
Abstract
A series of new thienyl-substituted pyrazoline benzenesulfonamides were synthesized and their carbonic anhydrase (CA, EC 4.2.1.1) inhibitory activities were tested on the human (h) isoforms hCA I and hCA II. The inhibition constant (Ki) of these sulfonamides were in the range of 232.16–637.70 nM toward the slow cytosolic isozyme hCA I, and in the range of 342.07–455.80 nM toward hCA II. Many of these compounds showed comparable inhibition with the reference sulfonamide acetazolamide, a clinically used drug. As the sulfonamide CA inhibitors (CAIs) show many therapeutic uses, these derivatives represent interesting examples of a novel class of such derivatives.
Introduction
Chalcones are open-chain flavonoids, being well-known intermediates for synthesizing various heterocyclic compounds, which are associated with an amazing range of biological activitiesCitation1,Citation2. Chalcones can be used as intermediate compounds for designing and synthesizing pharmacologically active heterocyclic structures such as pyrazolines. Pyrazolines have several biological activities including anticancer, antiviral, antimicrobial and anti-inflammatory activitiesCitation3–6.
Carbonic anhydrase (CA) is a superfamily of metalloenzymes that catalyzes the rapid conversion of CO2 to HCO3− and H+, being involved in many biochemical processesCitation7. CA isoforms are found in a variety of tissues where they participate in several important biological processes, such as acid–base balance, respiration, carbon dioxide and ion transport, bone resorption, ureagenesis, gluconeogenesis, lipogenesis and electrolyte secretionCitation7. Many CA isozymes involved in these processes are important therapeutic targets with the potential to be inhibited/activated for the treatment of a range of disorders, such as edema, glaucoma, obesity, cancer, epilepsy, diuretics, antiepileptics, anticancer and osteoporosisCitation8–14. Sulfonamide derivatives, especially aromatic and heterocyclic sulfonamides when the sulfonamido group is unsubstitutedCitation15, are specific and potent carbonic anhydrase inhibitors (CAIs) and still attract much interestCitation7–9. p-Hydrazinobenzenesulfonamide itself or its condensation derivatives with acetoacetic and levulinic acids had a definite inhibitory activity on carbonic anhydrases when there was no substituent on sulfonamido groupCitation16. It was reported that sulfonamide compounds having five-membered heterocycle system had better CA activity than six-membered ringsCitation17. So, the insertion of the thiophene ring into the chemical design can be a useful strategy to obtain compounds with impressive bioactivity.
The aim of this study was to synthesize 4-aryl-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl) benzenesulfonamides, 9–16, which have pyrazoline, sulfonamide and thiophene pharmacophores, to investigate their CA inhibitory activities.
Materials and methods
Melting points were determined on Buchi 530 (Buchi Labortechnik AG, Flawil, Switzerland). 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained using a Varian Mercury Plus spectrometer (Varian Inc., Palo Alto, CA). Chemical shifts (δ) were reported in parts per million (ppm). Liquid chromatography ion trap-time of flight tandem mass spectrometer (Shimadzu, Kyoto, Japan) equipped with an electrospray ionization (ESI) source, operating in both positive and negative ionization mode. Shimadzu’s LCMS Solution software was used for data analysis.
General procedure for the synthesis of chalcones (1–8)
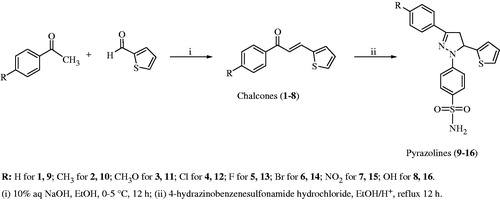
An aqueous solution of NaOH (10%, 10 mL) was added into the ethanol (6 mL) solution of 2-thiophene carbaldehyde (20.0 mmol) and a suitable acetophenone (20.0 mmol) (Scheme 1). The mixture was stirred overnight at room temperature and it was then poured on ice-water (100 ml). The mixture was neutralized with a solution of HCl (10%). The colored precipitate formed was filtered and crystallized from methanol–water (1–8). The yields of the chalcones were in the range of 32–59% [1 (50%), 2 (43%), 3 (46%), 4 (55%), 5 (59%), 6 (44%), 7 (41%), 8 (32%)]Citation18.
