Abstract
Selective PDE3 inhibitors improve cardiac contractility and may be used in congestive heart failure. However, their proarrhythmic potential is the most important side effect. In this research we designed, synthesized and evaluated the potential cardiotonic activity of thirteen PDE3 inhibitors (4-[(4-methyl-2-oxo-1,2-dihydro-6-quinolinyl)oxy]butanamide analogs) using the spontaneously beating atria model. The design strategy was based on the structure of cilostamide, a selective PDE3 inhibitor. In each experiment, atrium of reserpine-treated rat was isolated and the contractile and chronotropic effects of a synthetic compounds were assessed. All experiments were carried out in comparison with IBMX, amrinone and cilostamide as standard compounds. The results showed that, among the new compounds, the best pharmacological profile was obtained with the compound 6-[4-(4-methylpiperazine-1-yl)-4-oxobutoxy]-4-methylquinolin-2(1H)-one, 4j, which displayed selectivity for increasing the force of contraction (165 ± 4% change over the control) rather than the frequency rate (115 ± 7% change over the control) at 100 μM and potent inhibitory activity of PDE3 with IC50 = 0.20 μM.
Introduction
Congestive heart failure (CHF) is a major cause of death in patients with heart disease. For many years, digitalis glycosides have been used for the treatment of CHF. However, the application of these agents is limited because of their narrow therapeutic window and their propensity to cause life-threatening arrhythmias (arrhythmogenic lability). The search for orally active ‘non-glycoside’ cardiotonic drugs displaying a greater safety profile and improved efficacy on patient survival resulted in establishing the selective inhibitors of cyclic nucleotide phosphodiesterase (PDE) enzymes as a new class of cardiotonic agents.
PDE enzymes specifically hydrolyze cAMP (cyclic adenosine monophosphate) and cGMP (cyclic guanosine monophosphate) in the cells. On the basis of their amino-acid sequence homology, biochemical properties and inhibitor profiles, 11 PDE families have been recognized in mammalian tissues, PDE1, PDE2 to PDE11. Each PDE isozyme has a C-terminal catalytic domain conserved throughout the family, and an N-terminal regulatory domain unique for each isozyme. They are expressed in tissue and cell-specific distribution patterns, and they show different substrate affinities and inhibitor sensitivities [Citation1]. Most cell types express one or more PDE isozymes, each regulating intracellular cAMP and/or cGMP concentrations in different cellular compartments and in different manners. In cardiovascular tissues, PDE3 and PDE4 are well established as the dominant cAMP-hydrolysis isozymes [Citation2]. PDE1, PDE3, PDE4, and PDE5 are expressed in aortic smooth muscle cells [Citation3,Citation4]; PDE1, PDE2, PDE3, and PDE4 are expressed in the heart [Citation5], whereas PDE2, PDE3, and PDE5 are found in platelets [Citation2,Citation6,Citation7]. PDE3 is thus a unique cAMP-regulating isozyme expressed in all of mentioned tissues. These tissues contribute significantly to the pathogenesis of arteriosclerosis obliterans and restenosis after angioplasty. The inhibition of PDE3 activity in cardiovascular tissues results in increased levels of cAMP with consequent reduction in platelet aggregation and smooth muscle cell proliferation in vitro, and induction of a cardiotonic effect [Citation7,Citation8]. The PDE3 subfamily consists of two closely related subtypes: PDE3A and PDE3B. PDE3A is mostly expressed in cardiac tissue, platelets, and vascular smooth muscle cells, PDE3B is prevalently expressed in hepatocytes and adipose tissue [Citation9,Citation10].
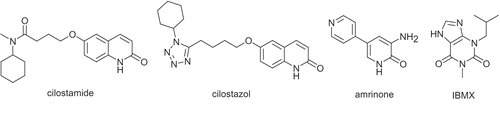
In a rat balloon-induced injury model of intimal hyperplasia, oral administration of cilostamide (OPC-3689), has been found to suppress the intimal proliferation [Citation11]. Recently, it was reported that local administration of the cardiotonic PDE3 inhibitor amrinone also inhibits neointima formation in this model [Citation12]. Therefore, PDE3 inhibitors may possess therapeutic potential for ischemic disease involving thrombosis and vascular smooth muscle cell proliferation. Indeed, cilostazol (OPC-13013) has been available clinically for a long time as a vasodilator and anti-platelet drug () [Citation13–15]. Cilostamide (potent and selective PDE3 inhibitors: IC50 = 70 nM [Citation16,Citation17]; IC50 (PDE3A) = 27 nM and IC50 (PDE3B) = 50 nM [Citation18]) possesses both antithrombotic and antiintimal hyperplastic actions [Citation7,Citation11,Citation19]. In summary, cilostamide is a highly functionalized compound which shows potent activity of PDE3 inhibitory [Citation3,Citation16,Citation20,Citation21] lead to cardiotonic effects [Citation22], inhibition of platelet aggregation [Citation7,Citation18,Citation23] and increase in the secretion of Insulin-stimulated glucose [Citation24]. Unfortunately, this compound shows the side effect of tachycardia [Citation19]. Since last three decades, too many related analogs of cilostamide have been synthesized and biologically evaluated [Citation19,Citation21,Citation25,Citation26].
Figure 1. Chemical structure of some PDE inhibitors.
Despite considerable research efforts towards providing insights into PDE3-ligand interactions [Citation27–30], progress in this area still relies heavily on information on Structure Activity Relationships (SAR) that is collected from identification of new ligands that were discovered by trial and error. Therefore, we designed and synthesized a series of new cardiotonic agents on the basis of structure of cilostamide using a computerized study of PDE3-ligand interaction.
In order to give further proof to the mechanism of action of designed inhibitors, molecular models of the complex enzyme-inhibitor were generated for both ligands and cilostamide, using PDE3B co-crystalized with MERCK1 [Citation30] and the site-directed mutagenesis data available for PDE3A and PDE3B [Citation28, Citation31–34].
There is reasonable homology between the PDE3A and PDE3B (identities: 48%, positives: 67%, extracted from NCBI-BLAST [Citation35–37]). This homology increases (∼95%) within 15å in the active site pocket ().
Figure 2. Multiple alignment of PDE3A (blue) and PDE3B (green). The residues in the active site of PDE3B (within 15å) are highlighted in yellow (generated by Clustal X (1.81)).

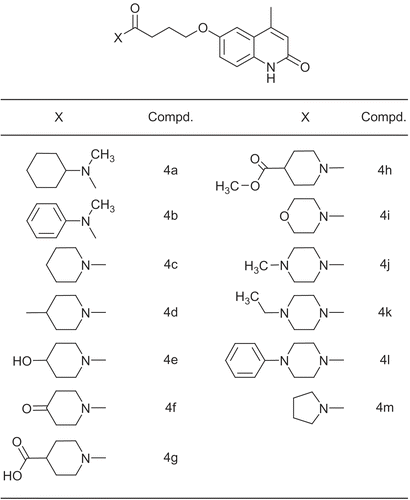
The lactamic portion of cilostamide was confirmed by Scapin et al as the primary binding moiety and as common anchor point [Citation30]. It displayed hydrogen bond with the amide group of Gln988 side chain in PDE3B while the N-cyclohexyl-N-methylbutanamide moiety, as the second binding part of cilostamide interact with pocket formed by residues: Tyr736, His737, His825, Thr829, Leu895, Ile938, Ile955, Glu958, Phe959, Leu987 and Phe991. As it will be explained later and in order to obtain new cilostamide analogues with a better cardiotonic activity, we planned the synthesis of 4-methyl substituted of 6-hydroxycarbostiryl moiety with changing N-cyclohexyl-N-methylamine into piperidine and piperazine derivatives (4a-m, ). Then, the cardiac activities of synthetic compounds were measured in the rat isolated atria preparations. All experiments were carried out in comparison with IBMX, a non-selective PDE inhibitor, amrinone and cilostamide, selective PDE3 inhibitors.
