
Abstract
A novel series of substituted pyrimidine compounds bearing N-phenylpyrazole and terminating with aryl and cyclic sulfonamido moiety were designed, synthesized, and evaluated in vitro as antiproliferative agents against a panel of 53 cell lines of different tissues at the NCI. Among them, compound 1d with p-chlorobenzenesulfonamido terminal moiety, ethylene spacer, and 4-chloro-3-methoxyphenyl ring at position 3 of the pyrazole nucleus showed the highest mean percentage inhibition value over the whole cancer cell line panel at 10 μM concentration. It showed broad-spectrum antiproliferative activity over many cell lines of different cancer types. For instance, compound 1d inhibited the growth of HL-60 (TB), SR leukemia, and T-47D and MCF-7 breast cancer cell line by 135.92%, 119.44%, 95.32%, and 82.03% at 10 μM, respectively. And it inhibited the growth of COLO 205 colon, HT29 colon, BT-549 breast, and ACHN renal cancer cell lines by more than 80% at the same test concentration. However, testing compound 1d upon determining its IC50 against the most sensitive cell lines showed to good extent selectivity against HT29 colon cancer cell line than HL-60 leukemia and MRC-5 lung fibroblasts (normal cells). Compound 1d was further tested against 12 kinases of different kinase families, and the highest inhibitory effect was exerted against RAF1, V600E-B-RAF, and V600K-B-RAF kinases.
Introduction
Cancer is considered the second leading cause of death after cardiovascular diseases all over the worldCitation1,Citation2. It is a collection of serious diseases caused by uncontrolled growth of cells invading surrounding tissues and spread to other parts of the body. In the statistics according to new report from the World Health Organization’s International Agency for Cancer Research (IARC) suggests that the incidence of cancer worldwide will grow by 75%, nearly doubling in some of the developing countriesCitation3. Those increases will add much more burden on the poorly developed health-care systems in such countries. That is why intensive research should be done in this field for getting more potent drugs with less toxic side effects.
Many research articles have recently reported the potential antiproliferative activity of sulfonamide and cyclic sulfonamide moieties against a variety of cancer cell linesCitation4–10.
In addition, much attention has been paid to the chemistry and biological activities of 1,3,4-triarylpyrazole scaffoldCitation11–15. In the present study, a new series of 1,3,4-triarylpyrazole derivatives containing terminal arylsulfonamido or cyclic sulfamide moieties were designed with similarity to SB203580 and Encorafenib (LGX818) (). SB203580 was reported to bind to P38/MAP kinase via hydrophobic and hydrogen bonding interactions of the fluorophenyl ring with a hydrophobic region, hydrogen bonding of the pyridyl and imidazole rings with the kinase hinge region, and the methylsulfinylphenyl ring with the phosphate binding regionCitation16,Citation17. There is another hydrophobic region below the pyridyl ring which was not occupied by SB203580Citation16. That’s why the terminal chain on the pyridyl ring was important. Our target compounds were also designed similar to Encorafenib through ligand-based design approach. The N-phenyl ring on the pyrazole ring mimics the N-isopropyl group of Encorafenib and estimated to undergo similar interaction. The two aryl rings at positions 3 and 4 of the pyrazole ring mimic the two aryl rings at the same positions on the Encorafenib structure. And the side chain on the pyridyl ring mimics that of Encorafenib with replacement of the terminal methoxycarbonyl moiety of Encorafenib with arylsulfonyl or cyclic sulfamide moiety.
Figure 1. Structures of SB203580, Encorafenib (LGX818), and rationale design to the target compounds 1a–l and 2a–l.
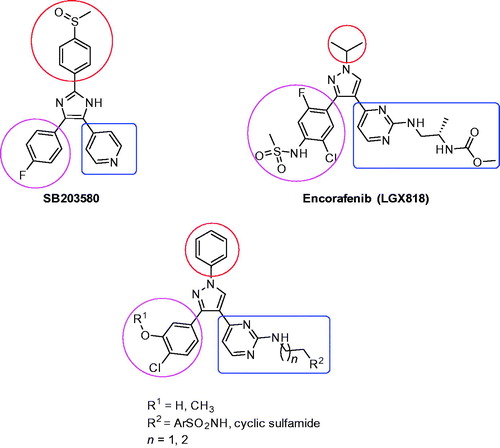
In the present study, we report synthesis of a new series of 1,3,4-triarylpyrazole scaffold having amino pyrimidine moiety similar to that part present in compound B which have been reported as an important fragment presented in many antiproliferative agentsCitation18–20 instead of pyridine and the aminopyrimidine was substituted with ethylene or propylenediamino aryl or cyclic sulfonamide derivatives to test their anticancer activities as illustrated in (. Their in vitro antiproliferative activities against NCI-60 cancer cell line panel are reported.
Experimental
Chemistry
General
The target compounds were purified by column chromatography using silica gel (0.040–0.063 mm, 230–400 mesh) and technical grade solvents. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 400 or 300 spectrometer (Bruker Bioscience, Billerica, MA) using tetramethylsilane as an internal standard. IR analysis (KBr discs, New Jersey, NJ) was carried out using a Bruker FT-IR instrument (Bruker Bioscience, Billerica, MA). Mass spectra (MS) were determined by LC-MS analysis using the following system: Waters 2998 photodiode array detector, Waters 3100 mass detector, Waters SFO system fluidics organizer, Waters 2545 binary gradient module, Waters reagent manager, Waters 2767 sample manager, SunfireTM C18 column (4.6 × 50 mm, 5 μm particle size) (Waters, UK); solvent gradient = 95% A at 0 min, 1% A at 5 min; solvent A: 0.035% trifluoroacetic acid (TFA) in water; solvent B: 0.035% TFA in CH3OH; flow rate = 3.0 mL/min. Solvents and liquid reagents were transferred using hypodermic syringes. Purity percentage of all the target compounds were determined by LC-MS and found to be >95%. The high-resolution mass spectrometry was done on positive mode with capillary: 3.1 KV, desolvation gas: 800 L/h, and extraction cone 4.0. Chromatography was performed on ACQUITY UPLC BEH C18 (1.6 μm, 2.1 × 100 mm) (Waters, Milford, MI) at 25 °C using acetonitrile and water (containing 0.1% formic acid as mobile phase in gradient elution mode at a flow rate of 0.4 mL/min, and injection volume: 5 μL. The data acquisition was under the control of Mass Hunter workstation software. Melting points were obtained on a Walden Precision Apparatus Electrothermal 9300 apparatus (Barnstead, UK) and are uncorrected. Solvents and liquid reagents were transferred using hypodermic syringes. All solvents and reagents were commercially available and used without further purification.
Synthesis of the ethylenediamine and propylenediamine sulfonamides side chain compounds 3a–l was carried out through a pathway as previously reportedCitation19.