Scheme 1. Synthesis of 1,3,5-trisubstituted pyrazoline-bearing benzenesulfonamides, 9–16.
General procedure for the synthesis of pyrazolines (9–16)
The mixture of a suitable chalcone (1.0 mmol) and 4-hydrazino benzenesulfonamide hydrochloride (1.1 mmol) was dissolved in ethanol, and then catalytic amount of glacial acetic acid was added (Scheme 1). The mixture was refluxed for 12 h (9–16). Reactions were followed by thin-layer chromotography (TLC). After the reaction was stopped, some of the solvent was removed under vacuum and the mixture was stirred for 12 h at room temperature. The obtained solid was filtered, dried at room temperature and crystallized from methanol–ether (9–16)Citation6.
4-(3-Phenyl-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (9)
M.p. 207–209 °C. Yield: 77%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 7.79 (d, 2H, J = 8.1 Hz), 7.60 (d, 2H, J = 8.8 Hz), 7.47–7.40 (m, 3H), 7.36 (d, 1H, J = 5.0 Hz), 7.19 (d, 2H, J = 8.8 Hz), 7.14 (d, 1H, J = 3.4 Hz), 7.03 (s, 2H, NH2), 6.94 (dd, 1H, J = 5.0, 3.4 Hz), 5.99 (dd, 1H, J = 11.5, 4.7 Hz), 3.92 (dd, 1H, J = 17.6, 11.5 Hz), 3.32 (dd, 1H, J = 17.6, 4.7 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 150.7, 146.7, 145.2, 134.2, 132.4, 130.1, 129.4, 127.7, 127.6, 126.8, 126.3, 126.2, 113.2, 59.2, 43.7; HRMS (ESI-MS): calcd. for C19H18N3O2S2 [M + H]+ 384.0835; found 384.0822.
4-(5-(Thiophen-2-yl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (10)
M.p. 206–208 °C. Yield: 66%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 7.68 (d, 2H, J = 8.6 Hz), 7.60 (d, 2H, J = 8.4 Hz), 7.35 (d, 1H, J = 5.0 Hz), 7.25 (d, 2H, J = 8.4 Hz), 7.17 (d, 2H, J = 8.6 Hz), 7.13 (d, 1H, J = 3.0 Hz), 7.03 (s, 2H, NH2), 6.93 (dd, 1H, J = 5.0, 3.0 Hz), 5.96 (dd, 1H, J = 11.3, 4.5 Hz), 3.89 (dd, 1H, J = 17.5, 11.3 Hz), 3.29 (dd, 1H, J = 17.5, 4.5 Hz), 2.33 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 150.8, 146.8, 145.2, 139.9, 134.0, 130.0, 129.6, 127.7, 127.6, 126.8, 126.26, 126.21, 113.1, 59.1, 43.8, 21.7; HRMS (ESI-MS): calcd. for C20H20N3O2S2 [M + H]+ 398.0991; found 398.0984.
4-(3-(4-Methoxyphenyl)-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (11)
M.p. 219–221 °C. Yield: 71%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 7.73 (d, 2H, J = 8.8 Hz), 7.58 (d, 2H, J = 8.8 Hz), 7.35 (d, 1H, J = 5.2 Hz), 7.16–7.12 (m, 3H), 7.01–6.99 (m, 4H), 6.93 (dd, 1H, J = 5.2, 3.3 Hz), 5.94 (dd, 1H, J = 11.6, 4.0 Hz), 3.88 (dd, 1H, J = 17.6, 11.6 Hz), 3.79 (s, 3H, OCH3), 3.29 (dd, 1H, J = 17.6, 4.0 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 161.0, 150.7, 146.9, 145.3, 133.8, 128.5, 127.7, 127.6, 126.22, 126.16, 124.9, 114.9, 112.9, 59.0, 56.0, 43.9; HRMS (ESI-MS): calcd. for C20H20N3O3S2 [M + H]+ 414.0941; found 414.0925.
