Abstract
A quantitative structure-activity relationship (QSAR) study has been carried out on growth hormone secretagogue receptor antagonistic activity of the derivatives of 2,4-diaminopyrimidine. To obtain significant QSARs, the approaches involving the non-parametric such as Fujita-Ban, and the parametric based on physicochemical and DRAGON descriptors in Hansch type of analysis have been employed. The Fujita-Ban approach, however, was constrained to 18 compounds only due to a limited number of substituents appeared at varying positions. The derived contributions of different substituents and the parent moiety were used to identify the potential congeners. The physicochemical model of Hansch was subsequently used to interpret the type of interactions involved between the receptor sites and varying positions of these compounds. The study, employing DRAGON descriptors in Hansch approach was also carried out on this data set to discuss the prevailing interactions in terms of topological descriptors. The derived highest significant model was discussed to delineate the type of interactions involved and suggestions have been made for different alterations to lead to further potential compounds of the series.
Keywords::
- Quantitative structure-activity relationship (QSAR)
- antagonists of growth hormone secretagogue receptor (GHS-R)
- binding activity to GHS-R
- derivatives of 2, 4-diaminopyrimidine
- Fujita-Ban approach
- Hansch type of analysis
- combinatorial protocol in multiple linear regression (CP-MLR) analysis
- physicochemical parameters and DRAGON descriptors
Introduction
Many chronic disorders such as insulin resistance, diabetes, dyslipidemia, hypertension, coronary heart disease, ischemic stroke, pulmonary disease, gout, osteoarthritis, cancer, neurological diseases, cataracts, gastrointestinal disease, and genitourinary disease (in women) are associated with, a worldwide problem, obesity. It has been shown that the progression of these disorders are reversed or slowed by weight loss [Citation1]. Body weight may be maintained by balancing caloric intake with the energy expenditure. Thus, diet and exercise are crucial to control the weight. However, these may be assisted by appetite reducing oral agents. Ghrelin, produced in stomach and hypothalamus [Citation2], is a 28 amino acid containing orexigenic hormone. It is an endogenous ligand of the growth hormone secretagogue receptor (GHS-R). GHS-R is a G-protein-coupled receptor which is expressed primarily in the pituitary gland and the brain and to a lesser extent in the periphery. The unique post-translational acylation, an octanoyl group at the Ser-3 hydroxyl group, of active ghrelin is essential for orexigenic function and also necessary for its penetration of the blood-brain barrier [Citation2,Citation3]. The intake of nutrient regulates the level of ghrelin in blood. In rats and human subjects, intracerebroventricular and intravenous administration of ghrelin, respectively, increased appetite and food intake [Citation4–8]. It has been observed that the weight gain, induced by ghrelin, is primarily from adipose tissue and not from muscle or bone mass [Citation9]. The inhibition of ghrelin actions with peptidyl GHS-R antagonists [Citation7,Citation9], ghrelin antibody [Citation10], and antisense oligonucleosides [Citation11] resulted in weight loss and food intake decrease in rodents [Citation7, Citation9–11]. The isoxazole and tetralin carboxamide based GHS-R antagonists [Citation12–15], displaced 125I-ghrelin binding and antagonized ghrelin-induced intracellular Ca2+ flux in functional assay. However, these compounds did not exhibit an effect when tested in in vivo animal models. More recently, a synthetic study was performed [Citation16] on the derivatives of 2,4-diaminopyrimidine and to identify them as effective GHS-R antagonists. These compounds were first run in the GHS-R binding assays and were further investigated in an intracellular Ca+2 flux activity assay.
The variation in the chemical space of these analogues is mainly focused around two phenyl rings, attached to the 2,4-diaminopyrimidine core. In order to investigate the scope of such chemical space around 2, 4-diaminopyrimidine moiety, a high dimensional quantitative structure-activity relationship (QSAR) study has been undertaken on these analogues to rationalize their GHS-R activity profiles. For this, it is necessary to characterize the molecules or their varying structural fragments from different perspectives. Among different methods, graph theoretical approaches provide large number of structural indices characteristic to the molecules and/or their functional units [Citation17–21]. Moreover, when dealing with a large number of descriptors, for the optimum utilization of information content of the generated data sets, it is necessary to follow a typical protocol(s) to identify the best models as well as information rich descriptors corresponding to the phenomenon under investigation. The Combinatorial Protocol in Multiple Linear Regression (CP-MLR), developed recently [Citation22–27], is an approach among many others to address the model evolution in high-dimensional QSAR studies. The aim of present communication is therefore, to establish the QSAR between the reported GHS-R antagonistic activity of 2,4-diaminopyrimidine derivatives and the molecular descriptors, obtained from graph theory.