Chloro-2-methoxy-4-methylbenzene (5)
To a solution of 2-chloro-5-methylphenol (4, 21.5 g, 150 mmol), dimethyl sulfate (20.8 g, 165 mmol) in acetone (250 mL), anhydrous K2CO3 (51.8 g, 375 mmol) was added. The reaction mixture was heated under reflux for 3 h. The mixture was filtered, and the filtrate was evaporated under reduced pressure. Water (150 mL) was added to the residue, and the resulting mixture was carefully extracted with ethyl acetate. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and filtered. The organic solvent was evaporated under reduced pressure, and the residue was used in the next step without further purification. MS m/z: 157.5 [M++ H].
Chloro-3-methoxybenzoic acid (6)
A stirred mixture of compound 5 (11.17 g, 71 mmol), potassium permanganate (35.0 g, 221 mmol), pyridine (36 mL), and water (107 mL) was heated at 50 °C for 24 h. The mixture was then stirred at room temperature for 13 h. The mixture was filtered and MnO2 was suspended in hot water and again filtered off. The combined aqueous filtrates were washed with ethyl acetate (3 × 75 mL), and then acidified with 2 N H2SO4. The precipitate was filtered off, washed with water, and dried to give the title compound (11.98 g, 90%). mp: 215–216 °C (lit. mp: 217–219 °CCitation21).
Methyl 4-chloro-3-methoxybenzoate (7)
To a solution of the acid 6 (1.0 g, 5.4 mmol) in MeOH (40 mL) at 0 °C, acetyl chloride (1.9 mL, 28.1 mmol) was added dropwise at this temperature and the reaction mixture was then stirred at room temperature for 15 h. After evaporation of the organic solvent, the residue was purified by flash column chromatography (silica gel, hexane–ethyl acetate 5:1 v/v) to give 4 (0.91 g, 85%) as a crystalline solid. mp: 49–50 °C; 1H NMR (400 MHz, DMSO-d6) d 7.62–7.55 (m, 3H), 3.94 (s, 3H), 3.88 (s, 3H).
1-(4-Chloro-3-methoxyphenyl)-2-(2-(methylthio)pyrimidin-4-yl)ethan-1-one (8)
To a solution of compound 7 (1.0 g, 5.0 mmol) and, 4-methyl-2-(methylthio)pyrimidine (0.78 g, 5.6 mmol) in THF (5 mL) in a cooled bath at −78 °C, LHMDS (3.7 mL, 1.0 M solution in THF,19.9 mmol) was slowly added to maintain the temperature at −78 °C. The resulting mixture was stirred overnight at room temperature. After disappearance of the starting material, the mixture was quenched with saturated aqueous NH4Cl. Ethyl acetate (20 mL) was added and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layer extracts were washed with brine and dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography (silica gel, hexane ethyl acetate 1:1 v/v then switching to hexane–ethyl acetate 1:5 v/v) to yield compound 6 (0.58 g, 45%). mp: 85–88 °C; 1H NMR (400 MHz, CDCl3) δ 8.24–8.23 (m, 1H), 7.65–7.60 (m, 1H), 7.36–7.21 (m, 4H), 6.95–6.93 (m, 1H), 6.84–6.82 (m, 1H), 3.95 (s, 5H), 2.65 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 194.4, 157.2, 156.4, 155.4, 155.1, 135.8, 134.9, 130.3, 118.8, 112.8, 109.5, 56.3, 47.9, 14.1.
4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)-2-(methylthio)-pyrimidine (9)
Compound 8 (1.1 g, 3.8 mmol) was added to DMF-DMA (5.14 mL, 38.2 mmol) and the mixture was stirred at 80 °C for 3 h. The resulting solution was concentrated to dryness to furnish oil which was used in the next step without purification. To a portion of the oil from the previous step (0.137 g, 0.457 mmol) in EtOH (3 mL) was added phenylhydrazine (98 mg, 0.9 mmol). The reaction mixture was stirred overnight at room temperature. Water (5 mL) was added to the reaction mixture and the organics were extracted with ethyl acetate (3 × 5 mL). The combined organic layer extracts were washed with brine and dried over anhydrous Na2SO4. After evaporation of the organic solvent, the residue was purified by column chromatography (silica gel, hexane–ethyl acetate 1:1 v/v then switching to hexane–ethyl acetate 1:5 v/v) to yield compound 9 (1.18 g, 81%). 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.26 (d, 1H, J = 5.2 Hz), 7.36–7.21 (m, 5H), 6.82–6.80 (m, 2H), 6.71 (d, 1H, J = 6.4 Hz), 3.68 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.5, 158.9, 156.9, 155.1, 141.2, 140.6, 139.1, 130.6, 129.4, 128.9, 128.0, 125.3, 123.7, 123.3, 120.0, 114.1, 112.5, 56.2, 13.7.
4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)-2-(methylsulfonyl)pyrimidine (10)
To a solution of compound 9 (2.4 g, 6 mmol) in methanol (250 mL), a solution of oxone (12.3 g, 18 mmol) in water (50 mL) was added. The mixture was stirred at room temperature for 16 h. The organic solvent was evaporated under reduced pressure and the remaining aqueous solution was extracted with CH2Cl2 (50 mL) and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (3 × 25 mL) and the combined organic layer extracts were washed with brine, dried over anhydrous MgSO4, and filtered. The organic solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography. 2.5 g of the title compound was obtained, yield 98%. 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 8.39 (d, 1H, J = 5.2 Hz), 7.46–7.29 (m, 5H), 6.92–6.85 (m, 2H), 6.79 (d, 1H, J = 6.0 Hz), 3.71 (s, 3H), 2.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 173.5, 159.1, 157.1, 156.1, 141.4, 140.8, 139.1, 130.7, 129.6, 128.9, 128.1, 125.3, 123.7, 123.3, 120.0, 114.1, 112.5, 56.5, 43.1.
General procedure for preparation of the target compounds 1a–l
A mixture of mesyl compound 10 (0.2 g, 0.5 mmol), amino side chain compound 3a–l (1.2 mmol), and diisopropylethylamine (0.29 mL, 1.7 mmol) in DMSO (5 mL) was stirred at 80 °C for 16 h. The mixture was cooled to room temperature, quenched with water (20 mL), then extracted with ethyl acetate (3 × 20 mL). The combined organic layer extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, appropriate ratio of hexane–ethyl acetate) to obtain the pure target compounds.
N-(2-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)-ethyl)benzenesulfonamide (1a)
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.02 (d, 1H, J = 5.2 Hz), 7.76 (d, 2H, J = 8.0 Hz), 7.51–7.21 (m, 9H), 6.81 (d, 1H, J = 8.0 Hz), 6.76 (s, 1H), 6.35 (brs, 1H), 5.85 (t, 1H, J = 6.4 Hz), 3.66 (s, 3H), 3.32 (brs, 2H), 3.05 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.3, 159.7, 157.4, 155.0, 141.0, 140.5, 140.0, 139.2, 132.4, 129.0, 128.9, 128.0, 126.9, 125.3, 120.5, 114.2, 107.7, 56.3, 43.6, 40.9.