4-(3-(4-Chlorophenyl)-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (12)
M.p. 184–186 °C. Yield: 41%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 7.80 (d, 2H, J = 8.6 Hz), 7.60 (d, 2H, J = 8.8 Hz), 7.51 (d, 2H, J = 8.8 Hz), 7.36 (d, 1H, J = 5.1 Hz), 7.19 (d, 2H, J = 8.6 Hz), 7.14 (d, 1H, J = 3.5 Hz), 7.04 (s, 2H, NH2), 6.93 (dd, 1H, J = 5.1, 3.5 Hz), 6.01 (dd, 1H, J = 11.6, 4.5 Hz), 3.91 (dd, 1H, J = 17.6, 11.6 Hz), 3.32 (dd, 1H, J = 17.6, 4.5 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 149.6, 146.5, 144.9, 134.6, 134.5, 131.3, 129.5, 128.5, 127.7, 127.6, 126.35, 126.32, 113.3, 59.4, 43.6; HRMS (ESI-MS): calcd. for C19H17ClN3O2S2 [M + H]+ 418.0445; found 418.0443.
4-(3-(4-Fluorophenyl)-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (13)
M.p. 200–202 °C. Yield: 85%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 7.84 (dd, 2H, J = 8.8, 5.5 Hz), 7.60 (d, 2H, J = 8.8 Hz), 7.36 (d, 1H, J = 5.1 Hz), 7.28 (t, 2H, J = 8.8 Hz), 7.18 (d, 2H, J = 8.8 Hz), 7.14 (d, 1H, J = 3.2 Hz), 7.03 (s, 2H, NH2), 6.93 (dd, 1H, J = 5.1, 3.2 Hz), 5.99 (dd, 1H, J = 11.6, 4.5 Hz), 3.91 (dd, 1H, J = 17.6, 11.6 Hz), 3.31 (dd, 1H, J = 17.6, 4.5 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 163.4 (d, 1J = 247 Hz), 149.9, 146.7, 145.1, 134.3, 129.1, 129.0, 127.7, 127.6, 126.3, 126.2, 116.5 (d, 2J = 22 Hz), 113.2, 59.3, 43.8; HRMS (ESI-MS): calcd. for C19H17FN3O2S2 [M + H]+ 402.0741; found 402.0734.
4-(3-(4-Bromophenyl)-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (14)
M.p. 197–199 °C. Yield: 58%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 7.73 (d, 2H, J = 8.6 Hz), 7.64 (d, 2H, J = 8.6 Hz), 7.60 (d, 2H, J = 8.6 Hz), 7.36 (d, 1H, J = 5.0 Hz), 7.19 (d, 2H, J = 8.6 Hz), 7.14 (d, 1H, J = 3.4 Hz), 7.04 (s, 2H, NH2), 6.93 (dd, 1H, J = 5.0, 3.4 Hz), 6.01 (dd, 1H, J = 11.7, 4.4 Hz), 3.91 (dd, 1H, J = 17.6, 11.7 Hz), 3.29 (dd, 1H, J = 17.6, 4.4 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 149.7, 146.5, 144.9, 134.5, 132.4, 131.6, 128.7, 127.7, 127.6, 126.4, 126.3, 123.3, 113.3, 59.3, 43.5; HRMS (ESI-MS): calcd. for C19H17BrN3O2S2 [M + H]+ 461.9940; found 461.9929.
4-(3-(4-Nitrophenyl)-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (15)
M.p. 209–211 °C. Yield: 78%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 8.27 (d, 2H, J = 8.8 Hz), 8.02 (d, 2H, J = 8.8 Hz), 7.64 (d, 2H, J = 8.8 Hz), 7.38 (d, 1H, J = 5.1 Hz), 7.26 (d, 2H, J = 8.8 Hz), 7.16 (d, 1H, J = 3.1 Hz), 7.07 (s, 2H, NH2), 6.94 (dd, 1H, J = 5.1, 3.1 Hz), 6.13 (dd, 1H, J = 11.8, 4.6 Hz), 3.98 (dd, 1H, J = 17.7, 11.8 Hz), 3.40 (dd, 1H, J = 17.7, 4.6 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 148.6, 147.8, 145.9, 144.6, 138.6, 135.2, 127.8, 127.7, 127.6, 126.6, 126.5, 124.7, 113.8, 59.7, 43.2; HRMS (ESI-MS): calcd. for C19H17N4O4S2 [M + H]+ 429.0686; found 429.0690.