Figure 3. The structure of amide moiety of compounds 4a-m.
Materials and methods
Animals, drugs, chemicals and instruments
Adult male Wistar rats (250–350g), obtained from animal house of Mashhad medical school, were kept in controlled environmental conditions (temperature: 23 ± 2˚C; light–dark cycle: 7 am to 7 pm). Animals had free access to a standard laboratory diet and water. In order to obtain atrial preparation, depleted in endogenous catecholamine, the animals were treated intraperitoneally with reserpine (5 mg/kg b.wt) 24h before euthanasia [Citation44]. In each experiment, the animals were anaesthetized with intraperitoneal injection of thiopental 80 mg/kg, and after midline thoracotomy, the heart was rapidly excised and placed in a dissection dish filled with oxygenated Krebs-Henseleit solution.
Reserpine, tyramine HCl, (–)isoprenaline HCl, amrinone, were bought from Sigma Chemical Co. (St. Louis, MO), and 3-isobutyl-1-methylxanthine was provided from Fluka Chemical Co. All other chemicals obtained from Merck Chemical Co.
Melting points were recorded on an Electrothermal type 9100 melting point apparatus. 1H NMR (500 MHz) were obtained by using a Bruker Avance DRX-500 Fourier transformer spectrometer. Chemical shifts are reported in ppm (δ) downfield from tetramethylsilane (TMS). Electron impact (EI) mass spectra were recorded on a Varian Match 7A spectrometer. The IR spectra were obtained on a 4300 Shimadzu Spectrometer. Elemental analysis was obtained on a Thermo Finnigan Flash EA microanalyzer. The developed tension was recorded isometrically by means of a high-sensitivity force transducer (type E. Zimmermann, Eipzig-Berlin) connected to powerlab 8/30 (model 870).
Chemistry
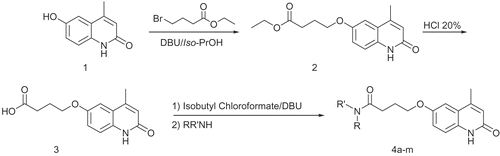
The synthesis of 6-hydroxy-4-methylquinolin-2(1H)-one (1) was reported in last literatures [Citation45,Citation46]. Condensation of 1 with ethyl-4-bromobutyrate in the presence of 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) afforded the corresponding ester 2 [Citation25]. Hydrolysis of 2 with HCl 20% gave the corresponding carboxylic acid 3. Various amide derivatives 4a-m were prepared from various secondary amines and an intermediate acid chloride which was formed from reacting of acid 3 and isobutyl chloroformate in the presence of DBU [Citation25] (). Cilostamide were prepared according to the method reported in previously published work [Citation21].
Scheme 1. General procedure for the synthesis of compounds 4a-m.
Ethyl 4-(1,2-dihydro-4-methyl-2-oxoquinolin-6-yloxy)butanoate 2
To a solution of 5 g (0.027 mol) of 6-hydroxy-4-methylquinolin-2(1H)-one 1 and 6 g of DBU in 80 ML of isopropyl alcohol was added ethyl 4-bromobutyrate (6.5 g, 0.033 mol) dropwise. The mixture was refluxed for 5 h, the solvent was evaporated off, and the residue was extracted with chloroform. After removal of the solvent, the residue was recrystallized from methanol to give 2 as colorless needles (6.1 g, 78%).
White crystal, mp: 159-160 °C; 1HNMR (DMSOd6): δ 1.19 (t, J = 7.1 Hz, 3H, CH3CH2O2C-), 1.99 (m, 2H, -OCH2CH2CH2CO2-), 2.41 (s, 3H, -CH3), 2.48 (t, J = 7.3 Hz, 2H, -CH2CO2-), 4.05 (t, J = 6.3 Hz, 2H, -CH2O-), 4.08 (q, J = 7.2 Hz, 2H, CH3CH2O2C-), 6.39 (s, 1H, H-3), 7.13 (d, J = 2.6 Hz, 1H, H-5), 7.16 (dd, J = 8.8 Hz, 2.6 Hz, 1H, H-7), 7.24 (d, J = 8.8 Hz, 1H, H-8), 11.48 (s, 1H, NHCO); MS m/z: 289 (M+), 115 (100%); Found: C, 66.37; H, 6.71; N, 4.87. C16H19NO4 requires: C, 66.42; H, 6.62; N, 4.84%.
4-(1,2-dihydro-4-methyl-2-oxoquinolin-6-yloxy)butanoic acid 3
A suspention of 3c (6 g, 0.021 mol) in 60 ML of 20% HCl was stirred at 85-90˚C for 2 h, and then cooled down to room temperature. The precipitated crystals were collected, and washed with water. The crystals were recrystallized from DMF-water, oven dried gave 3 (5.1 g, 93%,).
White solid, mp: 265-266 ˚C; 1HNMR (DMSO-d6): δ 1.96 (m, 2H, -OCH2CH2CH2CO2-), 2.41 (t, J = 7.2 Hz, 2H, -CH2CO2-), 2.42 (s, 3H, -CH3), 4.05 (t, J = 6.4 Hz, 2H, -CH2O-), 5.5-6.3 (br, 1H, -COOH), 6.42 (s, 1H, H-3), 7.16 (d, J = 2.1 Hz, 1H, H-5), 7.18 (dd, J = 8.5 Hz, 2.1 Hz, 1H, H-7), 7.27 (d, J = 8.5 Hz, 1H, H-8), 11.62 (s, 1H, NHCO); MS m/z: 261 (M+), 174 (100%); Found: C, 64.37; H, 5.71; N, 5.39. C14H15NO4 requires: C, 64.36; H, 5.79; N, 5.36%.