Material and methods
Data set
In present study 2, 4-diaminopyrimidine derivatives () have been taken from the literature report [Citation16] along with their antagonistic activity. The activity was expressed as the logarithm of the inverse of inhibitory concentration, pIC50, where IC50 represents the molar concentration, required to bring out 50% inhibition of GHS-R. Two different approaches, namely the non-parametric and the parametric, have been used to develop the important QSARs of titled compounds.
Table I. The structures of the 2, 4-diaminopyrimidine analogues included in training set along with their GHS-R binding activity.
The Fujita-Ban methodology [Citation28], based on additivity principle, is a non-parametric approach and requires certain group to occur two or more times at a given varying position in a molecule. The Hansch type of analysis [Citation29–31], on the other hand, is a parametric approach in which physicochemical and/or structural parameters are being used as the correlative parameters. This method is generally used to increase the understanding of the mechanisms of action of a set of congeners and to direct drug design in a congeneric series as well as to attempt to predict biological activities quantitatively. In general, the approach is to set up the equations involving different combinations of the substituents constants, then to allow the correlative methods to aid in the selection of the ‘best equation’ justifying it statistically and avoiding the chance correlations. For the present study, the physicochemical parameters were taken from the literature [Citation32]. The indicator variables, representative of the presence or absence of certain structural characteristic, have also been used. Both of these approaches have limitations as far as their use is concern for a new series of compounds. The Fujita-Ban methodology can not be employed if the appearance of certain substituent at a given position is less than two. Further, the study can not be extrapolated beyond the prevailing substituent’s pattern in the chemical space. Similarly the Hansch type of approach is found unsuitable, if the desired physicochemical parameters have not been determined for certain substituents. The use of structural descriptors, instead of physicochemical parameters, may then be proper to address the interaction involved at receptor sites.
In order to widen the scope of the study, the graph theory may be used as an alternative approach to obtain correlative descriptors. The highly informative descriptors, identified through certain mathematical procedure, may then be used to derive significant QSAR models to address the phenomenon under investigation. The DRAGON software [Citation20], developed recently, is able to compute a large number of descriptors belonging to 0D-, 1D-, 2D- and 3-D classes. Prior to the computation of molecular descriptors, the structures of the compounds have been drawn in ChemDraw using the standard procedure. These structures were converted into 3D objects using the default conversion procedure implemented in the CS Chem3D Ultra. These 3D-structures were then ported to DRAGON software and a total number of 478 descriptors, pertaining to 0D-, 1D-, and 2D-classes, have only been computed for the sake of simplicity to interpret them in terms of common physical phenomenon. The descriptor classes along with their definitions and scope in addressing the structural features have been given in . As the total number of descriptors involved in this study is very large, only the names of descriptor classes and the actual descriptor involved in the models have been addressed in the discussion. The QSAR model generation and validation have been done using the combinatorial protocol in multiple linear regression (CP-MLR) analysis.
Table II. Descriptor classes used for modeling the binding activity of the 2,4-diaminopyrimidine analogues and identified categories in modeling the activity.
CP-MLR
The CP-MLR is a ‘filter’-based variable selection procedure for the development of statistical models in high dimensional QSAR studies [Citation22–27]. It involves a combinatorial strategy with approximately placed ‘filters’ interfaced with MLR and extracts diverse models having unique combination of descriptors from the dataset. The filters set the thresholds for the descriptors in terms of inter-parameter correlation cutoff limits in subset regressions (filter-1), t-values of the regression coefficients (filter-2), internal explanatory power (filter-3; square root of adjusted multiple correlation coefficient of regression equation, r-bar), and the external consistency (filter-4; Q2 i.e. cross-validated R2 from the leave-one-out procedure). Throughout this study, the thresholds for the filters-1, 2 and 4 were assigned as 0.3, 2.0, and 0.3 ≤ Q2 ≤ 1.0, respectively while the filter-3 was assigned an initial value of 0.71. In order to collect the descriptors with higher information content, the threshold of filter-3 was successively incremented with increasing the number of descriptors (per model) by considering the r-bar value of the preceding optimum model as the new threshold for the next generation.