4-(3-(4-Hydroxyphenyl)-5-(thiophen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)benzenesulfonamide (16)
M.p. 249–251 °C. Yield: 83%. 1H NMR (400 MHz, DMSO-d6, ppm) δ = 9.88 (s, 1H, OH), 7.63 (d, 2H, J = 8.8 Hz), 7.57 (d, 2H, J = 8.8 Hz), 7.35 (d, 1H, J = 5.0 Hz), 7.13–7.11 (m, 3H), 7.01 (s, 2H, NH2), 6.92 (dd, 1H, J = 5.0, 3.6 Hz), 6.82 (d, 2H, J = 8.8 Hz), 5.91 (dd, 1H, J = 11.4, 4.3 Hz), 3.85 (dd, 1H, J = 17.6, 11.4 Hz), 3.24 (dd, 1H, J = 17.6, 4.3 Hz); 13C NMR (100 MHz, DMSO-d6, ppm) δ = 159.6, 151.0, 146.9, 145.4, 133.6, 128.6, 127.7, 127.5, 126.2, 126.1, 123.3, 116.3, 112.8, 58.9, 43.9; HRMS (ESI-MS): calcd. for C19H18N3O3S2 [M + H]+ 400.0784; found 400.0767.
Biological activity
Carbonic anhydrase inhibition assay
Both the CA isoenzymes (hCA I and II) were purified by Sepharose-4B-L-tyrosine-sulfanilamide affinity chromatography in a single purification step as decscribed previouslyCitation19. Thus, pH of the solution was adjusted to 8.7 using solid Tris. Then, supernatant was transferred to the previously prepared Sepharose-4B-L-tyrosine-sulphanilamide affinity columnCitation20. Subsequently, the proteins from the column were spectrophotometrically determined at 280 nmCitation21. For determination of the purity of the hCA isoenzymes, sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), having 10% and 3% acrylamide as an eluent and packing gel, respectively, with 0.1% SDSCitation22, was performed, through which a single band was observed for each isoenzyme.
CA isoenzyme activities were determined following the methods described by Verpoorte et alCitation23 and the methods reported previouslyCitation24. Absorbance change at 348 nm from p-nitrophenylacetate (NPA) to p-nitrophenolate (NP) was recorded in 3 min intervals at the room temperature (25 °C) using a spectrophotometer (Shimadzu, UV-VIS Spectrophotometer, UVmini-1240, Kyoto, Japan)Citation25,Citation26. Quantity of the protein was measured spectrophotometrically at 595 nm during the purification steps according to the Bradford methodCitation27. As reported previously, bovine serum albumin was used as a standard protein. An activity (%)–[benzenesulfonamides] graph was depicted to determine the inhibitory effect of each benzenesulfonamides derivative. For Ki values, three different concentrations of 9–16 were tested. NPA was used as a substrate at five different concentrations, and Lineweaver–Burk curvesCitation28 were drawn as described in previous studiesCitation29,Citation30.
Results and discussion
Compounds 9–16 were successfully synthesized by starting from suitable chalcone and their chemical structures were confirmed by 1H NMR, 13C NMR and HRMS. The detailed interpretation of the spectra are presented in the “Materials and methods” section. CA inhibitory activities of the compounds were tested on hCA I and II isoenzymes and the results are shown in .
Table 1. Human CA isoenzymes (hCA I and II) inhibition value of the compounds (9–16) by the esterase method with 4-nitrophenyl acetate as substrate.
When IC50 values of the compounds were considered, all compounds [compounds(times): 9 (1.9), 10 (2.9), 11 (2.4), 12 (2.0), 13 (1.9), 14 (3.3), 15 (2.9), 16 (2.5)] had 1.9–3.3 times more potent inhibitory potential than acetazolamide (AZA) toward hCA I. The most effective compound toward hCA I in terms of IC50 value was 14, which has bromine substituent, while the least effective ones were fluorine-substituted compound 13 and nonsubstituted compound 9.