N-(2-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-4-methylbenzenesulfonamide (1b)
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.03 (d, 1H, J =5.2 Hz), 7.65 (d, 2H, J = 8.0 Hz), 7.33–7.20 (m, 8H), 6.81 (dd, 1H, J = 8.0, 1.2 Hz) 6.76 (d, 1H, J = 1.2 Hz), 6.35 (brs, 1H), 5.77 (t, 1H, J = 6.4 Hz), 3.67 (s, 3H), 3.31 (brs, 2H), 3.05 (brs, 2H), 2.37 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.3, 159.7, 157.8, 155.0, 143.2, 141.0, 140.5, 139.2, 130.5, 129.8, 129.6, 128.9, 128.0, 126.9, 125.3, 123.4, 120.5, 114.2, 107.7, 56.2, 43.6, 40.8, 21.5; LC-MS (m/z): 574.8 (M+).
N-(2-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-4-methoxybenzenesulfonamide (1c)
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.03 (d, 1H, J = 5.2 Hz), 7.70 (dd, 2H, J = 9.2, 2.0 Hz), 7.34–7.21 (m, 6H), 6.90–6.87 (m, 2H), 6.82 (dd, 1H, J = 8.4, 2.0 Hz), 6.35 (brs, 1H), 5.77 (t, 1H, J = 6.4 Hz), 3.81 (s, 3H), 3.67 (s, 3H), 3.33 (brs, 2H), 3.05 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.7, 162.3, 159.7, 157.8, 155.0, 141.0, 140.5, 139.3, 130.5, 129.8, 129.1, 128.9, 128.0, 125.3, 123.4, 120.5, 114.2, 114.1, 107.7, 56.2, 55.6, 43.6, 40.8.
4-Chloro-N-(2-((4-(3-(4-chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)benzenesulfonamide (1d)
IR (KBr disc, cm−1): 3378, 3264, 3088, 2931, 2855, 1577, 1499; 1H NMR (400 MHz, CDCl3) δ 8.36 (brs, 1H), 8.09 (d, 1H, J = 5.2 Hz), 7.76 (d, 3H, J = 8.0 Hz), 7.62 (d, 2H, J = 8.4 Hz), 7.41–7.28 (m, 6H), 6.82 (d, 1H, J = 1.2 Hz), 6.92–6.86 (m, 2H), 6.47 (brs, 1H), 5.76 (s, 1H), 3.67 (s, 3H), 3.07 (brs, 2H), 2.70 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.3, 161.1, 159.7, 157.8, 154.7, 141.3, 141.1, 140.5, 139.7, 139.6, 137.7, 130.4, 129.7, 129.4, 128.9, 128.5, 126.1, 124.0, 120.8, 115.5, 107.9, 56.7, 55.4, 42.2; LC-MS (m/z): 594.8 (M+).
N-(2-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-3-fluorobenzenesulfonamide (1e)
1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 8.02 (d, 1H, J = 5.2 Hz), 7.54 (d, 2H, J = 8.0 Hz), 7.43–7.18 (m, 9H), 6.82 (d, 1H, J = 8.0 Hz), 6.77 (s, 1H), 6.38 (brs, 1H), 5.87 (t, 1H, J = 6.4 Hz), 3.67 (s, 3H), 3.31 (brs, 2H), 3.05 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 163.6, 162.3, 161.1, 159.7, 157.8, 155.0, 142.1, 141.1, 140.5, 139.2, 130.8, 130.4, 129.7, 128.9, 128.0, 126.9, 125.3, 123.4, 122.0, 120.3, 119.7, 119.5, 114.3, 114.1, 107.8, 56.2, 43.8, 40.8.
N-(3-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)benzenesulfonamide (1f)
1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 8.06 (d, 1H, J = 5.2 Hz), 7.68 (brs, 2H), 7.51–7.24 (m, 9H), 6.85–6.80 (m, 2H), 6.40 (brs, 1H), 5.85 (t, 1H, J = 6.4 Hz), 3.67 (s, 3H), 3.20 (brs, 2H), 2.89 (brs, 2H), 1.58 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.5, 159.8, 157.9, 154.9, 141.3, 140.3, 140.2, 139.3, 132.3, 130.3, 129.6, 128.9, 128.1, 126.8, 125.6, 123.3, 120.5, 114.6, 107.6, 56.3, 39.9, 37.3, 30.1.
N-(3-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-4-methylbenzenesulfonamide (1g)
1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 8.06 (d, 1H, J = 5.2 Hz), 7.71 (d, 2H, J = 8.0 Hz), 7.37–7.25 (m, 8H), 6.85 (dd, 1H, J = 8.0, 1.2 Hz), 6.72 (d, 1H, J = 1.2 Hz), 6.40 (brs, 1H), 5.67 (t, 1H, J = 6.4 Hz), 3.66 (s, 3H), 3.08 (brs, 2H), 2.89 (brs, 2H), 2.34 (s, 3H), 1.58 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.2, 159.8, 157.9, 154.9, 143.1, 141.2, 140.4, 139.1, 130.4, 129.8, 129.6, 128.9, 128.0, 126.9, 125.3, 123.4, 120.5, 114.2, 107.7, 56.2, 43.6, 40.8, 30.3, 21.5.
N-(3-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-4-methoxybenzenesulfonamide (1h)
IR (KBr disc, cm−1): 3380, 3275, 3091, 2928, 1580, 1500; 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 8.08 (d, 1H, J = 5.2 Hz), 7.72 (dd, 2H, J = 9.2, 2.0 Hz), 7.33–7.19 (m, 6H), 6.92–6.82 (m, 3H), 6.41 (brs, 1H), 5.67 (t, 1H, J = 6.4 Hz), 3.80 (s, 3H), 3.65 (s, 3H), 3.08 (brs, 2H), 2.88 (brs, 2H), 1.58 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.5, 162.1, 159.8, 157.7, 155.0, 141.1, 140.4, 139.2, 130.5, 129.8, 129.1, 128.9, 128.0, 125.3, 123.4, 120.5, 114.2, 114.1, 107.7, 56.3, 55.7, 39.6, 37.3, 29.2; LC-MS (m/z): 604.9 (M+).
4-Chloro-N-(3-((4-(3-(4-chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)benzenesulfonamide (1i)
1H NMR (400 MHz, CDCl3) δ 8.34 (brs, 1H), 8.11 (d, 1H, J = 4.8 Hz), 7.76 (d, 3H, J = 8.4 Hz), 7.66 (d, 1H, J = 8.4 Hz), 7.41–7.28 (m, 6H), 6.82 (s, 1H), 6.92–6.86 (m, 2H), 6.55 (brs, 1H), 5.76 (s, 1H), 3.66 (s, 3H), 2.88 (brs, 2H), 2.72 (brs, 2H), 1.45 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.6, 161.1, 159.7, 157.8, 154.7, 141.3, 141.1, 140.5, 139.8, 139.6, 137.7, 130.4, 129.7, 129.4, 128.9, 128.5, 126.1, 124.0, 120.8, 115.5, 107.9, 56.7, 55.2, 42.0, 29.5.