Descriptor classification protocol
The Three-stage descriptor classification protocol [Citation23] is implemented with two-descriptor combinations (baseline models), as these are the simplest to understand and to explain the activity. In the first stage of the classification protocol, the correlations of the activity with two descriptor combinations from the individual descriptor classes (DCs) of the dataset were used to sort them into four categories. They are primary contributors (category I: a DC forms a model with its constituent descriptors), collective contributors (category II: a DC unable to form a model with its constituent descriptors, but forms model(s) in combination with a descriptor from another such DC), secondary contributors (category III: a DC from a model(s) only in combination with category I) and noncontributors (category IV: a DC unable to form a model(s) in any manner like that of category I, II, and III). The sorted DCs were collated in the second stage to identify all the 3-descriptor models across the categories. In the last stage, the individual descriptors of all three-descriptor models were pooled to discover the higher models for the activity.
All the identified models have been put to the randomization test [Citation24,Citation33] by repeated randomization of the activity to discover the chance correlations, if any, associated with them. For this every model has been subjected to 100 simulation runs with scrambled activity. The scrambled activity models with regression statistics better than or equal to that of the original activity model have been counted to express the percent chance correlation of the model under scrutiny. The model development procedure has been finally validated externally by creating test set from complete data set.
Results and discussion
Initially, the GHS-R Ca+2 flux activity was correlated to the GHS-R binding activity of all the 35 congeners to confer whether the two activities are inter-correlated to each other. The derived correlation between them is as in Equation (1)
Here and in the follow up discussion, n, r, s, F and p are respectively the number of data-points considered in the analysis, the regression coefficient, the standard error of estimate, the Fischer ratio and the significance of the model. Above correlation has divulged significant statistical parameters, which ensures that the GHS-R binding activity and the GHS-R intracellular Ca+2 flux activity are inter-related to each other. The subsequent elaboration is, therefore, based on the consideration of only the pIC50(Binding) as the dependent variable.
In formulation of the Fujita-Ban matrix, eighteen compounds of were retained in the training set, with compound 1 as the parent congener. However, nine compounds (3, 6, 15–18, 25–27) from this Table were not included in this set as certain substituents, at a given position, in these congeners occurred only once. The matrix consisting of 18 compounds (rows) and 9 substituents (including parent contribution) related to varying positions of the parent moiety (columns) is not documented here for the sake of brevity. The matrix was subjected to MRA and the derived statistical parameters of the study corresponding to the binding activity were:
All these statistical parameters tune to the highly significant results as F-value remained significant at 99% level while r-value has accounted for 98% of variance in observed activities. The calculated activity values () of the compounds considered in the study were in close agreement with the observed ones. The activity contributions of different substituents and parent moiety are given in . From this Table, the substituents that make higher positive contribution to activity, relative to parent moiety, may easily be identified. Thus, the substituent OCH2C6H5 present at R1 and the substituents SO2CH3, NO2, SO2CF3, COCH3 and CN, in that order, present at position Y are advantageous. The appropriate substituents for varying positions, which make highest positive contribution to parent moiety may be selected for the further design of more active analogues of the series.
Table III. Observed, calculated and predicted (LOO) pIC50 values of training set compounds.
Table IV. The Fujita-Ban contributions of substituents and parent moiety to the GHS-R binding activity of the 2,4-diaminopyrimidine analogues.