The inhibitory activity of the halogen-bearing compounds toward hCA I was inversly correlated with electronegativity of the halogen [14 with bromine (IC50 = 299.48 nM) > 13 with fluorine (IC50 = 521.84 nM) > 12 with chlorine (IC50 = 502.90 nM)] by considering IC50 values. Any type of substituent on the phenyl ring (except fluorine substituent) was useful modification to increase the inhibitory potential of the compounds by decreasing IC50 value toward hCA I.
When the methyl (10)- and methoxy (11)-substituted compound’s IC50 values were compared, introduction of oxygen atom in 11 decreased the inhibitory potential by increasing IC50 of 11. When oxygen-bearing methoxylated compound 11 was compared with compound 16 that is hydroxy-substituted one, 16 was more potent inhibitor than 11. This may suggest that decrease in steric hindrance on oxygen may be helpful to increase the inhibitory potential toward hCA I. On the other hand, when the substituent was nitro (15) in which two oxygen atoms are available on nitrogen, inhibitory potential of 15 was more potent than 16. The order of potency of the compounds toward hCA I in terms of IC50 was 15 (nitro) > 16 (hydroxy) > 11 (methoxy).
It was interesting that compound 10 having methyl group on phenyl ring, which is an electron-donating substituent, had similar inhibitory effect (similar IC50 values) with 15, which has an electron-attracting substituent. This may suggest that the activity is not dependent on the electonegativity of the substituents on the phenyl ring toward hCA I isoenzyme.
On the other hand, when IC50 value of the compounds toward hCA II were considered, the compounds had similar IC50 to AZA. When IC50 values of the compounds were considered, the best inhibitor was nonsubstituted compound 9 toward hCA II. Other substituents than hydrogen on phenyl ring decreased the inhibitory activity by increasing IC50 values of the compounds. The least effective compound was hydroxy-substituted compound 16. Increasing steric hindrance on oxygen atom by the replacement of hydrogen with methyl in methoxy-substituted compound 11 increased the activity toward hCA II isoenzyme, while the introduction of nitro substituent instead of hydroxy group was a useful modification to increase the acitivity. IC50 values of the halogen-bearing compounds were not dependent on the electronegativities of the substituents.
When Ki value of the compounds toward hCA isoenzymes were considered: Ki values were in the range of 232.16 ± 18.17–637.70 ± 310.30 nM toward hCA I and 342.07 ± 94.07–455.80 ± 128.40 nM toward hCA II, while Ki values of AZA were 278.76 ± 44.28 nM and 293.43 ± 46.41 nM toward hCA I and hCA II, respectively. According to Ki values of the compounds toward hCA I, the most effective compound, which has the lowest Ki value, was methyl-substituted compound 10 and the least effective one was chlorine-substituted 12. Compound 10 and hydroxy-substituted compound 16 had more potent Ki values than AZA, while bromine-substituted 14 has Ki value similar to AZA toward hCA I.
When the Ki values of the compounds toward hCA II were considered, the most effective one was fluorine-substituted compound 13 and the least effective one was nitro-susbtituted compound 15. All compounds were less effective than AZA toward hCA II isoenzyme.
Compounds 10, 12, 13, 14 and 16 are reported here for the first time, by detailed spectral analysis and bioactivities. When the IC50 values were considered, the most effective compounds were bromine-substituted 14, which are 3.3 times more potent than AZA, and nonsubstituted compound 9 toward hCA I and II, respectively. On the other hand, according to Ki values, methyl-substituted compound 10 toward hCA I and nitro-substituted compound 15 toward hCA II can be considered as leader compounds for further studies.
In conclusion, we report the synthesis and CA inhibitory activity of a new class of sulfonamides, which showed medium potency against the cytosolic isoforms hCA I and II, presumably due to the very bulky scaffolds present in their molecules. However, such compounds may show interest for the inhibition of other CA isoforms that possess a wider active site, such as hCA IX, XII and XIVCitation31.
Declaration of interest
The authors report no conflicts of interest. The authors are responsible for the content and writing of this article. This research work was supported by Ataturk University Research Found, Turkey (Project No. BAP: 2013/289).
