereas its bromine atom interacts with Ile524, Tyr237 and Cys223 [Citation7]. In the minimized structure (see Materials and Methods) all key interactions are conserved with the exception of the hydrogen bond between the protonated nitrogen and the Asp455 carboxylate, this hydrogen bond being replaced by an ionic interaction between the same atoms (3.10 Å in crystal data and 3.97 Å in the model).
Compounds 1, 2, 3 and 4 were submitted to a molecular docking analysis to compare the interaction pattern of the four inhibitors with the pattern of reference compound Ro 48-8071.
Firstly, inhibitor 2 will be discussed because its aliphatic chain is the same as the reference compound. The docking results show that the aliphatic chains of compound 2 and Ro 48-8071 occupy the same region of the binding site (); thus, the basic nitrogen atom of compound 2 shows the same interactions pattern of Ro 48-8071. The umbelliferone ring is roughly superposed to the benzophenone moiety of the reference compound and it is stacked with the side chains of Phe696 and His232. The heteroatom in the lactone ring replaces carbonylic oxygen of Ro 48-8071, which suggests that it may form a hydrogen bond with the water molecule bound to Ile338. Finally, the umbelliferone ring cannot fit well the portion of the binding site where the Ro 48-8071 bromophenyl moiety is located.
Figure 3. OSC residues interacting with the compound 2. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.
![Figure 3. OSC residues interacting with the compound 2. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.](/cms/asset/56deca6e-b39b-45a3-897e-3ce3c29fd929/ienz_a_332035_f0003_b.gif)
In compound 1 the replacement of an allyl with a methyl group and the elongation of the alkyl chain cause a shift of the ligand in the binding site, bringing the nitrogen atom close enough to Asp455 for an ionic bond to form (). The aliphatic chain is roughly superposed to the corresponding Ro 48-8071 chain, whereas the umbelliferone ring is essentially superposed to the same moiety in the compound 2 docked pose showing a similar interaction pattern.
Figure 4. OSC residues interacting with the compound 1. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.
![Figure 4. OSC residues interacting with the compound 1. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.](/cms/asset/6496ac9a-7218-4fac-80e4-b04a98dca16f/ienz_a_332035_f0004_b.gif)
In compound 3 the basic nitrogen lies closer to the corresponding moiety of the reference compound, whereas the aliphatic chain, the longest in all of the docked compounds, is superposed to the hexyloxy chain as well the fluorophenyl group of Ro 48-8071 with the loss of π-π interactions with Phe696 and His232 (). In such a position, the oxygen atom is able to form a hydrogen bond with the water molecule as registered for Ro 48-8071. Besides, the umbelliferone ring is superposed to the bromophenyl moiety, making a good fit in the binding site, although the oxygen atoms are located in an essentially hydrophobic region near the side chains of Trp192, Tyr237 and Val236.
Figure 5. OSC residues interacting with the compound 3. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.
![Figure 5. OSC residues interacting with the compound 3. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.](/cms/asset/aac65983-3b02-4908-be24-6f9112da7d56/ienz_a_332035_f0005_b.gif)
In compound 4 (the least active of the four inhibitors) the protonated tertiary amino group is found as the reference inhibitor and the aliphatic chain is superimposed to the aliphatic chain of Ro 48-8071 (). The aliphatic chain of inhibitor 4 replaces the fluorophenyl ring and the umbelliferone motif is roughly located in the space occupied by the benzophenone ring as in compound 3. The main differences compared to compound 3 are in the umbelliferone position; in this case, an oxygen atom of umbelliferone lies near the water molecule interacting with Asp455, but the umbelliferone hydroxyl and the carbonyl belonging to the aliphatic chain lie in a hydrophobic region near the aromatic side chains of residues Phe696 and Phe103. Finally, the umbelliferone hydroxyl can form a hydrogen bond with the side chain of Asp455.
Figure 6. OSC residues interacting with the compound 4. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.
![Figure 6. OSC residues interacting with the compound 4. (in sticks) in the docking model. For comparison, the reference inhibitor Ro 48-8071 in the minimized model, as derived from X-ray [Citation7], is also reported.](/cms/asset/58b03b0b-df0e-4b7e-b43e-79711e085343/ienz_a_332035_f0006_b.gif)
In conclusion, the aliphatic portions of the compounds 1 and 2 preserve the key interactions present in the reference compound. The stacking with the side chains of Phe696 and His232, and the hydrogen bond with the water molecule bound to Ile338 are not precluded, though the occupation of the binding site is not optimal. In compound 1 the substituents of the nitrogen group can modify slightly the interaction pattern respect to the reference compound.