It is important to note that the Fujita-Ban approach cannot extrapolate beyond the compounds, considered in the training set whereas the Hansch approach, discussed below can do so. Thus, in the latter approach the data set has been extended to include the compounds which were not considered in Fujita-Ban study. In order to carry out the Hansch type of analysis, a number of physicochemical parameters for the R1- and Y-substituents were selected and analyzed in a systematic manner for their correlations to GHS-R binding activity. The data-set, consisting of substituent constants such as hydrophobicity, π, hydrogen-bond donor, HD, hydrogen-bond acceptor, HA, electronic, σ (meta and para), field, F, resonance, R, dipole moment, μ, molar refraction, MR, molecular weight, MW and van der Waals volume Vw for each of the varying positions of the compounds were considered by permuting them to derive all possible models. Due to limited variations at incision X, the indicator variable was only considered. The highest significant correlation that could be emerged from these substituents constants is given by Equation (2)
where the indicator variables IX and IY highlighting the presence of an -OCH2- at X-incision in between two phenyl rings and NO2-substituent at Y-position, respectively. The values 1 or 0 for these variables indicate, respectively, the presence or absence of aforesaid structural features. The statistical indices, Q2LOO and Q2L5O have been obtained, respectively, by leave-one-out (LOO) and leave-five-out (L5O) procedures [Citation34–36]. The derived statistical parameters have indicated highly significant results and Equation (2) as such reflected the parametric requirement of various substituents present at different positions of said analogues. The r2-value has accounted for 85% of variance in the observed activity values while F-value remained significant at 99% level. The sufficiently high values of cross-validated indices, Q2LOO and Q2L5O, are both in favor of a robust and a highly predictable QSAR model. The calculated activity values using Equation (2) have remained in close agreement with the observed ones (). From Equation (2), it appeared that a R1-substituent having hydrogen-bond acceptor character and a Y-substituent being more bulky/polar are advantageous in improving the binding activity of a compound. Additionally, the presence of NO2-substituent at Y and absence of -OCH2- at X are also essential.
To widen the scope of present study, a high dimensional QSAR analysis has also been performed on these congeners. Twenty seven analogues, considered above, have been investigated to correlate their the GHS-R binding activity with a variety of 0D-, 1D- and 2D-descriptors computed from DRAGON software. The significant correlation, derived in such descriptors, may then be used to interpret a topological model in relation to commonly employed physicochemical properties. The emerged topological model(s), in this way, will have the wider perspective to describe GHS-R binding activity in relation with DRAGON descriptors. A total number of ten classes pertaining to 0D-, 1D, and 2D-descriptors have been implemented in this software. For present study, however, only seven classes were found important. A large number of descriptors from these classes have yielded significant correlations. These descriptors were identified as the primary contributors (category I) in modeling the inhibitory activity of these compounds. Nine significant models (Equations 3-11), identified in these descriptors and their statistical parameters, have been listed in . From listed models in this Table, the inhibition activities of the compounds have been best explained by CONS, TOPO, BCUT, GVZ and 2DAUTO descriptor classes involving one- and two-descriptors. The CONS descriptors appeared in Equation (4), favors higher number of nitrogen and oxygen atoms (evinced, respectively, through nN and nO descriptors) in a molecular structure for enhanced activity. The TOPO class descriptor, S3K (3-path Kier alpha-modified shape index; information about centrality of branching) and T(N..O) (sum of topological distances between N and O) in Equation (6), favor the higher value of Kier alpha-modified shape index and topological distance between nitrogen and oxygen atom for improving the activity. These descriptors, collectively, have explained 70 percent of variance in the activity. The appeared descriptors from BCUT class in Equation (7) have revealed the importance of BEHv8, (highest eigenvalue n. 8 of Burden Marix/weighted by atomic van der Waals volumes) and BELp2 (lowest eigenvalue n. 2 of Burden Marix/weighted by atomic polarizabilities). The higher values of such descriptors have been found advantageous in improving the activity of a compound. In Equation (8), the descriptors, GGI8 (topological charge index of order 8) and JGI3 (mean topological charge index of order 3), both from GVZ class, have explained the involvement of charge indices of order 8 and 3 to augment the activity. Similarly the 2D-AUTO descriptors in Equation (9), the MATS7v (Moran autocorrelation of lag 7/weighted by atomic van der Waals volumes) and GATS1e (Geary autocorrelation of lag 1/weighted by atomic Sanderson electronegativities) have suggested the importance of lags 7 and 1 weighted by aforesaid properties. The significance of various emerged models () may be ascertained through the statistical parameters, r, s, and F. The cross-validated index Q2, obtained from LOO procedure, may further assist in identifying the robustness of these models. The physical interpretation of resultant descriptors has been described briefly in the footnotes under .
Table V. The selected QSAR models, emerged in one and two descriptors from different descriptor classes belonging to category-I.
In order to have statistical significant correlations, the higher models, involving the descriptors from two different pools, have been derived further. The first pool comprise of 14 descriptors () which have been identified earlier as primary contributors under the category analysis. These descriptors were subjected to CP-MLR and the resulting two highly significant models, in three descriptors, are shown in Equation (12) and (13)
In above models, the descriptor nN is accounting for the number of nitrogen atoms in the structure under consideration. The positive regression coefficient of this descriptor recommends for more number of nitrogen atoms in a compound. Likewise, the higher values of the topological charge index of order 8, GGI8 and mean topological charge index of order 3, JGI3, are also essential for the improvement of activity of a compound. A higher value of sum of topological distances between N and O atoms [T(N..O)] and a lower positive value of MATS7v (Moran autocorrelation of lag 7/weighted by atomic van der Waals volumes) would enhance the binding activity.