N-(3-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-3-fluorobenzenesulfonamide (1j)
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.08 (d, 1H, J = 5.2 Hz), 7.45–7.16 (m, 9H), 6.82–6.80 (m, 2H), 6.40 (brs, 1H), 5.47 (t, 1H, J = 6.4 Hz), 3.68 (s, 3H), 3.20 (brs, 2H), 2.89 (brs, 2H), 1.59 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 163.8, 162.4, 161.0, 159.8, 157.4, 155.0, 142.1, 141.1, 140.5, 139.2, 130.8, 130.4, 129.7, 128.9, 128.0, 126.9, 125.3, 123.4, 122.0, 120.3, 119.7, 119.5, 114.3, 114.1, 107.8, 56.2, 39.8, 37.4, 30.3.
2-(2-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-5-ethyl-1,2,5-thiadiazolidine 1,1-dioxide (1k)
1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.06 (brs, 1H), 7.28–7.14 (m, 6H), 6.76 (dd, 1H, J = 8.0, 2.0 Hz), 6.68 (d, 1H, J = 2.0 Hz), 6.38 (brs, 1H), 5.62 (t, 1H, J = 6.0 Hz), 3.61 (s, 3H), 3.46 (brs, 2H), 3.38–3.21 (m, 4H), 3.12 (brs, 2H), 3.00 (m, 2H), 1.18 (t, 3H, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 161.3, 159.9, 157.4, 155.0, 141.0, 140.6, 130.5, 129.8, 128.9, 128.0, 125.3, 123.5, 120.7, 114.1, 107.7, 56.2, 47.1, 46.1, 45.1, 42.6, 39.3, 13.0.
2-(3-((4-(3-(4-Chloro-3-methoxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-5-ethyl-1,2,5-thiadiazolidine 1,1-dioxide (1l)
1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 8.09 (d, 1H, J =5.2 Hz), 7.36–7.21 (m, 6H), 6.81(dd, 1H, J = 8.4, 2.0 Hz), 6.76 (d, 1H, J = 2.0 Hz), 6.33 (brs, 1H), 5.31 (t, 1H, J = 6.0 Hz), 3.68 (s, 3H), 3.40 (brs, 2H), 3.30 (brs, 4H), 3.14–3.08 (m, 4H), 1.85 (brs, 2H), 1.26 (t, 3H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 162.4, 159.6, 157.9, 154.9, 140.8, 140.6, 139.4, 130.4, 129.9, 128.9, 128.0, 127.9, 125.3, 123.5, 120.9, 114.2, 107.5, 56.2, 45.5, 45.0, 42.5, 38.7, 27.5, 12.9.
General procedure for preparation of the target compounds 2a–l
A solution of the appropriate methoxy compound 1a–l (0.2 mmol) in anhydrous dichloromethane (3 mL) was cooled to −78 °C, and BBr3 (1M in DCM solution, 3 mL, 2.1 mmol) was added dropwise thereto under nitrogen over a period of 10 min. The reaction mixture was stirred at the same temperature for 1 h, then the temperature was allowed to reach room temperature and kept overnight at same temperature. 10% Na2CO3 aqueous solution (15 mL) and ethyl acetate (15 mL) were added, and the alkaline aqueous layer was extracted with ethyl acetate (3 × 20 mL). The combined organic layer extracts were washed with brine, separated, and dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure, and the residue was purified by column chromatography to obtain the purified title compounds.
N-(2-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)benzenesulfonamide (2a)
1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 7.85 (d, 1H, J = 5.2 Hz), 7.75 (d, 2H, J = 7.2 Hz), 7.47–7.15 (m, 9H), 6.85 (d, 1H, J = 1.6 Hz), 6.59 (dd, 1H, J = 8.4, 5.0 Hz), 6.35 (brs, 1H), 5.74 (t, 1H, J = 6 Hz), 3.26 (brs, 2H), 2.99 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 161.8, 159.9, 157.2, 152.4, 141.1, 140.4, 139.8, 139.0, 132.5, 130.0, 129.8, 128.9, 128.0, 126.9, 125.2, 122.8, 121.7, 120.3, 118.8, 107.7, 43.2, 40.9.
N-(2-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-4-methylbenzenesulfonamide (2b)
1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 8.04 (d, 1H, J = 5.2 Hz), 7.71 (d, 2H, J = 8.0 Hz), 7.29–7.19 (m, 8H), 7.08 (s, 1H), 6.56 (dd, 2H, J = 8.0, 2.0 Hz), 6.26 (brs, 2H), 3.34 (brs, 2H), 3.01 (brs, 2H), 2.32 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.6, 159.7, 158.9, 152.2, 143.3, 140.9, 138.8, 136.7, 129.9, 129.7, 128.3, 127.1, 125.3, 122.7, 121.9, 119.6, 118.9, 107.5, 41.1, 30.9, 21.5.
N-(2-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-4-hydroxybenzenesulfonamide (2c)
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.03 (d, 1H, J = 5.2 Hz), 7.70 (dd, 2H, J = 9.2, 2.0 Hz), 7.34–7.21 (m, 6H), 6.90–6.87 (m, 2H), 6.82 (dd, 1H, J = 8.4, 2.0 Hz), 6.35 (brs, 1H), 5.77 (t, 1H, J = 6.4 Hz), 3.35 (brs, 2H), 3.09 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.9, 162.4, 159.8, 157.9, 154.9, 141.4, 140.7, 139.8, 130.5, 129.8, 129.4, 128.9, 128.0, 125.3, 123.4, 120.5, 114.2, 114.2, 107.7, 41.6, 30.9.
4-Chloro-N-(2-((4-(3-(4-chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)benzenesulfonamide (2d)
1H NMR (400 MHz, CDCl3) δ 8.15 (brs, 1H), 7.85 (d, 1H, J = 5.2 Hz), 7.65 (d, 2H, J = 8.8 Hz), 7.32–7.15 (m, 8H), 6.85 (d, 1H, J = 1.6 Hz), 6.60 (d, 1H, J = 8.0 Hz), 6.36 (brs, 1H), 5.79 (t, 1H, J = 6.0 Hz), 3.23 (brs, 2H), 2.97 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 161.1, 159.9, 157.1, 152.5, 141.2, 140.4, 138.9, 138.8, 138.3, 130.0, 129.8, 129.3, 128.9, 128.4, 125.5, 122.8, 121.8, 120.2, 118.8, 107.6, 43.1, 40.8.
N-(2-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-3-fluorobenzenesulfonamide (2e)
1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 8.09 (d, 1H, J = 5.2 Hz), 7.58 (d, 2H, J = 8.0 Hz), 7.73–7.28 (m, 9H), 6.92 (d, 1H, J = 8.0 Hz), 6.83 (s, 1H), 6.57 (brs, 1H), 5.96 (t, 1H, J = 6.4 Hz), 3.35 (brs, 2H), 3.09 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 163.9, 162.8, 161.5, 159.9, 157.7, 155.6, 142.4, 141.3, 140.7, 139.7, 131.3, 130.0, 129.8, 128.8, 128.3, 126.9, 125.3, 123.4, 122.0, 120.3, 119.7, 119.5, 114.3, 114.2, 107.5, 41.2, 30.8.