General procedure for the synthesis of 4a-m
Isobuthyl chloroformate (1.5 g, 0.011 mol) was added dropwise to a solution of 3 (0.010 mol) and 1.7 g of DBU in 50 ML chloroform while stirring in ice-water. After removing the ice-bath, the reaction mixture was stirred at room temperature for 1 h. the required amine (0.010 mol) was then added dropwise and stirring continued for 3 h. The resulting solution was washed with 0.5 N NaOH (2 × 50 ML), dilute HCl (2 × 50 ML) and water (2 × 50 ML). The organic layer was dried over sodium sulfate. After removing of the solvent under reduced pressure, the residue was recrystallized from acetone to give 4a-m:
4-(1,2-dihydro-4-methyl-2-oxoquinolin-6-yloxy)-N- cyclohexyl-N-methylbutanamide 4a:
White solid, mp: 132-133 °C; 1HNMR (CDCl3): δ 1.1-1.88 (m, 10H, -CH2- (cyclohexyl)), 2.22 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.56 (t, J = 7 Hz, 1H, -CH2CO-), 2.62 (t, J = 7 Hz, 1H, -CH2CO-), 2.86 (s, 1.5H, NCH3), 2.89 (s, 1.5H, NCH3), 3.64 (m, 0.5H, NCH (cyclohexyl)), 4.15 (t, J = 5.7 Hz, 2H, -CH2O-), 4.50 (m, 0.5H, NCH (cyclohexyl)), 6.62 (s, 1H, H-3), 7.15 (d, J = 1.9 Hz, 1H, H-5), 7.19 (dd, J = 7.5 Hz, 1.9 Hz, 1H, H-7), 7.40 (d, J = 7.5 Hz, 1H, H-8), 11.62 (br, 1H, NHCO); MS m/z: 356 (M+), 156 (100%); Found: C, 70.84; H, 8.01; N, 7.91. C21H28N2O3 requires: C, 70.76; H, 7.92; N, 7.86%.
4-(1,2-dihydro-4-methyl-2-oxoquinolin-6-yloxy)-N-methyl-N-phenylbutanamide 4b:
White crystal, mp: 164-165 ˚C; 1HNMR (CDCl3): δ 2.14 (m, 2H, -OCH2CH2CH2CO-), 2.34 (t, J = 6.8 Hz, 2H, -CH2CO-), 2.51 (s, 3H, -CH3), 3.32 (s, 3H, NCH3), 4.05 (t, J = 5.8 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.06 (d, J = 1.9 Hz, 1H, H-5), 7.08 (dd, J = 7.5 Hz, 1.9 Hz, 1H, H-7), 7.19 (d, J = 7.5 Hz, 1H, H-8), 7.36-7.45 (m, 5H, CH (phenyl)), 12.40 (br, 1H, NHCO); MS m/z: 350 (M+), 176 (100%); Found: C, 71.84; H, 6.31; N, 7.93. C21H22N2O3 requires: C, 71.98; H, 6.33; N, 7.99%.
6-(4-(piperidin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4c:
White crystal, mp: 197-198˚C; 1HNMR (CDCl3): δ 1.59 (m, 4H, -CH2CH2CH2- (piperidine)), 1.66 (m, 2H, -CH2CH2CH2- (piperidine)), 2.21 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.59 (t, J = 7.1 Hz, 2H, -CH2CO-), 3.47 (t, J = 5.4 Hz, 2H, -CH2NCOCH2- (piperidine)), 3.61 (t, J = 4.8 Hz, 2H, -CH2NCOCH2- (piperidine)), 4.14 (t, J = 5.9 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.13 (d, J = 2.5 Hz, 1H, H-5), 7.19 (dd, J = 8.9 Hz, 2.5 Hz, 1H, H-7), 7.39 (d, J = 8.9 Hz, 1H, H-8), 12.26 (s, 1H, NHCO); MS m/z: 328 (M+), 154 (100%); Found: C, 69.41; H, 7.39; N, 8.36. C19H24N2O3 requires: C, 69.49; H, 7.37; N, 8.53%.
6-(4-(4-methylpiperidin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4d:
White crystal, mp: 166-167 °C; 1HNMR (CDCl3): δ 0.97 (d, J = 6.5, 3H, CH3- (piperidine)), 1.12 (m, 2H, -CH2CHCH2- (piperidine)), 1.62 (m, 1H, -CH2CHCH2- (piperidine)), 1.71 (m, 2H, -CH2CHCH2- (piperidine)), 2.20 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.59 (t, J = 7.0 Hz, 2H, -CH2CO-), 2.60 (m, 1H, -CH2NCOCH2- (piperidine)), 3.02 (m, 1H, -CH2NCOCH2- (piperidine)), 3.90 (m, 1H, -CH2NCOCH2- (piperidine)), 4.13 (t, J = 6.0 Hz, 2H, -CH2O-), 4.63 (m, 1H, -CH2NCOCH2- (piperidine)), 6.63 (s, 1H, H-3), 7.12 (d, J = 2.6 Hz, 1H, H-5), 7.18 (dd, J = 8.9 Hz, 2.6 Hz, 1H, H-7), 7.42 (d, J = 8.9 Hz, 1H, H-8), 12.59 (s, 1H, NHCO); MS m/z: 342 (M+), 168 (100%); Found: C, 69.91; H, 7.59; N, 8.23. C20H26N2O3 requires: C, 70.15; H, 7.65; N, 8.18%.
6-(4-(4-hydroxypiperidin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4e:
White crystal, mp: 205-206 °C; 1HNMR (DMSOd6): δ 1.25 (m, 1H, -CH2CHCH2- (piperidine)), 1.31 (m, 1H, -CH2CHCH2- (piperidine)), 1.66-1.73 (m, 2H, -CH2CHCH2- (piperidine)), 1.95 (m, 2H, -OCH2CH2CH2CO-), 2.40 (s, 3H, -CH3), 2.48 (t, J = 7.3 Hz, 2H, -CH2CO-), 3.00 (m, 1H, -CH2NCOCH2- (piperidine)), 3.15 (m, 1H, -CH2NCOCH2- (piperidine)), 3.68 (m, 2H, -CH2NCOCH2- (piperidine)), 3.92 (m, 1H, -CHOH- (piperidine)), 4.04 (t, J = 6.4 Hz, 2H, -CH2O-), 4.73 (s, 1H, -OH), 6.39 (s, 1H, H-3), 7.14 (d, J = 2.6 Hz, 1H, H-5), 7.17 (dd, J = 8.8 Hz, 2.6 Hz, 1H, H-7), 7.24 (d, J = 8.8 Hz, 1H, H-8), 11.47 (s, 1H, NHCO); MS m/z: 344 (M+), 170 (100%); Found: C, 66.40; H, 7.09; N, 8.11. C19H24N2O4 requires: C, 66.26; H, 7.02; N, 8.13%.
6-(4-(4-piperidon-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4f:
White crystal, mp: 198-199 °C; 1HNMR (DMSOd6): δ 2.00 (m, 2H, -OCH2CH2CH2CO-), 2.35 (t, J = 6.1, -CH2COCH2- (piperidone)), 2.41 (s, 3H, -CH3), 2.43 (t, 2H, J = 6.0, -CH2COCH2- (piperidine)), 2.60 (t, J = 7.2 Hz, 2H, -CH2CO-), 3.75 (t, 4H, J = 6.2 -CH2NCOCH2- (piperidone)), 4.07 (t, J = 6.4 Hz, 2H, -CH2O-), 6.39 (s, 1H, H-3), 7.15 (d, J = 2.5 Hz, 1H, H-5), 7.18 (dd, J = 8.8 Hz, 2.5 Hz, 1H, H-7), 7.25 (d, J = 8.8 Hz, 1H, H-8), 11.46 (s, 1H, NHCO); MS m/z: 342 (M+), 168 (100%); Found: C, 66.47; H, 6.41; N, 8.15 C19H22N2O4 requires: C, 66.65; H, 6.48; N, 8.18%.