The second pool of descriptors was formulated from all 10 DRAGON classes considered collectively. A total number of 478 descriptors have been analyzed, through CP-MLR analysis, to generate 87 models. The participating descriptors (the number being 44), in these models along with their average regression coefficients, and total incidences are listed in . From 87 such models, the highly significant two models are shown in Equation (14) and (15)
Table VI. Descriptors identified for modeling the binding affinities of 2,4-diaminopyrimidine derivatives at the growth hormone secretagogue receptors along with the average regression coefficients and the total incidences.
where Q2LOO have addressed to a robust model and r-value accounted for 83 and 82 % of variances, respectively, in the observed activities. The newly appeared descriptors in above equations, nHDon (number of donor atoms for H-bonds with N and O) and nNHRPh (number of secondary aromatic amines) belong to FUNC class of descriptors. A higher value of these descriptors in addition to the higher value of topological distances between N and O atoms would increase the binding activity. The other emerged descriptor, from ACF class, C-002, is representative of the fragment ‘CH2R2’ in which two valences of a carbon atom are satisfied by hydrogen atoms and other two by alkyl groups. The negative regression coefficient of this descriptor advocates the absence of such fragments in a compound to have its improved activity. The descriptors of hold scope for even higher models. The best model, involving four descriptors, has been selected out of 41 models evolved by CP-MLR and is given in Equation (16)
The statistical parameters of Equation (16) have now improved over to that of Equation (14) and (15), justifying its superiority over to that of previously derived models. The TOPO class descriptor, MAXDP (the maximal electrotopological positive variation), which contributes positively to the activity and hence recommends maximum positive field effects for improvement of activity. The other FUNC class descriptor, nNO2Ph (number of aromatic nitro groups) recommends nitro substituted aromatic ring to augment the activity. A more number of donor atoms for H-bonds (nHDon) and absence of ‘CH2R2’ fragment (C-002) in a structure are essential to improve the binding activity of titled compounds.
All the descriptors appeared in above models had no mutual correlation is shown in . The calculated (using models) and predicted activities (through LOO procedure) have been found in close agreement to the observed ones (). The graphical representation of the same is given in for the sake of clarity and goodness of fit.
Table VII. Cross-correlation matrixa amongst predictor variables in models.
Figure 1. Plot of observed versus calculated and predicted pIC50 values.
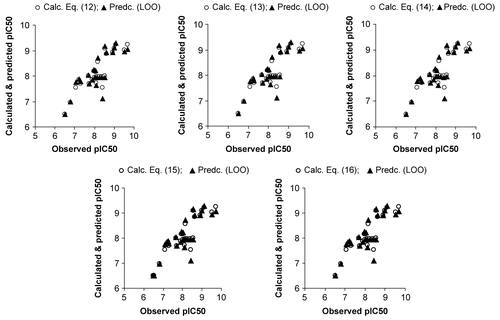
Above model Equations (12–16) were further subjected to randomization process, where 100 simulations per model were carried out but none of the identified models has shown any chance correlation. Additionally, the above model Equations have been concomitantly validated with the external test set of structurally dissimilar (bearing heteroaryl group at R3) eight compounds listed in . They have shown the test set r2 values in the range of 0.456 to 0.563. The predictions of the test set compounds obtained with the models (12-16) and the corresponding predictive r2 have also been given in the same Table.
Table VIII. The structuresa of the 2, 4-diaminopyrimidine analogues included in test set and the predictions with corresponding test set r2.
Conclusions
Fujita-Ban and Hansch type of analyses, for 27 congeners of , have revealed the results which are complementary to each other. For example, the Hansch type of study has shown the importance of a hydrogen-bond acceptor substituent at R1; the Fujita-Ban study, in conformity with it, has assigned highest substituent’s contribution to a similar substituent such as OCH2C6H5. Similarly, in Hansch type of analysis the substituent such as NO2 having higher refraction parameter, accounting for bulk and polar effect, at Y has been identified advantageous. In agreement to this, the Fujita-Ban study has emphasized the importance of substituents such as SO2CH3, NO2, SO2CF3, and COCH3. Both these studies have favored a –CH2NH- type of substitution at incision X and H at R2.