N-(3-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)benzenesulfonamide (2f)
IR (KBr disc, cm−1): 3366, 3269, 3071, 2955, 2853, 1577, 1499; 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.89 (d, 1H, J = 5.2 Hz), 7.81 (d, 2H, J = 7.2 Hz), 7.51–7.18 (m, 9H), 6.87 (d, 1H, J = 1.6 Hz), 6.47 (dd, 1H, J = 8.4, 5.0 Hz), 6.35 (brs, 1H), 5.74 (t, 1H, J = 6.0 Hz), 3.26 (brs, 2H), 2.99 (brs, 2H), 1.84 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 161.9, 159.8, 157.4, 152.6, 141.4, 140.5, 139.7, 138.8, 132.6, 130.0, 129.8, 128.9, 128.0, 126.9, 125.2, 122.8, 121.7, 120.3, 118.8, 107.7, 43.2, 40.9, 29.5; LC-MS (m/z): 559.9 (M+).
N-(3-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-4-methylbenzenesulfonamide (2g)
1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 8.06 (d, 1H, J = 5.2 Hz), 7.61 (d, 2H, J = 7.2 Hz), 7.31–7.21 (m, 10H), 6.97 (s, 1H), 6.73 (d, 2H, J = 8.0 Hz), 6.38 (brs, 1H), 3.28 (brs, 2H), 2.92 (brs, 2H), 2.38 (s, 3H), 1.62 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.6, 159.7, 158.9, 153.4, 142.9, 141.2, 140.5, 139.5, 138.1, 130.6, 129.4, 128.4, 128.0, 125.7, 122.5, 120.9, 118.8, 106.5, 43.3, 41.0, 29.5, 21.4.
N-(3-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-4-hydroxybenzenesulfonamide (2h)
1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 8.11 (d, 1H, J = 5.2 Hz), 7.82 (dd, 2H, J = 9.2, 2.0 Hz), 7.36–7.21 (m, 6H), 6.96–6.88 (m, 3H), 6.41 (brs, 1H), 5.67 (t, 1H, J = 6.4 Hz), 3.12 (brs, 2H), 2.92 (brs, 2H), 1.68 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 162.5, 162.1, 159.8, 157.7, 155.0, 141.1, 140.4, 139.2, 130.5, 129.8, 129.1, 128.9, 128.0, 125.3, 123.4, 120.5, 114.2, 114.1, 107.7, 37.4, 35.3, 29.2.
4-Chloro-N-(3-((4-(3-(4-chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)benzenesulfonamide (2i)
1H NMR (400 MHz, DMSO-d6) δ 8.36 (brs, 1H), 8.14 (d, 1H, J = 5.2 Hz), 7.79 (d, 3H, J = 8.0 Hz), 7.70 (d, 1H, J = 8.4 Hz), 7.47–7.32 (m, 6H), 6.89 (s, 1H), 6.98–6.89 (m, 2H), 6.65 (brs, 1H), 5.86 (s, 1H), 2.95 (brs, 2H), 2.80 (brs, 2H), 1.61 (brs, 2H); 13C NMR (100 MHz, DMSO-d6) δ 162.6, 161.1, 159.9, 157.1, 153.4, 141.2, 140.6, 139.9, 139.6, 137.6, 129.8, 129.4, 128.9, 128.4, 128.1, 125.7, 122.5, 121.8, 120.9, 118.8, 107.6, 41.1, 31.2, 29.5.
N-(3-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-3-fluorobenzenesulfonamide (2j)
1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 8.13 (d, 1H, J = 5.2 Hz), 7.75–7.29 (m, 11H), 7.00–6.72 (m, 3H), 6.51 (brs, 1H), 3.21 (brs, 2H), 2.81 (brs, 2H), 1.55 (brs, 2H); 13C NMR (100 MHz, DMSO-d6) δ 163.5, 162.6, 161.0, 159.2, 158.5, 153.4, 143.1, 141.2, 140.6, 139.6, 132.1, 130.5, 129.4, 128.4, 125.7, 123.2, 122.5, 120.9, 120.0, 119.8, 118.8, 114.1, 113.8, 106.6, 41.1, 38.3, 29.6.
2-(2-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)ethyl)-5-ethyl-1,2,5-thiadiazolidine 1,1-dioxide (2k)
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 8.06 (d, 1H, J = 5.2 Hz), 7.32–7.18 (m, 6H), 6.77 (dd, 1H, J = 8.0, 2.0 Hz), 6.72 (d, 1H, J = 2.0 Hz), 6.38 (brs, 1H), 5.35 (t, 1H, J = 6.0 Hz), 3.48 (brs, 2H), 3.34 (brs, 2H), 3.13–3.05 (m, 4H), 1.87 (brs, 2H), 1.31 (t, 3H, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 162.5, 159.8, 157.7, 155.1, 141.1, 140.5, 139.5, 130.5, 129.8, 128.9, 128.0, 127.9, 125.3, 123.6, 120.9, 114.2, 107.7, 42.5, 41.9, 41.3, 36.5, 12.4.
2-(3-((4-(3-(4-Chloro-3-hydroxyphenyl)-1-phenyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propyl)-5-ethyl-1,2,5-thiadiazolidine 1,1-dioxide (2l)
1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 8.11 (d, 1H, J = 5.2 Hz), 7.46–7.23 (m, 6H), 6.84 (dd, 1H, J = 8.0, 2.0 Hz), 6.86 (d, 1H, J = 2.0 Hz), 6.36 (brs, 1H), 5.47 (t, 1H, J = 6.0 Hz), 3.43 (brs, 2H), 3.35 (brs, 4H), 3.18–3.12 (m, 4H), 1.89 (brs, 2H), 1.37 (t, 3H, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 162.6, 159.8, 157.7, 155.0, 141.2, 140.2, 139.5, 130.5, 129.8, 128.9, 128.0, 127.9, 125.3, 123.7, 120.9, 114.2, 107.5, 42.5, 42.0, 41.5, 36.6, 25.4, 12.5.
Cancer cell line screening at the NCI
Screening against the cancer cell lines was carried out at the National Cancer Institute (NCI), Bethesda, MD, applying the standard protocol of the NCI (DTP Human Tumor Cell Line Screen Process: http://www.dtp.nci.nih.gov/branches/btb/ivclsp.html). Single-dose protocol: compounds submitted to the NCI 60 Cell screen are tested initially at a single high dose (10−5 M) in the full panel of 53 cancer cell line. The single-dose data will be reported as a mean graph of the percentage growth of treated cells and will be similar in appearance to mean graphs from the 5-dose assay. The number reported for the single-dose assay is growth relative to the no-drug control and to the time zero number of cells. This allows detection of both growth inhibition (values between 0 and 100) and lethality (values less than 0). The percentage growth is calculated at each of the drug concentrations levels. Percentage growth inhibition is calculated as:
[(Ti − Tz)/(C − Tz)] × 100 for concentrations for which Ti ≥ Tz
[(Ti − Tz)/Tz] × 100 for concentrations for which Ti < Tz.