6-(4-(4-piperidincarboxylic-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4g:
White crystal, mp: 201-202 °C; 1HNMR (DMSOd6): δ 1.36 (m, 1H, -CH2CHCH2- (piperidine)), 1.47 (m, 1H, -CH2CHCH2- (piperidine)), 1.82 (m, 2H, -CH2CHCH2- (piperidine)), 1.95 (m, 2H, -OCH2CH2CH2CO-), 2.41 (s, 3H, -CH3), 2.47 (m, 1H, -CH2CHCH2- (piperidine)), 2.49 (t, J = 7.2 Hz, 2H, -CH2CO-), 2.72 (m, 1H, -CH2NCOCH2- (piperidine)), 3.01 (m, 1H, -CH2NCOCH2- (piperidine)), 3.81 (m, 1H, -CH2NCOCH2- (piperidine)), 4.04 (t, J = 6.4 Hz, 2H, -CH2O-), 4.23 (m, 1H, -CH2NCOCH2- (piperidine)), 6.39 (s, 1H, H-3), 7.14 (d, J = 2.5 Hz, 1H, H-5), 7.17 (dd, J = 8.8 Hz, 2.5 Hz, 1H, H-7), 7.24 (d, J = 8.8 Hz, 1H, H-8), 11.47 (s, 1H, NHCO), 12.23 (s, 1H, COOH); MS m/z: 372 (M+), 198 (100%); Found: C, 64.63; H, 6.44; N, 7.60. C20H24N2O5 requires: C, 64.50; H, 6.50; N, 7.52%.
6-(4-(4-methylpiperidincarboxylate-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4h:
White crystal, mp: 147-148 °C; 1HNMR (CDCl3): δ 1.69 (m, 2H, -CH2CHCH2- (piperidine)), 1.98 (m, 2H, -CH2CHCH2- (piperidine)), 2.21 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.57 (m, 1H, -CH2CHCH2- (piperidine)), 2.60 (t, J = 7.2 Hz, 2H, -CH2CO-), 2.87 (m, 1H, -CH2NCOCH2- (piperidine)), 3.17 (m, 1H, -CH2NCOCH2- (piperidine)), 3.73 (s, 3H, CH3OOC), 3.91 (m, 1H, -CH2NCOCH2- (piperidine)), 4.13 (t, J = 5.9 Hz, 2H, -CH2O-), 4.47 (m, 1H, -CH2NCOCH2- (piperidine)), 6.63 (s, 1H, H-3), 7.13 (d, J = 2.6 Hz, 1H, H-5), 7.18 (dd, J = 8.9 Hz, 2.6 Hz, 1H, H-7), 7.38 (d, J = 8.9 Hz, 1H, H-8), 12.09 (s, 1H, NHCO; MS m/z: 386 (M+), 212 (100%); Found: C, 65.41; H, 6.83; N, 7.30. C21H26N2O5 requires: C, 65.27; H, 6.78; N, 7.25%.
6-(4-morpholino-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4i:
White crystal, mp: 209-210 °C; 1HNMR (CDCl3): δ 2.22 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.60 (t, J = 7.1 Hz, 2H, -CH2CO-), 3.54 (m, 4H, -CH2NCOCH2- (morpholine)), 3.69 (m, 6H, -CH2NCOCH2- & -CH2OCH2- (morpholine)), 4.14 (t, J = 5.9 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.12 (d, J = 2.6 Hz, 1H, H-5), 7.18 (dd, J = 8.9 Hz, 2.6 Hz, 1H, H-7), 7.41 (d, J = 8.9 Hz, 1H, H-8), 12.42 (s, 1H, NHCO); MS m/z: 330 (M+), 156 (100%); Found: C, 65.67; H, 6.69; N, 8.41. C18H22N2O4 requires: C, 65.44; H, 6.71; N, 8.48%.
6-(4-(4-methylpiperazin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4j:
White crystal, mp: 181-182 °C; 1HNMR (CDCl3): δ 2.21 (m, 2H, -OCH2CH2CH2CO-), 2.32 (s, 3H, -NCH3), 2.41 (dd, J = 9.5 Hz, 4.8 Hz, 4H, -CH2NCH2- (piperazine)), 2.52 (s, 3H, -CH3), 2.60 (t, J = 7.1 Hz, 2H, -CH2CO-), 3.54 (t, J = 4.8 Hz, 2H, -CH2NCOCH2- (piperazine)), 3.69 (t, J = 4.8 Hz, 2H, -CH2NCOCH2- (piperazine)), 4.13 (t, J = 5.9 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.12 (d, J = 2.5 Hz, 1H, H-5), 7.17 (dd, J = 8.9 Hz, 2.5 Hz, 1H, H-7), 7.41 (d, J = 8.9 Hz, 1H, H-8), 12.47 (s, 1H, NHCO); MS m/z: 343 (M+), 169 (100%); Found: C, 66.41; H, 7.39; N, 12.31. C19H25N3O3 requires: C, 66.45; H, 7.34; N, 12.24%.
6-(4-(4-ethylpiperazin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4k:
White crystal, mp: 173-174 °C; 1HNMR (CDCl3): δ 1.12 (t, 3H, J = 7.2 Hz, CH3CH2- ) 2.21 (m, 2H, -OCH2CH2CH2CO-), 2.43-2.48 (m, 6H, -NCH3 & -CH2NCH2- (piperazine)), 2.52 (s, 3H, -CH3), 2.60 (t, J = 7.2 Hz, 2H, -CH2CO-), 3.56 (t, J = 4.9 Hz, 2H, -CH2NCOCH2- (piperazine)), 3.70 (t, J = 5.0 Hz, 2H, -CH2NCOCH2- (piperazine)), 4.14 (t, J = 5.9 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.13 (d, J = 2.6 Hz, 1H, H-5), 7.17 (dd, J = 8.9 Hz, 2.6 Hz, 1H, H-7), 7.41 (d, J = 8.9 Hz, 1H, H-8), 12.31 (s, 1H, NHCO); MS m/z: 357 (M+), 183 (100%): Found: C, 66.98; H, 7.63 N, 11.69. C20H27N3O3 requires: C, 67.20; H, 7.61; N, 11.76%.
6-(4-(4-phenylpiperazin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4l:
White crystal, mp: 163-164 °C; 1HNMR (CDCl3): δ 2.24 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.66 (t, J = 7.1 Hz, 2H, -CH2CO-), 3.20 (m, 4H, -CH2NCH2- (piperazine)), 3.70 (t, J = 5.0 Hz, 2H, -CH2NCOCH2- (piperazine)), 3.84 (t, J = 5.0 Hz, 2H, -CH2NCOCH2- (piperazine)), 4.16 (t, J = 5.8 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.14 (d, J = 2.6 Hz, 1H, H-5), 7.19 (dd, J = 8.9 Hz, 2.6 Hz, 1H, H-7), 7.32 (m, 5H, Ph-), 7.39 (d, J = 8.9 Hz, 1H, H-8), 12.13 (s, 1H, NHCO); MS m/z: 405 (M+), 231 (100%); Found: C, 71.22; H, 6.63; N, 10.29. C24H27N3O3 requires: C, 71.09; H, 6.71; N, 10.36%.