Inhibitors 3 and 4 reproduce the key interactions of the aliphatic portion of Ro 48-8071 but the length of their chains is responsible for the loss of π-π interactions with Phe696 and His232. Nevertheless, they fit better the space occupied by the bromophenyl moiety. Some drawbacks arise from the location of the umbelliferone oxygen atoms; in particular, in compound 4 the oxygen atoms in the umbelliferone ring and the hydroxyl groups are unfavourably located in a hydrophobic region that in the complex with Ro 48-8071 is occupied by the bromophenyl ring.
The docking results suggest for the umbelliferone moiety of inhibitor 4 an interaction pattern different from that of the other compounds, in accordance with the different activity (the lowest) of the compound, whereas compounds 1, 2 and 3 are able to reproduce, although partially, the key interactions of the reference compound with some drawbacks that justify a similar, though lower activity compared to the reference compound.
Discussion
A series of aminoalkyl umbelliferone derivatives were tested as inhibitors of mouse and human OSC. The study branches out from a more general project addressed to search for novel scaffold for designing new inhibitors of oxidosqualene cyclases. Most of the compounds of the present study proved to be good inhibitors of OSCs from Trypanosoma cruzi, Pneumocystis carinii and Saccharomyces cerevisiae, and have been also investigated as inhibitors of oxidosqualene-cycloartenol synthase from Arabidopsis thaliana [Citation9]. In this work we explored a novel scaffold aimed to design new cholesterol-lowering agents targeting cholesterol pathway beyond farnesylpyrophosphate formation. All the compounds shared a scaffold consisting of a coumarin nucleus bearing a side chain with a terminal tertiary amino function.
Inhibitors have been designed considering that the most known effective inhibitors of enzymes belonging to the family of squalene and oxidosqualene cyclases share a structure consisting in two well characterized moieties: a moiety bearing a properly shaped tertiary amino function capable of interacting with a catalytically critical Asp455 residue (human numbering) of the enzyme [Citation4,Citation32], and a moiety shaped so as to productively interact with the aromatic residues of the active site. The latter moiety is an isoprenic structure mimicking the squalene skeleton in azasqualene derivatives [Citation26,Citation33], whereas in other series of inhibitors is an electron-deficient aromatic system [Citation4]. Crystallographic studies carried on both squalene-hopene cyclase from Alicyclobacillus acidocaldarious and human OSC provided excellent explanations of the inhibitory properties of different compounds bearing an amino function [Citation4,Citation7,Citation32,Citation33].
As the inhibitors of the present study belong to the amino compounds containing an aromatic nucleus, they have been compared with Ro 48-8071, a known aromatic-type inhibitor of OSCs [Citation3].
The effect of the inhibitors on sterol biosynthesis appeared specifically addressed to lanosterol synthase both in human keratinocytes and in mouse 3T3 fibroblasts, as indicated by the accumulation of radioactive oxido- and dioxidosqualene in non-saponifiable lipids extracted from cells incubated with radiolabeled acetate in the presence of inhibitors. Keratinocytes and fibroblasts are both highly susceptible to inhibitor 3, whereas the effect caused by the inhibitor 4 was negligible. Two significant differences in the response to inhibitors between human keratinocytes and mouse 3T3 fibroblasts were observed: (i) the ratio between radiolabeled dioxido- and oxidosqualene is generally higher in keratinocytes than in fibroblasts and (ii) in fibroblasts the most active compounds were 3 and 2 whereas in keratinocytes the most active compounds were 3 and 1. The former difference could result from a different metabolism in two types of cells: squalene monoepoxide, accumulated following the inhibition of OSC, might be a better substrate for a second epoxidation by squalene epoxidase in keratinocytes. The latter difference may depend on a subtle difference in the active site cavity between human and murine enzyme/s. Docking experiments could not exhaustively explain the above difference as only structural data from human enzyme are available. What distinctly emerges from docking models of inhibitor-enzyme complexes is that the aliphatic chains of 1, 2 and 3, differing in length and type of amino group, are hosted by human OSC active site in a similar fashion as that of Ro 48-8071, and the amino group, whether dimethyl- or methylallyl substituted, is able to form an ionic bond with the catalytic residue Asp455.