References
- Yamali C, Tugrak M, Gul HI, et al. The inhibitory effects of phenolic Mannich bases on carbonic anhydrase I and II isoenzymes. J Enzyme Inhib Med Chem 2016. [Epub ahead of print]. doi: 10.3109/14756366.2015.1126715
- Sahu NK, Balbhadra SS, Choudhary J, Kohli DV. Exploring pharmacological significance of chalcone scaffold: a review. Curr Med Chem 2012;19:209–25
- Havrylyuk D, Zimenkovsky B, Vasylenko O, Lesyk R. Synthesis and anticancer and antiviral activities of new 2-pyrazoline-substituted 4-thiazolidinones. J Heterocyclic Chem 2013;50:E55–62
- Siddiqui ZN, Musthafa TN, Ahmad A, Khan AU. Thermal solvent-free synthesis of novel pyrazolyl chalcones and pyrazolines as potential antimicrobial agents. Bioorg Med Chem Lett 2011;21:2860–5
- Johnson M, Younglove B, Lee L, et al. Design, synthesis, and biological testing of pyrazoline derivatives of combretastatin-A4. Bioorg Med Chem Lett 2007;17:5897–901
- Sharma PK, Kumar S, Kumar P, et al. Synthesis of 1-(4-aminosulfonylphenyl)-3,5-diarylpyrazoline derivatives as potent antiinflammatory and antimicrobial agents. Med Chem Res 2012;21:2945–54
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49
- Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704
- Hisar O, Beydemir S, Gulcin I, et al. Effects of low molecular weight plasma inhibitors of rainbow trout (Oncorhynchus mykiss) on human erythrocyte carbonic anhydrase-II isozyme activity in vitro and rat erythrocytes in vivo. J Enzyme Inhib Med Chem 2005;20:35–9
- Senturk M, Gulcin I, Dastan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11
- Vedani A, Meyer EF. Jr Structure-activity relationships of sulfonamide drugs and human carbonic anhydrase C: modeling of inhibitor molecules into the receptor site of the enzyme with an interactive computer graphics display. J Pharm Sci 1984;73:352–8
- Lespagnol A, Osteux R, Bar D. Research on carbonic anhydrase inhibitors. Bull Soc Chim Biol 1961;43:789–99
- Davenport HW. The inhibition of carbonic anhydrase by thiophene-2-sulfonamide and sulfanilamide. J Biol Chem 1945;158:567–71
- Dimmock JR, Kandepu NM, Hetherington M, et al. Cytotoxic activities of Mannich bases of chalcones and related compounds. J Med Chem 1998;41:1014–26
- Yildirim A, Atmaca U, Keskin A, et al. N-acylsulfonamides strongly inhibit human carbonic anhydrase isoenzymes I and II. Bioorg Med Chem 2015;23:2598–605
- Gulcin I, Kufrevioglu OI, Oktay M. Purification and characterization of polyphenol oxidase from nettle (Urtica dioica L.) and inhibitory effects of some chemicals on enzyme activity. J Enzyme Inhib Med Chem 2005;20:297–302
- Boztas M, Cetinkaya Y, Topal M, et al. Synthesis and carbonic anhydrase isoenzymes I, II, IX, and XII inhibitory effects of dimethoxybromophenol derivatives incorporating cyclopropane moieties. J Med Chem 2015;58:640–50
- Sarikaya SBO, Sisecioglu M, Cankaya M, et al. Inhibition profile of a series of phenolic acids on bovine lactoperoxidase enzyme. J Enzyme Inhib Med Chem 2015;30:479–83
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9
- Gocer H, Akincioglu A, Goksu S, et al. Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J Enzyme Inhib Med Chem 2015;30:316–20
- Senturk M, Gulcin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9
- Taslimi P, Gulcin I, Oztaskın N, et al. The effects of some bromophenols on human carbonic anhydrase isoenzymes. J Enzym Inhib Med Chem 2016;31:603–7
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934;56:658–66
- Goksu S, Naderi A, Akbaba Y, et al. Carbonic anhydrase inhibitory properties of novel benzylsulfamides using molecular modeling and experimental studies. Bioorg Chem 2014;56:75–82
- Topal M, Gulcin I. Rosmarinic acid: a potent carbonic anhydrase isoenzymes inhibitor. Turk J Chem 2014;38:894–902
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60