6-(4-(pyrrolidin-1-yl)-4-oxobutoxy)-4-methylquinolin-2(1H)-one 4m:
White crystal, mp: 213-214 °C; 1HNMR (CDCl3): δ 1.89 (m, 2H, -CH2CH2- (pyrrolidine)), 1.98 (m, 2H, -CH2CH2- (pyrrolidine)), 2.23 (m, 2H, -OCH2CH2CH2CO-), 2.52 (s, 3H, -CH3), 2.54 (t, J = 7.1 Hz, 2H, -CH2CO-), 3.47 (t, J = 6.9 Hz, 2H, -CH2NCOCH2- (pyrrolidine)), 3.52 (t, J = 6.9 Hz, 2H, -CH2NCOCH2- (pyrrolidine)), 4.15 (t, J = 6.0 Hz, 2H, -CH2O-), 6.63 (s, 1H, H-3), 7.13 (d, J = 2.6 Hz, 1H, H-5), 7.18 (dd, J = 8.9 Hz, 2.6 Hz, 1H, H-7), 7.39 (d, J = 8.9 Hz, 1H, H-8), 11.84 (s, 1H, NHCO); MS m/z: 314 (M+), 140 (100%).
(Found: C, 68.57; H, 7.11; N, 8.83. C18H22N2O3 requires: C, 68.77; H, 7.05; N, 8.91%)
Molecular modeling, docking and SAR study
Multiple alignment
Highly conserved amino acids were identified through multiple alignment in clustalX 1.81 [Citation47]. Sequences of all human phosphodiesterases were selected from blasted sequences via ExPASY proteomics server [Citation48]. Multiple alignment process was then carried out on the selected sequences (protein weight matrix: BLOSUM series, opening gap penalty = 10).
Structure optimization
Structures 4a-m and cilostamide were simulated in chem3D professional; Cambridge software; using MM2 method (RMS gradient = 0.05 kcal/mol) [Citation49]. Output files were minimized under semi-empirical AM1 method in the second optimization (Convergence limit = 0.01; Iteration limit = 50; RMS gradient = 0.05 kcal/mol; Fletcher-Reeves optimizer algorithm) in HyperChem7.5 [Citation50,Citation51].
Crystal structure of human PDE3B complex with MERCK1 was retrieved from RCSB Protein Data Bank (PDB entry: 1ISO).
Molecular docking
Automated docking simulation was implemented to dock 4a-m and cilostamide into the active site of PDE3B with AutoDockTools version 1.4 [Citation52] using Lamarckian genetic algorithm [Citation53]. This method has been previously shown to produce bonding models similar to the experimentally observed models [Citation27,Citation51,Citation53,Citation54]. The torsion angles of the ligands were identified, hydrogens were added to the macromolecule, bond distances were edited and solvent parameters were added to the enzyme 3D structure. Partial atomic charges were then assigned to the macromolecule as well as ligands (Gasteiger for the ligands and Kollman for the protein).
The regions of interest of the enzyme were defined by considering Cartesian chart 58.4, 2.4 and 10.0 as the central of a grid size of 40, 50 and 40 points in X, Y and Z axises. The docking parameter files were generated using Genetic Algorithm and Local Search Parameters (GALS) while number of generations was set to 100. Compound 4a-m and cilostamide were each docked into the active site of PDE enzyme and the simulations were composed of 100 docking runs, each of 50 cycles containing a maximum of 10,000 accepted and rejected steps. The simulated annealing procedure was started at high temperature (RT = 616 kcal/mol, where R is the gas constant and T is the steady state temperature) and was decreased by a fraction of 0.95 on each cycle [Citation27]. The 100 docked complexes were clustered with a root-mean-square deviation tolerance of 0.2 å. Autodock generated 100 docked conformers of 4a-m and cilostamide-corresponding to the lowest-energy structures. After docking procedure in AD3, docking results were submitted to Weblab Viewerlite 4.0 [Citation55] and Swiss-PdbViewer 3.7 (spdbv) [Citation56] for further evaluations. The results of docking processing (ΔGb: Estimated Free Energy of Bonding, Ed: Final Docked Energy and Ki: Estimated Inhibition Constant) are outlined in .
Table 3. Docking analysis data of consensus conformers.
Assessment of inotropic and chronotropic activities
The whole atrium was separated from ventricle and mounted vertically in a 50 ml organ-bath containing Krebs-Henseleit solution constantly gassed by 95% O2 and 5% CO2, 35-37 °C, pH of 7.35-7.45. The bathing solution was made up (in mM): NaCl 118, KCl 4.5, CaCl2 1.36, MgSO4 1.21, NaH2PO4 1.22, NaHCO3 25, and glucose 11 [Citation29]. Resting tension was adjusted to about 0.5 g in the whole atria [Citation24] and the initial equilibration period was 40–50 min for each preparation. Since the atria were isolated from reserpine-pretreated animals, depletion of catecholamine was verified by lack of any positive inotropic effect induced by tyramine (1.5 μM) [Citation29]. Experiments were performed only in preparations that did not response to tyramine. IBMX, and the test compounds were added cumulatively (1-100 μM) and the responses of each concentration were recorded up to the maximum.
Assessment of PDE3 inhibitory activity
Rat ventricular PDE3 was isolated and purified on HPLC according to the procedure by Bode et al [Citation57]: The soluble fraction from whole rat ventricle [Citation57] (4 mg protein in 1.0 ML) was loaded onto a MonoQ HPLC column (5 3 0.5 cm; bed volume, 1.0 ML), and the proteins were eluted with a linear gradient of NaCl from 75 mM to 500 mM (in 25 mM Bis-Tris, pH 6.5) at a flow rate of 1 ML/min. The fractions of the last peak (peak 4) were collected (2.5 ML), of which 25 μL was assayed for PDE activity. PDE3 activity was assayed by the two-step radiometric method of Thompson et al [Citation58]: 0.4 ml medium containing 40 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 1 mM EGTA, 1 μM [3H]cAMP, PDE inhibitors in 1% DMSO (0.01 – 10 μM) and 25 μL of isolated ventricular PDE3 [Citation57] placed in 37˚C water bath and incubated for 40 min. The similar amount of DMSO was added to the control samples. The reaction was quenched by heating the tubes in boiling water for 2 min and the tubes were then placed on ice followed by snake venom (Naja Naja Oxiana) addition. The tubes were returned to the water bath (37 °C), incubated for 10 min, treated by 30% slurry Dowex1-Cl resin suspension, consecutively vortexed for 15 min and centrifuged for 2 min at 14000 rpm. Finally, the supernatant was collected for β-scintillation counter. The activity of our preparations of PDE3 was 0.35 ± 0.004 pmol hydrolyzed cAMP/μL of protein/min.
Statistical analysis
Data were expressed as mean ± standard error of mean (S.E.M). Student’s unpaired t-test was used to evaluate the effect of an intervention between two experimental groups. Whenever more than two groups have been examined; we used One-way analysis of variance (ANOVA) and the Tukey’s post hoc test. Differences between means were considered significant if p < 0.05. All data passed a normality test.
Results and discussions
The newly synthesized analogues (4a-m), were tested for their effects on the force of contraction and frequency rate of rat spontaneously beating atria in comparison with cilostamide, amrinone and IBMX. To avoid bias of catecholamine release, we used preparations obtained from reserpine-pretreated animals.