All the other 7-aminoalkyl coumarin derivatives, bearing a more rigid side chain (5) or a cyclic amino group (6, 7 and 8), proved to be less effective than compounds 1, 2 and 3 as inhibitors of OSC in cultures of murine fibroblasts. They were also poorly effective on homogenates prepared from yeast cells expressing human OSC [Citation9]. Thus, the main route to design effective coumarin-type inhibitors of human OSC appeared traced out from our study: a good inhibitor consists of a coumarin nucleus bearing a 7-linked hexyl- to decyl-carbon flexible chain with a N,N-dimethyl- or a N,N-methylallylamino terminal group. Studies are in progress to further optimise this scaffold. Substituting amino groups with the corresponding N-oxides in 1, 2, 4 and 5 (an alteration already explored for compound 3 [Citation9]), and increasing the electron-deficiency of the coumarin nucleus are among the future alterations designed for this series of compounds.
Acknowledgements
We thank the University of Turin, the regional government (Regione Piemonte) and the MIUR (PRIN 2004; prot. 2004037895) the for financial support of this research.
The authors are very grateful to Dr. H. Dehmlow (Hoffmann-La Roche, Basel, Switzerland) for a generous gift of Ro 48-8071. Thanks are due to Professor Seiichi Matsuda (Rice University Houston, Texas-USA) for providing the clone pOTB7 containing human OSC cDNA and the lanosterol synthase mutant SMY8.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- ES Istvan, and J Deisenhofer. (2001). Structural mechanism for statin inhibition of HMG-CoA reductase. Science 292:1160–1164.
- A Chugh, A Ray, and JB Gupta. (2003). Squalene epoxidase as hypocholesterolemic drug target revisited. Prog Lipid Res 42:37–50.
- OH Morand, JD Aebi, H Dehmlow, YH Ji, N Gains, H Langsfeld, and J Himber. (1997). Ro48-8071, a new 2,3-oxidosqualene:lanosterol cyclase inhibitor lowering plasma cholesterol in hamsters, squirrel monkeys, and minipigs: comparison to simvastatin. J Lipid Res 38:373–390.
- H Dehmlow, JD Aebi, S Jolidon, YH Ji, EM von der Mark, J Himber, and OH Morand. (2003). Synthesis and structure-activity studies of novel orally active non-terpenoic 2,3-oxidosqualene cyclase inhibitors. J Med Chem 46:3354–3370.
- MW Huff, and DE Telford. (2005). Lords of the rings – the mechanism for oxidosqualene:lanosterol cyclase becomes crystal clear. Trends Pharmacol Sci 26:335–340.
- AH Rowe, CA Argmann, JY Edwards, CG Sawyez, OH Morand, RA Hegele, and MW Huff. (2003). Enhanced synthesis of the oxysterol 24(S),25-epoxycholesterol in macrophages by inhibitors of 2,3-oxidosqualene:lanosterol cyclase: a novel mechanism for the attenuation of foam cell formation. Circ Res 93:717–725.
- R Thoma, T Schulz-Gasch, B D'Arcy, J Benz, J Aebi, H Dehmlow, M Hennig, M Stihle, and A Ruf. (2004). Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature 432:118–122.
- S Oliaro-Bosso, F Viola, SPT Matsuda, G Cravotto, S Tagliapietra, and G Balliano. (2004). Umbelliferone aminoalkyl derivatives as inhibitors of oxidosqualene cyclases from Saccharomyces cerevisiae, Trypanosoma cruzi and Pneumocystis carinii. Lipids 39:1007–1012.
- S Oliaro-Bosso, F Viola, S Taramino, S Tagliapietra, A Barge, G Cravotto, and G Balliano. (2007). Inhibitory effect of umbelliferone aminoalkyl derivatives on oxidosqualene cyclases from Saccharomyces cerevisiae, Trypanosoma cruzi, Pneumocystis carinii, Homo sapiens and Arabidopsis thaliana: a structure-activity study. Chem Med Chem 2:226–233.
- M Ceruti, G Balliano, F Viola, L Cattel, N Gerst, and F Schuber. (1987). Synthesis and biological activity of azasqualenes, bis-azasqualenes and derivatives. Eur J Med Chem 22:199–208.
- G Cravotto, G Balliano, S Tagliapietra, S Oliaro-Bosso, and GM Nano. (2004). Novel squalene-hopene cyclase inhibitors derived from hydroxycoumarins and hydroxyacetophenones. Chem Pharm Bull 52:1171–1174.
- G Cravotto, G Balliano, S Tagliapietra, G Palmisano, and A Penoni. (2004). Umbelliferone aminoalkyl derivatives, a new class of squalene-hopene cyclase inhibitors. Eur J Med Chem 39:917–924.