MTT cell viability assay
Materials and methods
Compound 1d was submitted to Reaction Biology Corporation (http://www.reactionbiology.com) for MTT assay. The targeted compound was dissolved in DMSO at 10 mM stock. Control compound was purchased from Sigma-Aldrich (Saint Louis, MO). CellTiter 96® Non-Radioactive Cell Proliferation Assay (MTT) reagent was purchased from Promega (Madison, WI). HL-60, HT29 and MRC-5 cell lines were purchased from American Type Culture Collection (Manassas, VA). HL-60 cells were grown in IMDM medium, HT-29 in McCoy’s 5α medium, and MRC-5 cells in EMEM medium. 100 μg/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum were added to all of the culture media. Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Compound 1d and the reference compound staurosporine were diluted with DMSO in 10-dose and 3-fold serial dilutions. 225 nL of 1d or 25 nL of staurosporine was delivered from the source plate to the wells of a 384-well cell culture plate by Echo 550 Liquid Handler. 25 μL of culture medium containing 2000 of cells was added to the wells of the 384-well culture plate. The plate was incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air for 72 h. 5 μL of MTT Dye Solution was added to each well. The plate was incubated at 37 °C in a CO2 incubator for 6 h. After incubation, the formazan was produced in the cells. 25 μL of Solubilization/Stop Solution was added to each well and the plate was incubated for another 1 h at 37 °C in a CO2 incubator to dissolve the formazan crystals. The absorbance of each sample was measured at 590 nm using Envision 2104 Multilabel Reader (PerkinElmer, Santa Clara, CA). The cell viability was determined based on the quantification of the color intensity in each culture well. The IC50 curves were plotted and the IC50 values were calculated using the GraphPad Prism 4 program based on a sigmoidal dose–response equation.
In vitro kinase screening
Reaction Biology Corp. Kinase HotSpotSM service (http://www.reactionbiology.com) was used for screening of compound 1d. Assay protocol: in a final reaction volume of 25 μL, kinase (5–10 mU) is incubated with 25 mM Tris pH 7.5, 0.02 mM EGTA, 0.66 mg/mL relevant substrate, and 10 mM magnesium acetate and [γ33P-ATP] (specific activity approx. 500 cpm/pmol, concentration 1 μM). The reaction is initiated by the addition of the Mg-ATP mix. After incubation for 40 min at room temperature, the reaction is stopped by the addition of 5 μL of a 3% phosphoric acid solution. 10 μL of the reaction is then spotted onto a P30 filtermat and washed thrice for 5 min in 75 mM phosphoric acid and once in methanol prior to drying and scintillation counting.
Results and discussion
Chemistry
Synthesis of the target designed compounds 1a–l and 2a–l was achieved through the pathway shown in Scheme 1. Methylation of the phenolic hydroxyl group of 2-chloro-5-methylphenol (4) using dimethyl sulfate gave 1-chloro-2-methoxy-4-methylbenzene (5)Citation22. Oxidation of the methyl group of 5 using KMnO4 produced 4-chloro-3-methoxybenzoic acid (6)Citation21. Esterification of the carboxylic acid 6 using methanol in the presence of acetyl chloride produced the corresponding methyl ester 7. 4-Methyl-2-(methylthio)pyrimidine was prepared by methylation of 4-methylpyrimidine-2-thiol with iodomethane in the presence of aqueous NaOH. Upon reacting the ester with the pyrimidine methyl thioether in the presence of lithium bis(trimethylsilyl)amide (LiHMDS), the ketide intermediate 8 was formed. Reaction of compound 8 with dimethylformamide dimethyl acetal (DMF-DMA) followed by subsequent treatment with phenylhydrazine yielded the N-phenylpyrazole intermediate 9Citation23. Oxidation of the methylsulfide moiety of compound 9 using oxoneCitation19 produced the corresponding mesyl compound 10. Heating compound 10 with the appropriate amino reagents 3a–l in the presence of diisopropylethylamine led to displacement of the mesyl group of compound 10 by the amino group and formation of the target compounds 1a–l. Demethylation of the methoxy group of 1a–l using BBr3 afforded the corresponding hydroxyl derivatives 2a–l. Structures of the target compounds, their yield percentages, and melting points are illustrated in .
Scheme 1. Reagents and conditions: (a) (i) triethylamine, CH2Cl2, 0 °C, RT, overnight; (ii) N-ethyl ethylenediamine, C5H5N, reflux, 3 h; (iii) benzyloxycarbonyl chloride, TEA, CH2Cl2, 0 °C; (iv) methanesulfonyl chloride, TEA, CH2Cl2, 0 °C; (v) IV, NaH (60% dispersion in mineral oil), DMF; (vi) H2/Pd-C, MeOH, RT, 1 h; (b) (CH3)2SO4, K2CO3, acetone, reflux, 3 h; (c) KMnO4, C5H5N, H2O, 50 °C, 24 h, then RT, 13 h; (d) acetyl chloride, CH3OH, RT, 15 h; (e) 4-methyl-2-(methylthio)pyrimidine, LiHMDS, THF, −78 °C, 2 h then RT, overnight; (f) (i) DMF-DMA, 80 °C, 3 h; (ii) phenylhydrazine, C2H5OH, RT, overnight; (g) oxone, MeOH, H2O, RT, 16 h; (h) 3a–l, diisopropylethylamine, DMSO, 80 °C, 16 h; (i) BBr3, CH2Cl2, −78 °C, 1 h; RT, overnight.
Table 1. Structures of the target compounds 1a–l and 2a–l, their yield percentage, and melting points.
In vitro antiproliferative activity against NCI-60 cancer cell line panel (single dose)
Structures of the target synthesized compounds were submitted to National Cancer Institute (NCI, Bethesda, MD), and the 11 derivatives shown in were selected on the basis of degree of structural variation technique for testing their antiproliferative activity. The selected compounds were subjected to an in vitro anticancer assay against a full panel of 53 cancer cell lines of nine different cancer types; leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancers. The compounds were tested at a single-dose concentration of 10 μM, and the percentages of growth inhibition over the 53 tested cell lines were determined. The mean percentage growth of the NCI-60 cancer cell line panel after treatment with each of the tested compound is illustrated in .
Table 2. Mean percentage growth of the 53 cancer cell line panel after treatment with the tested target compounds (10 μM concentration).