Since the descriptors in DRAGON software have been computed for the structure of a molecule as a whole, therefore, it may be improper to rationalize the individual variations in terms of evolved descriptors. Thus, for the data set in , the descriptors appeared in highest significant model have advocated the importance of maximum positive field effects (MAXDP), more nitro substituted phenyl ring (nNO2Ph) in addition to a larger number of donor atoms for H-bonds containing substituents (nHDon). Likewise, the descriptor C-002 favored the absence of a particular fragment ‘CH2R2’ in a structure to enhance the GHS-R binding activity of titled compounds.
The present study may, therefore, provide further scope for substitutional modifications of 2, 4-diaminopyrimidine scaffold to bring out high GHS-R binding potential compounds. The substitutions having increased chain lengths, such as OCH2CH2Ph, at R1 position and hydrogen donor, such as N(OH)CH2, at X may be explored.
Acknowledgments
We are thankful to our Institutions for providing necessary facilities to complete this work.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Klein S, Romijn JP. Chapter 33-Obesity. In: Williams Textbook of Endocrinology. 10th ed.: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS (Eds.), WB Saunders Co., St. Louis: MO; 2003. p 1619–1635.
- Kojima M, Hosoda H, Date Y, Nakazato H, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999;402:656–660.
- Banks WA, Tschöp M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther. 2002;302:822–827.
- Murikami N, Hayashida T, Kuroiwa T, Nakahara K, Ida T, Mondal MS, M. Nakazato M, Kojima M, Kangawa K. Role for central ghrelin in food intake and secretion profile of stomach ghrelin in rats. J Endocrinol 2002;174:283–288.
- Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 2001;86:5992–5995.
- Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, Batterham RL, Taheri S, Stanley SA, Ghatei MA, Bloom SR. Ghrelin causes hyperphagia and obesity in rats. Diabetes 2001;50:2540–2547.
- Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S. A role for ghrelin feeding in the central regulation of feeding. Nature 2001;409:194–198.
- Bagnasco M, Tulipano G, Melis MR, Argiolas A, Cocchi D, Muller EE. Endogenous ghrelin is an orexigenic peptide acting in the arcuate nucleus in response to fasting. Regul Pept 2003;111:161–167.
- Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, Kasuga M. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut 2003;52:947–952.
- Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagouge receptor. Proc Natl Acad Sci USA 2004;101:4679–4684.
- Shuto Y, Shibasaki T, Otagiri A, Kuriyama H, Ohata H, Tamura H, Kamegai J, Sugihara H, Oikawa S. Hypothalamic growth hormone secretagouge receptor regulates growth hormone secretion. J Clin Invest 2002;109:1429–1436.
- Liu B, Liu G, Xin Z, Serby M, Zhao H, Schaefer V, Falls D, Kaszubska W, Collins C, Sham H. Novel isoxazole carboxamides as growth hormone secretagouge receptor antagonists. Bioorg Med Chem Lett 2004;14:5223–5226.
- Xin Z, Zhao H, Serby M, Liu B, Schaefer V, Falls D, Kaszubska W, Collins C, Sham H, Liu G. Synthesis and structure-activity relationships of isoxazole carboxamides as growth hormone secretagouge receptor antagonists. Bioorg Med Chem Lett 2005;15:1201–1204.
- Zhao H, Xin Z, Liu G, Schaefer V, Falls D, Kaszubska W, Collins C, Sham H. Discovery of tetralin carboxamide growth hormone secretagouge receptor antagonists via scaffold manipulation. J Med Chem 2004;47:6655–6657.
- Zhao H, Xin Z, Patel JR, Nelson LTJ, Liu B, Szczepankiewicz BG, Schaefer V, Falls D, Kaszubska W, Collins C, Sham H, Liu G. Structure-activity relationships on tetralin carboxamide growth hormone secretagouge receptor antagonists. Bioorg Med Chem Lett 2005;15:1825–1828.
- Serby MD, Zhao H, Szczepankiewicz BG, Kosogof C, Xin Z, Liu B, Liu M, Nelson LTJ, Kaszubska W, Falls HD, Schaefer V, Bush EN, Shapiro R, Droz BA, Knourek-Segel VE, Fay TA, Brune ME, Beno DWA, Turner TM, Collins CA, Jacobson PB, Sham HL, Liu G. 2,4-Diaminopyrimidine derivatives as potent growth hormone secretagouge receptor antagonists. J Med Chem 2006;49:2568–2578.