All the test compounds increased force of contraction of atria in a concentration-dependent way (from 1 μM to 100 μM), except for 4e-g (). None of the compounds affected the atrial frequency significantly even at 100 μM (). Washing out the myocardial preparations, completely retained the contractility to the pre-drug state. This makes us to suppose the effects of our compounds as reversible.
Table 1. Effects of the test compounds upon force of contractility of whole atria from reserpine-pretreated rats: comparison with IBMX, amrinone and cilostamide.
Table 2. Effects of the test compounds upon frequency rate of whole atria from reserpine-treated rats: comparison with IBMX amrinone and cilostamide.
Compound 4j was the most active inotropic agent, increasing atrial contractility to an extent significantly higher than that of amrinone and cilostamide. The inotropic effect of the highest concentration of 4j (65 ± 4% increase at 100 μM), was comparable with the maximum effect of IBMX (). However, the potency of 4j was lower than that of IBMX. During experiment, 4j did not cause any arrhythmias. IBMX at 100 μM increased heart rate 64 ± 8% while 4j seems to increase the heart rate 15 ± 7% at the same concentration (not significant). Amrinone and cilostamide also increased heart rate by 20 ± 4 and 68 ± 4% respectively at high concentration (100 μM).
In similar conditions, isoprenaline, β1-adrenoceptor agonist, produced maximum effect at a concentration of 0.3 μM which caused an increase in the contraction 192 ± 8% and an increase in heart rate 84 ± 8% over the basal values.
The in vitro data acquired for this class of heterocycles (4a-m) showed that PDE3 inhibition can be increased by introducing methyl group at position 4 of quinolinone in comparison with cilostamide and it can be manipulated by changing the N-substituents of butanamide moiety.
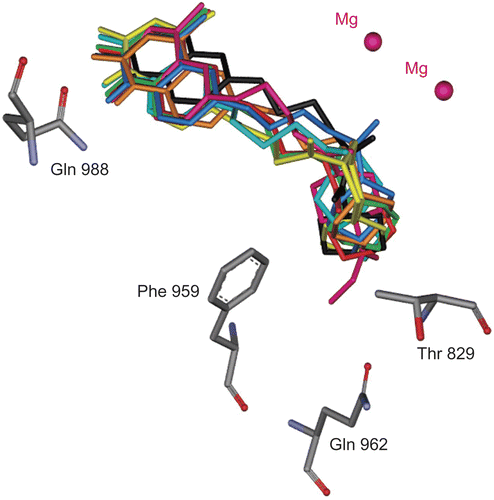
We generated 100 docked conformers of 4a-m in ADT software. A detailed assessment of each inhibitor conformers (all of the 100 docked models) revealed that more than 40% of docking results had nearly identical orientations in which: 1) quinolinone group of inhibitors oriented toward Glu988, 2) propyl chain of butanamide moiety was flanked by the hydrophobic portion of the Phe991 side chain and 3) amide groups surrounded by Thr829, Phe959 and Gln962 side chains like cilostamide [Citation30]. The most mimic conformer with cilostamide (RMSD = 50.001 å, ) at lowest Ki from each 100 docked models was adopted as the “consensus” structure and used for further analysis ().
Figure 4. Superimposition of the consensus bonding conformations of cilostamide (black), 4c-d and 4h-m in green stick in the PDE3B active site. (Protein Data Bank: 11 SO)
To simplified analysis of docking results, the active site was divided into three regions A, B and C () similar to the work of Card et al on PDE5A, PDE4B and D [Citation38]. Thus the corresponding amino acids of the three regions are: His948, Thr952 and Gln988 for A; Tyr736, Leu895, Ile938, Ile955, Leu987 and Phe991 for B; His737, His825, Arg828, Thr829, Glu958, Phe959 and Gln962 for C. Multiple alignment, among above amino acids showed Tyr736, His737, His825, Leu895, Ile938, Ile955, Glu958, Phe959, Gln962, Gln988, Phe991 identical over all species (). Role of the mentioned amino acids in dephosphorylation activity of PDE3 and PDE4 were proved by mutagenesis studies [Citation28,Citation32,Citation33,Citation39].
Figure 5. Stick (above) and two view of solvent surface (below) of the interacted amino acids with 4j (colored stick with transparent surface). The three proposed regions A, B and C are distinguished by yellow, green and blue respectively.
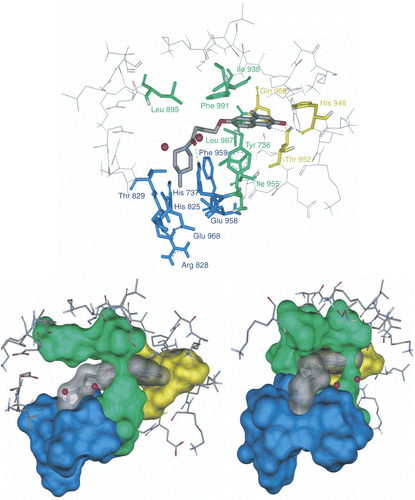
Figure 6. Clustal X (1.81) multiple alignment of human PDEs. The amino acids in the regions A, B and C are highlighted by yellow, green and blue respectively.

In proposed inhibitory model, based on docking results, the side chain of amino acids in region B, surround the butanamide moiety and aromatic part of quinolinone portion like a ring via lipophilic interactions (). All amino acids which form this lipophilic ring are highly conserved except Leu987. Essential role of Tyr736, Leu895 and Phe991 in bonding affinity of cGMP, cilostazol and cilostamide towards PDE3A and their effects in catalytic activity has been reported [Citation28,Citation33]. In PDE3A, Leu910(895)† → Ala, Tyr751(736) → Ala and Phe1004(991) → Ala mutants decreased catalytic efficiency (kcat/Km) by 15, 43 and 286 folds from WT (wild type) respectively [Citation33]. PDE4B mutation, Ile410(955) → Thr and Asn, increased rolipram IC50 288 and 1000 folds respectively [Citation40]. The mentioned mutations may reduce hydrophobic interaction between the protein and rolipram’s phenyl group.
IBMX, competitive inhibitor of most known PDEs except PDE8 and PDE9, is a purine-derived compound [Citation41]. In the co-crystal structure of PDE3B:IBMX complex [Citation30], IBMX makes two hydrogen bond with the side chain of the highly conserved Gln988. The xanthine ring is stacked against the side chains of Phe991 and Ile955. The side chain of Tyr736 contributes to the formation of the hydrophobic pocket containing the xanthine ring. The isobutyl chain lies in a wide pocket formed by the side chains of Tyr736, His737, Leu895, and Phe959. All consensus structures have hydrogen bonds with His948, Thr952 and Gln988 (pocket A) via amide group of their quinolinone portion (similar to xanthine ring of IBMX in IBMX:PDE3B complex). The hydrogen bond of ligands with highly conserved Gln988 within the pocket is very important. The catalytic role of aligned residue of the Thr952 in PDE4B (Thr407) has been studied by mutagenesis experiment In which the mutant Thr407 → Ala increased PDE4B Km and the rolipram IC50, 8 and 330 fold respectively [Citation42].