- M Ceruti, G Balliano, F Rocco, P Milla, S Arpicco, L Cattel, and F Viola. (2001). Vinyl sulfide derivatives of truncated oxidosqualene as selective inhibitors of oxidosqualene and squalene hopene cyclases. Lipids 36:629–636.
- G Popják. Steroids and terpenoid. In. RB Clayton, editor. Methods in enzymology. Vol. XV New York: Academic Press; (1969) p 438–443.
- RB Field, CE Holmund, and NF Whittake. (1979). The effects of the hypocholesteremic compound 3 beta-(beta-dimethylaminoethoxy)-androst-5-en-17-one on the sterol and steryl ester composition of Saccharomyces cerevisiae. Lipids 14:741–747.
- EJ Corey, SPT Matsuda, CH Baker, AY Ting, and H Cheng. (1996). Molecular cloning of a Schizosaccharomyces pombe cDNA encoding lanosterol synthase and investigation of conserved tryptophan residues. Biochem Biophys Res Commun 219:327–331.
- FM Ausubel, R Brent, RE Kingston, DD Moore, JD Seidman, J Smith, and K Struhl. Current protocols in molecular biology. NY: Wiley-Interscience; (1999).
- GL Peterson. (1977). A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83:346–356.
- RG Duggleby. (1981). A nonlinear regression program for small computers. Anal Biochem 110:9–18.
- BE Chan, and BR Knowles. (1976). A solvent system for delipidation of plasma or serum without protein precipitation. J Lipid Res 17:176–181.
- HM Berman, J Westbrook, Z Feng, G Gilliland, TN Bhat, H Weissig, IN Shindyalov, and PE Bourne. (2000). The protein data bank. Nucleic Acids Res 28:235–242.
- P Labute, C Williams, M Feher, E Sourial, and JM Schmidt. (2001). Flexible alignment of small molecules. J Med Chem 44:1483–1490.
- G Ermondi, M Lorenti, and G Caron. (2004). Contribution of ionization and lipophilicity to drug binding to albumin: a preliminary step toward biodistribution prediction. J Med Chem 47:3949–3961.
- TA Halgren. (1996). Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490–519.
- IT Christensen, and FS Jorgensen. (1997). Molecular mechanics calculations of proteins. Comparison of different energy minimization strategies. J Biomol Struct Dyn 5:473–488.
- F Viola, P Brusa, G Balliano, M Ceruti, O Boutaud, F Schuber, and L Cattel. (1995). Inhibition of 2,3-oxidosqualene cyclase and sterol biosynthesis by 10- and 19-azasqualene derivatives. Biochem Pharmacol 50:787–796.
- U Galli, S Oliaro-Bosso, S Taramino, S Venegoni, E Pastore, GC Tron, G Balliano, F Viola, and G Sorba. (2007). Design, synthesis and biological evaluation of new (2E,6E)-10-(dimethylamino)-3,7-dimethyl-2,6-decadien-1-ol ethers as inhibitors of human and Trypanosoma cruzi oxidosqualene cyclase. Biorg Med Chem Letters 17:220–224.
- CH Baker, SPT Matsuda, DR Liu, and EJ Corey. (1995). Molecular cloning of the human gene encoding lanosterol synthase from a liver cDNA library. Biochem Biophys Res Commun 213:154–160.
- CK Sung, M Shibuya, U Sankawa, and Y Ebizuka. (1995). Molecular cloning of cDNA encoding human lanosterol synthase. Biol Pharm Bull 18:1459–1461.
- E Roessler, L Mittaz, Y Du, HS Scott, J Chang, C Rossier, M Guipponi, SPT Matsuda, M Muenke, and SE Antonarakis. (1999). Structure of the human lanosterol synthase gene and its analysis as a candidate for holoprosencephaly (HPE1). Hum Genet 105:489–495.
- F Viola, M Ceruti, G Balliano, O Caputo, and L Cattel. (1990). 22,23-epoxy-2-aza-2,3-dihydrosqualene derivatives: potent new inhibitors of squalene 2,3-oxide-lanosterol cyclase. Il Farmaco 45:965–978.
- A Lenhart, DJ Reinert, JD Aebi, H Dehmlow, OH Morand, and GE Schulz. (2003). Binding structures and potencies of OSC inhibitors with the homologous squalene-hopene cyclase. J Med Chem 46:2083–2092.
- DJ Reinert, G Balliano, and GE Schulz. (2004). Conversion of squalene to the pentacarbocyclic hopene. Chem Biol 11:121–126.