Upon investigating the effect of linker length on antiproliferative activity, it was found that compound 1d with ethylene linker was more active than the corresponding propylene derivative 1i. The ethylene spacer might be optimal for appropriate fitting at the receptor site. Regarding the terminal moieties (arylsulfonamido and the cyclic sulfamide present in the terminal moieties), it was found that compounds 1a, d, h–j, 2f, and 2i possessing arylsulfonamido showed higher antiproliferative activity than cyclic ones 1k, 1l, 2k, and 2l. This may be attributed to that increased hydrophobicity by the aromatic moieties might enhance the compound penetration through the cell membrane to inside the cell and/or stronger affinity at the receptor site due to stronger hydrophobic interactions. Any or both of these effects might lead to stronger antiproliferative activity. This assumption could be supported by the slightly higher antiproliferative activity of the methoxy compounds 1k and 1l than the corresponding hydroxyl analogs 2k and 2l. The results also showed that the compounds with substituted arylsulfonamide moieties 1d and 1h–j were more active than those with unsubstituted phenyl ring 1a. The increased bulkiness and lipophilicity induced by those substituents might lead to enhanced activity due to more appropriate fitting at the receptor site and/or more penetration to inside the cancer cells.
The percentage inhibition values of the most active compounds over the most sensitive cell lines are summarized in . SR leukemia, MCF-7, and T-47D breast cancer cell lines were the most sensitive cell lines to this series of compounds. The most active compounds against SR leukemia were 1d, 2f, and 1h with percentage inhibitions of 119.44%, 58.02, and 56.01%, respectively. MCF-7 and T-47D breast cancer cell line were also sensitive to the most potent compounds. illustrates the percentage inhibition values of the tested target compounds against SR leukemia, MCF-7, and T-47D breast cancer cell line. It is obvious from the figure that compounds 1d then 2f are the most active among the 10 tested compounds against those three cell lines.
Figure 2. Percentage inhibition expressed by compounds 1d (a), 1h (b), and 2f (c) at 10 μM concentration over all cell lines of the NCI cancer cell line panel of nine different cancer types. The inhibition percentages are calculated by subtracting the growth percentages from 100.
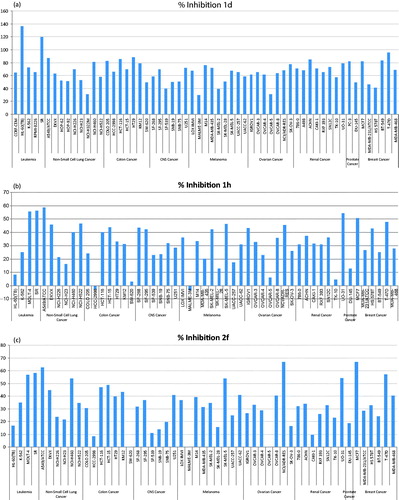
Table 3. IC50 (μM) results exerted by the compound 1d and staurosporine against HL-60 leukemia cell line, HT29 colon cancer cell line, and MRC-5 lung fibroblasts.
Among all the tested derivatives, compound 1d was the most active. It exerted broad-spectrum antiproliferative activity against different cell lines of different cancer types. It exerted the highest activities against HL-60 (TB) and SR leukemia cell lines.
Compounds 1d, 1h, and 2f showed the highest mean inhibitions. The percentages of inhibition of these three compounds over each tested cell line of the NCI-60 panel at 10 μM concentration are depicted in . The three compounds exerted broad-spectrum antiproliferative activities against different cell lines of different cancer types. In general, compound 1d was significantly more active than compounds 1h and 2f. The p-chlorophenylsulfonamido terminal moiety of compound 1d was more optimal than p-methoxyphenylsulfonamido and phenylsulfonamido moieties of compounds 1h and 2f, respectively. As mentioned previously, it might enhance the penetration into inside the cells and/or increase the affinity at the receptor site. The ethylene spacer of compound 1d seems to be more optimal for appropriate fitting at the receptor site than the propylene linker of compounds 1h and 2f. And the methoxy group on the aryl ring at position 3 of the pyrazole ring could be more favorable than hydroxyl due to hydrophobicity, steric, and electronic differences between them.
MTT assay
Compound 1d was further tested at Reaction Biology Corporation to identify its IC50 against HL-60 (TB) leukemia and HT-29 colon cancer against which our compound showed the most promising growth inhibition percentage and also were available in reaction biology cell line screening panels. It was also tested against MRC-5 lung fibroblasts normal cell line in order to investigate the selectivity index. The IC50 values are illustrated in . The compound showed higher potency against HT29 colon cancer cell line than HL-60 leukemia cell line, with good selectivity index toward cancer cells than normal cells. On the other hand, staurosporine, the reference standard drug, was much more potent against normal fibroblasts than cancer cells. So, compound 1d has the merit of higher safety than staurosporine.
In vitro kinase screening
In order to investigate the probable mechanism of action of compound 1d at molecular level, it was selected to be tested at a single-dose concentration of 10 μM over 12 kinases; from which V600E-B-RAF and RAF1 which are over-expressed in HT29 cancer cell lineCitation24–26 and others belong to different kinase families to investigate the selectivity. As illustrated in , compound 1d showed better inhibitory effect over three kinases at 10 μM. It exerted 78.04%, 74.47%, and 72.46% inhibitions against RAF1, V600E-B-RAF, and V600K-B-RAF kinases, respectively. But it demonstrated low activity against the other tested kinases. So, it was further tested in a 10-dose testing mode in order to determine its IC50 values over RAF1 and V600E mutated B-RAF. The IC50 values were 2.47 μM and 3.57 μM, respectively. As mentioned previously, we can deduce the selectivity of this compound against RAF1 and V600E-B-RAF kinases, and the corresponding cell line possessing the over-expressed kinases V600E-B-RAF and RAF1 as postulated () which helps us deduce that this compound might be working, at least in part, through inhibition of RAF kinases as antiproliferative agent. V600K-B-RAF kinase has been reported more in melanoma than in colon cancerCitation27. The moderate activity of compound 1d against the melanoma subpanel might be partially due to V600K-B-RAF inhibition.
Table 4. Percentage inhibition and IC50 (μM) results exerted by the most potent compounds over the selected kinases.
Conclusion
In this study, a novel series of pyrimidinylpyrazoles possessing terminal arylsulfonamido or cyclic sulfamide moiety separated by ethyl or propyl spacer was synthesized. The target compounds were tested for in vitro antiproliferative activities over NCI-60 cancer cell line panel of nine different cancer types. Among them, compound 1d possessing p-chlorobenzenesulfonamido terminal moiety, ethylene linker, and 4-chloro-3-methoxyphenyl ring at position 3 of the pyrazole nucleus showed the highest activity. So these moieties together may comprise potential pharmacophoric hits for this series of compounds. Compound 1d exerted broad-spectrum antiproliferative activities over different cell lines of different cancer types. The inhibition percentage values of compound 1d over HL-60 (TB) and SR leukemia, HT29 colon, and T-47D and MCF-7 breast cancer cell lines were 135.92%, 119.44%, 88.66%, 95.32%, and 82.03%, respectively. SR leukemia, MCF-7, and T-47D breast cancer cell lines were found to be highly sensitive to the most active compounds. Compound 1d gave 13.37 μM IC50 value upon testing against HT29 colon cancer cell line, but it showed lower potency against HL-60 leukemia and MRC-5 lung fibroblasts. It showed higher selectivity toward HT29 colon cancer cell line than the normal fibroblasts, compared to staurosporine. So despite of inferior potency than staurosporine against HT29 colon cancer cell line, it is much safer for the normal cells. Upon testing the compound against kinases, it showed selectivity against RAF1 and V600E-B-RAF kinases which are over-expressed in HT29 colon cancer cell line from which we can deduce that this compound might express its antiproliferative effect, at least in part, through inhibition of RAF kinases. Therefore, compound 1d could be utilized as a lead compound for future development of active anticancer agents inhibiting RAF kinases. Further modifications of this series of compounds in order to improve their anticancer activity are currently in progress.