- Basak SC, Harriss DK, Magnuson VR. POLLY, University of Minnesota, Duluth, MN, 1988.
- Molconn-Z, ver. 2.07, eduSoft Lc, a Virginia Corporation, Ashland, VA 23005 USA. www.edusoft-lc.com.
- (a) Katritzky AR, Lobnov V, Karelson M. CODESSA (Comprehensive descriptors for structural and statistical analysis), University of Florida, Gainesville, FL, 1994. (b) Katritzky AR, Perumal S, Petrukhin R, Kleinpeter E. CODESSA-based theoretical QSPR model for hydantoin HPLC-RT lipophilicities. J Chem Inf Comput Sci 2001;41:569–574.
- DRAGON software version 3.0-2003. By Todeschini R, Consonni V, Mauri A, Pavan M. Milano, Italy. http//disat.unimib.it/chm/Dragon.htm.
- Gonzalez MP, Helguera AM. TOPS-MODE verces DRAGON descriptors to predict permeability coefficients through low-density polymer. J Comput-Aided Mol Des 2003;17:665–672.
- Prabhakar YS. A combinatorial approach to the variable selection in multiple linear regression: Analysis of Selwood et al data set – A case study. QSAR Comb Sci 2003;22: 583–595.
- Gupta MK, Prabhakar YS. Topological descriptors in modeling the antimalarial activity of 4-(3’,5’-disubstituted aniline)quinolines. J Chem Inf Model. 2006;46;93–102.
- Prabhakar YS, Solomon VR, Rawal RK, Gupta MK, Katti SB. CP-MLR/PLS directed structure-activity modeling of the HIV-1 RT inhibitory activity of 2,3-diaryl-1,3-thiazolidin-4-ones. QSAR Comb Sci 2004;23:234–244.
- Prabhakar YS. A combinatorial protocol in multiple linear regression to model gas chromatographic response factor of organophosphonate esters. Internet Electron J Mol Des 2004;3:150–162. http://www.biochempress.com.
- Gupta MK, Sagar R, Shaw AK, Prabhakar YS. CP-MLR directed QSAR studies on the antimycobacterial activity of functionalized alkenols-topological descriptors in modeling the activity. Bioorg Med Chem 2005;13:343–351.
- Saquib M, Gupta MK, Sagar R, Prabhakar YS, Shaw AK, Kumar R, Maulik PR, Gaikwad, AN, Sinha S, Srivastava AK, Chaturvedi V, Srivastava R, Srivastava BS. C-3 alkyl/arylalkyl-2,3-dideoxy hex-2-enopyranosides as antitubercular agents: Synthesis, biological evaluation and QSAR study. J Med Chem 2007;50:2942–2950.
- Fujita T, Ban T. Structure-activity relation. 3. Structure-activity study of phenethylamines as substrates of biosynthetic enzymes of sympathetic transmitters. J Med Chem 1971;14:148–152.
- Hansch C, Fujita T. p-σ-π analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc 1964;86:1616–1626.
- Hansch C. Quantitative approach to biochemical structure-activity relationships. Acc Chem Res 1969;2:232–239.
- Hansch C. In: Drug Design, Vol. I, Ariens EJ (Ed.) New York, N Y-Academic Press, 1971. p 271.
- Hansch C, Leo A. Substituents constants for correlation analysis in chemistry and biology, New York, John Wiley, 1979.
- So SS, Karplus M. Three-dimensional quantitative structure-activity relationship from molecular similarity matrices and genetic neural networks. 2. Applications. J Med Chem 1997;40:4347–4359.
- Wold S, Eriksson L. Statistical validation of QSAR results. In: Chemometrics methods in molecular design, H. Van de Waterbeemed (Ed.), Wiley-VCH, Weinheim, 1995.
- Efron B. Estimating the error rate of a prediction rule: Some improvement on cross-validation. J Am Stat Assoc 1983;78:316–331.
- Efroymson MA. Multiple regression analysis. In: Mathematical methods for digital computers, Ralston A, Wilf HS (Eds.), Wiley-NewYork, 1960.
- SYSTAT, Version 7.0 SPSS Inc., 444 North Michigan Avenue, Chicago, IL, 60611.