The amino acids His737, His825, Arg828, Thr829, Glu958, Phe959 and Gln962 which form region C, surround the amide moiety of the synthetic inhibitors as a bowl. The side chain of Arg828 is not participating in formation of the bowl. Among the mentioned residues His737, His825, Glu958, Phe959 and Gln962 are highly conserved. Mutation of Thr844(829), Gln962(975), Phe(972)959, Glu971(958) and His752(737) to Ala in PDE3A decreased catalytic efficiency by 7, 10, 15, 461 and 1251 fold from WT respectively [Citation32–33]. It is notable that mutant His840(825)→ Ala in PDE3A lead to no catalytic activity even at high concentration of the mutated enzyme [Citation43]. These results indicate that the mentioned residues contribute significantly to catalysis. Because of the numerous hydrophilic and hydrophobic interaction sites in region C () we synthesized many compounds with different amide groups. The hydrophilic surface of pocket C is formed by His737, His825, Glu958 and Gln962 side chains whoever the hydrophobic interactions comes from the side chains of Phe959 and Thr829. Mutation of residues Thr829, Phe959 and Gln962 affect only bonding of larger inhibitors such as cilostamide and cilostazol, not cGMP or smaller inhibitors such as milrinone [Citation28]. In our inhibitory model the cyclohexylamide, phenylamide, piperidine, piperazine and pyrrolidine moiety of the compounds 4a-m are extended toward Thr829, Phe959 and Gln962. The Ki (estimated inhibition constant) of compounds 4c-d and 4h-l with substituted piperidine and piperazine (), showed a good linear relationship with the increase in force of contractility (). To study the mechanism of action of the synthetic compounds, 4j-m were candidate for studying of PDE3 inhibitory activity. Effect of the mentioned compounds and cilostamide on cAMP degradation by purified rat heart PDE3 was significantly blocked with IC50 values of 0.20, 0.41, 2.14, 1.13 and 0.28 μM respectively (). Among these compounds, 4j showed most potent activity to inhibition of PDE3. The Ki of proposed model of compound 4j-m and cilostamide, have good relation with IC50 results (). It is nearly in accordance with inotropic results of them.
Figure 7. Solvent surface (green) of pocket C conserved amino acids having polar and non-polar interactions with consensus structure of compounds 4l (red stick), 4m (green stick), 4j (yellow stick) and 4k (bleu stick). The amide groups of inhibitors have been drawn in contrasted colors.
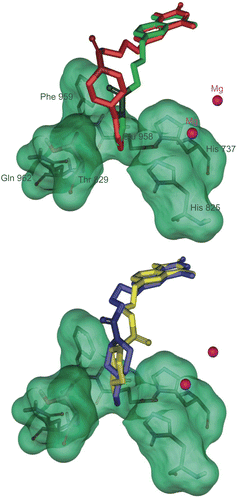
Figure 8. Diagram of increase in force of contractility versus estimated inhibition constant (Ki) for compounds 4c-l.
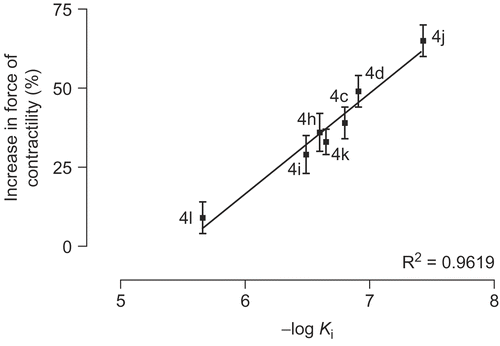
Figure 9. Concentration-effect curves for inhibition of the activity of PDE3 partially purified from rat ventricle by cilostamide and 4j-m. Results are means ± SEM from four experiments. Data are expressed as percent inhibition of the basal enzyme activity (0.35 ± 0.004 pmol hydrolyzed cAMP/μL of protein/min).
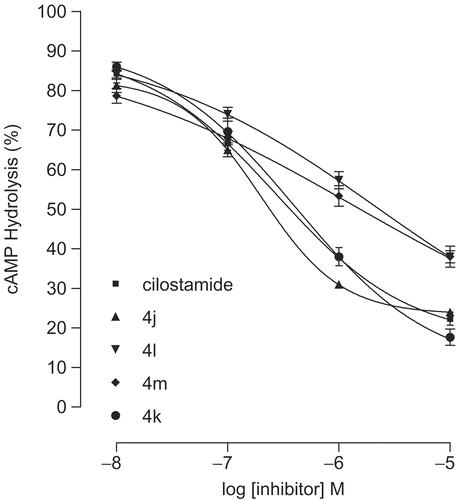
Figure 10. Diagram of -log IC50 versus -log Ki for compounds 4j-m and cilostamide (cil.).
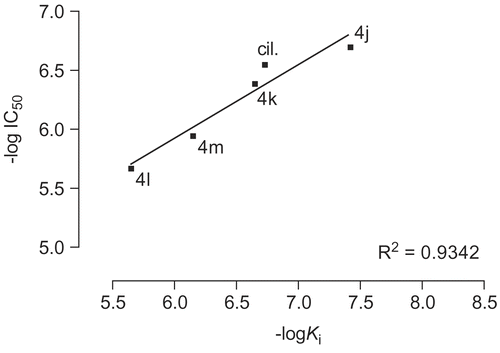
We propose hydrophobic and hydrophilic interactions as well as hydrogen bonding affinity along with the tendency of the amide group for filling the pocket C, as the main reason for the presented positive results. The force of contractility is seemed to be related to the size of the amide moiety. As shown in and compounds 4l and 4m show the less activity among the active compounds (∼10%), where the size of their amide moiety is too large and too small respectively. It is interesting to view 4l and 4m as weak inhibitors of PDE3 in comparison with cilostamide and 4j-k. Similar result is being drawn for compounds 4j and 4k (). Compound 4j with methyl piperazine moiety shows higher incensement in force of contractility (65%) and PDE3 inhibitory potency (IC50 = 0.20 μM) than compound 4k with ethyl substituent in its piperazine moiety (34%; IC50 = 0.41 μM).
It is interesting to see 4-hydroxypiperidine, 4-piperidinecarboxylic acid and 4-piperidinone analogs (4e-g) as non-inotropic agents. A probable reason might be present of hydrogen donor group in all three compounds which disturb amide interaction with the pocket C. It is notable that in aqueous solution, 4-piperidinone mainly exists as 4-dihydroxypiperidine form which converts compound 4f as hydrogen donor.
In summary we have carried out the SAR comparative studies on 4-[(4-methyl-2-oxo-1,2-dihydro-6-quinolinyl)oxy]butanamides 4a-m as inotropic agents. The comparison method we used in this study may be useful to link SAR properties and inotropic results of compounds with partly similar size, hydrophobic and hydrophilic portions. Finally, we could conclude that 4j with high inhibitory potency of PDE3 may be a good candidate for treatment of CHF since; in spite of high efficacy it may introduce less proarrhythmic side effects.
Acknowledgements
We express our sincere gratitude to Ferdowsi University of Mashhad and Mashhad University of Medical Sciences for financial support of this work.
Declaration of interest: The authors report no conflicts of interest.
References
- Beavo JA. Physiol Rev 1995;75:725–748.