Declaration of interest
The authors have declared no conflict of interest.
This work was supported by Korea Institute of Science and Technology (KIST), KIST Project (2E25260). We would like to thank the National Cancer Institute (NCI), Bethesda, Maryland, USA, for performing the in vitro anticancer testing over the cell lines.
Supplementary material available online
IENZ_1190715_Supplementary_Material.pdf
Download PDF (1.3 MB)Related Research Data
References
- Frankish H. 15 million new cancer cases per year by 2020, says WHO. Lancet 2003;361:1278
- Belpomme D, Irigaray P, Sasco AJ, et al. The growing incidence of cancer: role of lifestyle and screening detection (Review). Int J Oncol 2007;30:1037–49
- Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer 2013;132:1133–45
- Bano S, Javed K, Ahmad S, et al. Synthesis and biological evaluation of some new 2-pyrazolines bearing benzene sulfonamide moiety as potential anti-inflammatory and anti-cancer agents. Eur J Med Chem 2011;46:5763–8
- Ghorab MM, Shaaban MA, Refaat HM, et al. Anticancer and radiosensitizing evaluation of some new pyranothiazole-Schiff bases bearing the biologically active sulfonamide moiety. Eur J Med Chem 2012;53:403–7
- Lal J, Gupta SK, Thavaselvam D, Agarwal DD. Biological activity, design, synthesis and structure activity relationship of some novel derivatives of curcumin containing sulfonamides. Eur J Med Chem 2013;64:579–88
- Wang ZC, Qin YJ, Wang PF, et al. Sulfonamides containing coumarin moieties selectively and potently inhibit carbonic anhydrases II and IX: design, synthesis, inhibitory activity and 3D-QSAR analysis. Eur J Med Chem 2013;66:1–11
- Wang XM, Xu J, Li YP, et al. Synthesis and anticancer activity evaluation of a series of [1,2,4]triazolo[1,5-a]pyridinylpyridines in vitro and in vivo. Eur J Med Chem 2013;67:243–53
- Xu F, Xu H, Wang X, et al. Discovery of N-(3-((7H-purin-6-yl)thio)-4-hydroxynaphthalen-1-yl)-sulfonamide derivatives as novel protein kinase and angiogenesis inhibitors for the treatment of cancer: synthesis and biological evaluation. Part III. Bioorg Med Chem 2014;22:1487–95
- Gamal El-Din MM, El-Gamal MI, Abdel-Maksoud MS, Oh CH. Synthesis, in vitro antiproliferative activity, and in silico studies of new 1,3,4-oxadiazole derivatives possessing sulfonamide moiety. Eur J Med Chem 2015;90:45–52
- Choi WK, El-Gamal MI, Choi HS, et al. New diarylureas and diarylamides containing 1,3,4-triarylpyrazole scaffold: synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur J Med Chem 2011;46:5754–62
- Choi WK, El-Gamal MI, Choi HS, et al. Design, synthesis, and preliminary cytotoxicity evaluation of new diarylureas and diarylamides possessing 1,3,4-triarylpyrazole scaffold Bull. Korean Chem Soc 2012;33:98–2991
- El-Gamal MI, Park YS, Chi DY, et al. New triarylpyrazoles as broad-spectrum anticancer agents: design, synthesis, and biological evaluation. Eur J Med Chem 2013;65:315–22
- El-Gamal MI, Choi HS, Cho HG, et al. Design, synthesis, and antiproliferative activity of 3,4-diarylpyrazole-1-carboxamide derivatives against melanoma cell line. Arch Pharm (Weinheim) 2011;344:745–54
- El-Gamal MI, Choi HS, Yoo KH, et al. Antiproliferative diarylpyrazole derivatives as dual inhibitors of the ERK pathway and COX-2. Chem Biol Drug Des 2013;82:336–47
- Dietrich JD. The design, synthesis, and evaluation of novel DFG-out allosteric kinase inhibitors. Available from: http://gradworks.proquest.com/33/38/3338842.html [last accessed 22 Apr 2016]
- Honndorf VS, Coudevylle N, Laufer S, et al. Angew Chem Int Ed 2008;47:3548–51
- Gavilan MD, Gomez-Vidal JA, Serrano FR. Anticancer activity of (1,2,3,5-tetrahydro-4,1-benzoxazepine-3-yl)-pyrimidines and -purines against the MCF-7 cell line: preliminary cDNA microarray studies. Bioorg Med Chem Lett 2008;18:1457–60
- Abdel-Maksoud MS, Kim MR, El-Gamal MI, et al. Design, synthesis, in vitro antiproliferative evaluation, and kinase 10 inhibitory effects of a new series of imidazo[2,1-b]thiazole derivatives. Eur J Med Chem 2015;95:453–63
- Abdel-Mohsen HT, Ragab FAF, Ramla MM, Diwani HIE. Novel benzimidazole-pyrimidine conjugates as potent antitumor agents. Eur J Med Chem 2010;45:2336–44
- Claudi F, Giorgioni GD, Stefano A, et al. Synthesis and pharmacological characterization of 2-(4-chloro-3-hydroxyphenyl)ethylamine and N,N-dialkyl derivatives as dopamine receptor ligands. J Med Chem 1992;35:4408–14
- Weinstock J, Gaitanopoulos D, Oh HJ, et al. Synthesis and dopaminergic activity of some halogenated mono- and dihydroxylated 2-aminotetralins. J Med Chem 1986;29:1615–27
- Lee J, Kim H, Yu H, et al. Discovery and initial SAR of pyrimidin-4-yl-1H-imidazole derivatives with antiproliferative activity against melanoma cell lines. Bioorg Med Chem Lett 2010;20:1573–7
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54
- Wickenden JA, Jin H, Johnson M, et al. Colorectal cancer cells with the BRAFV600E mutation are addicted to the ERK1/2 pathway for growth factor-independent survival and repression of BIM. Oncogene 2008;27:7150–61
- Subramanian RR, Yamakawa A. Combination therapy targeting Raf-1 and MEK causes apoptosis of HCT116 colon cancer cells. Int J Oncol 2012;41:1855–62
- Thiel A, Ristimaki A. Toward a molecular classification of colorectal cancer: the role of BRAF. Front Oncol 2013;3:281