- Nicholson CD, Challiss RAJ, Shahid M. Trends Pharmacol Sci 1991;12:19–27.
- Polson JB, Strada SJ. Annu Rev Pharmacol Toxicol 1996;36:403–427.
- Rose RJ, Liu H, Palmer D, Maurice DH. Br J Pharmacol 1997;122:233–240.
- Reeves ML, Leigh BK, England PJ. Biochem J 1987;241:535–541.
- Hidaka H, Asano T. Biochim Biophys Acta 1976;429:485–497.
- Hidaka H, Hayashi H, Kohri H, Kimur H, Hosokawa T, Igawa T, Saitoh Y. J. Pharmacol Exp Ther 1979;211:26–30.
- Souness JE, Hassall GA, Parrott DP. Biochem Pharmacol 1992;44:857–866.
- Reinhardt RR, Chin E, Zhou J, Taira M, Murata T, Manganiello VC, Bondy CA. J Clin Invest 1995;95:1528–1538.
- Palmer D, Maurice DH. Mol Pharmacol 2000;58:247–252.
- Inoue Y, Toga K, Tachibana K, Kimura Y, Hidaka H. Atherosclerosis 1994;109:253–261.
- Indolfi C, Avvedimento EV, Lorenzo ED, Esposito G, Rapacciuolo A, Giuliano P, Grieco D, Cavuto L, Stingone AM, Ciullo I, Condorelli G, Chiariello M. Nat Med 1997;3:775–779.
- Umekawa H, Tanaka T, Kimura Y, Hidaka H. Biochem Pharmacol 1984;33:3339–3344.
- Kimura Y, Tani T, Kanbe T, Watanabe K. Arzneimittelforschung 1985;35:1144–1149.
- Tanaka T, Ishikawa T, Hagiwara M, Onoda K, Itoh H, Hidaka H. Pharmacology 1988;36:313–320.
- Degerman E, Belfrage P, Newman AH, Rice KC, Manganiello VC. J Biol Chem 1987;262:5797–5808.
- Tang KM, Jang EK, Haslam RJ. Eur J Pharmacol 1994;268:105–114.
- Sudo T, Tachibana K, Toga K, Tochizawa S, Inoue Y, Kimura Y, Hidaka H. Biochem Pharmacol 2000;59:347–356.
- Koga Y, Kihara Y, Okada M, Inoue Y, Tochizawa S, Toga K, Tachibana K, Kimura Y, Nishi T, Hidaka H. Bioorg Med Chem Lett 1998;8:1471–1476.
- Lugnier C, Stierle A, Beretz A, Schoeffter P, Lebec A, Wermuth CG, Cazenave JP, Stoclet JC. Biochem Biophys Res Comm 1983;113:954–959.
- Jones GH, Venuti MC, Alvarez R, Bruno JJ, Berks AH, Prince A. J Med Chem 1987;30:295–303.
- Inoue Y, Toga K, Sudo T, Tachibana K, Tochizawa S, Kimura Y, Yoshida Y, Hidaka H. Br J Pharmacol 2000;130:231–241.
- Nishikawa M, Komada F, Morita K, Deguchi K, Shirakawa S. Cell Signal 1992; 4:453–463.
- Juan-Fita MJ, Vargas ML, Hernandez J. Eur J Pharmacol 2005;512:208–211.
- Nishi T, Tabusa F, Tanaka T, Ueda H, Shimizu T, Kanbe T, Kimura Y, Nakagawa K. Chem Pharm Bull 1983;31:852–860.
- Fujioka T, Teramoto S, Mori T, Hosokawa T, Sumida T, Tominaga M, Yabuuchi Y. J Med Chem 1992;35:3607–3612.
- Dym O, Xenarios I, Ke H, Colicelli J. Mol Pharmacol 2002;61:20–25.
- Zhang W, Ke H, Colman RW. Mol Pharmacol 2002;62:514–520.
- Fossa P, Menozzi G, Dorigo P, Floreani M, Mosti L. Bioorg Med Chem 2003;11:4749–4759.
- Scapin G, Patel SB, Chung C, Varnerin JP, Edmondson SD, Mastracchio A, Parmee ER, Singh SB, Becker JW, Van der Ploeg L, Tota MR. Biochemistry 2004;43:6091–6100.
- Cheung PP, Yu L, Zhang H, Colman RW. Arch Biochem Biophys 1998;360:99–104.
- Zhang W, Colman RW. Blood 2000;95:3380–3386.
- Zhang W, Ke H, Tretiakova AP, Jameson B, Colman RW. Protein Sci 2001;10:1481–1489.
- Chung C, Varnerin J, Morin NR, MacNeil DJ, Singh SB, Patel S, Scapin G, Van der Ploeg L, Tota MR. Biochem Biophys Res Commun 2003;307:1045–1050.
- http://www.ncbi.nlm.nih.gov/BLAST/Blast.cgi
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman D. J. Nucleic Acids Res 1997;25:3389–3402.
- Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin E V, Altschul SF. Nucleic Acids Res 2001;29:2994–3005.
- Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillete S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim SH, Schlessinger J, Zang KYJ. Structure 2004;12:2233–2247.
- Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RE. J Mol Biol 2004;337:355–365.
- Atienza JM, Susanto D, Huang C, McCarty AS, Colicelli J. J Biol Chem 1999;274:4839–4847.
- Francis SH, Turko IV, Corbin JD. Prog Nucleic Acid Res Mol Biol 2001;65:1–52.
- Pillai R, Kytle K, Reyes A, Colicelli J. Proc Natl Acad Sci USA 1993;90:11970–11974.
- Cheung PP, Zhang LYH, Colman RW. Arch Biochem Biophys 1998;360:99–104.
- Martinez E, Penaafiel R, Collado MC, Hernaandez J. Eur J Pharmacol 1995;282: 169–175.
- Campbell KN, Tipson RS, Elderfield RC, Campbell BK, Clapp MA, Gensler YJ, Morrison D, Moran WJ. J Org Chem 1946;11:803–811.
- Holmes RR, Conrady J, Guthrie J, Mckay R. J Am Chem Soc 1954;76:2400–2407.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. Nucleic Acids Research 1997;24:4876–4882.
- http://us.expasy.org/
- ChemDraw® Ultra, Chemical Structure Drawing Standard, CambridgeSoft Corporation, 100 Cambridge Park Drive, Cambridge, MA 02140 USA, http://www.cambrigesoft.com
- HyperChem® Release 7, Hypercube Inc., http://www.hyper.com/
- Sadeghian H, Seyedi SM, Saberi MR, Arghiani Z, Riazi M. Bioorg Med Chem 2008;16:890–911.
- Auto Dock Tools (ADT), the Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037-1000, USA; Python MFS. J Mol Graphics Mod 1999;17:57–61.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem 1998;19:1639–1662.
- Sippl W. J Comput Aided Mol Des 2000;14:559–572.
- http://sunfire.vbi.vt.edu/gcg/seqweb-guides/WebLab_Viewer.html
- Swiss-pdbViewer 3.6, Glaxo Wellcome Experimental Research, http://www.expasy.org/spdbv.
- Bode DC, Kanter JR, Brunton LL. Circ Res 1991;68:1070–1079.
- Thompson W, Appleman MM. Biochemistry 1971;10:311–316.